

Summary
Deposition of intracellular tau fibrils has been the focus of research on the mechanisms of neurodegeneration in Alzheimer’s disease (AD) and related tauopathies. Here, we developed a new class of tau ligands, phenyl/pyridinyl-butadienyl-benzothiazoles/benzothiazoliums (PBBs), for visualizing diverse tau inclusions in brains of living patients with AD or non-AD tauopathies and animal models of these disorders. In vivo optical and positron emission tomographic (PET) imaging of a transgenic mouse model demonstrated sensitive detection of tau inclusions by PBBs. A pyridinated PBB, [11C]PBB3 was next applied in a clinical PET study, and its robust signal in the AD hippocampus wherein tau pathology is enriched contrasted strikingly with that of a senile plaque radioligand, [11C]Pittsburgh Compound-B ([11C]PIB). [11C]PBB3-PET data were also consistent with the spreading of tau pathology with AD progression. Furthermore, increased [11C]PBB3 signals were found in a corticobasal syndrome patient negative for [11C]PIB-PET.
INTRODUCTION
Hallmark pathologies of Alzheimer’s disease (AD) are extracellular senile plaques consisting of aggregated amyloid β peptide (Aβ) and intraneuronal neurofibrillary tangles (NFTs) composed of pathological tau fibrils, while similar tau lesions in neurons and glia are also characteristic of other neurodegenerative disorders such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) that are collectively referred to as tauopathies (Ballatore et al., 2007). The discovery of tau gene mutations in a familial form of tauopathy, known as frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), and subsequent studies of transgenic (Tg) mice expressing human tau with or without these mutations clearly implicate pathological tau in mechanisms of neurodegeneration in AD and related tauopathies (Ballatore et al., 2007). Thus, there is an urgent need for tau imaging techniques to complement Aβ amyloid imaging methods that now are widely used.
In vivo imaging modalities, as exemplified by positron emission tomography (PET) (Klunk et al., 2004; Small et al., 2006; Kudo et al., 2007; Maeda et al., 2007), optical scanning (Bacskai et al., 2003; Hintersteiner et al., 2005) and magnetic resonance imaging (MRI) (Higuchi et al., 2005), have enabled visualization of Aβ deposits in humans with AD and/or AD mouse models, and there has been a growing expectation that low-molecular-weight ligands for β-pleated sheet structures would also serve as molecular probes for tau amyloids. While the majority of plaque imaging agents used for clinical PET studies do not bind to tau lesions (Klunk et al., 2003), at least one radiolabeled β-sheet ligand, [18F]FDDNP, enables PET imaging of AD NFTs (Small et al., 2006). However, a relatively low contrast of in vitro autoradiographic and in vivo PET signals for [18F]FDDNP putatively reflecting tau lesions does not allow a simple visual inspection of images for the assessment of tau pathologies in living subjects (Small et al., 2006; Thompson et al., 2009). Thus, better tau radioligands with higher affinity for tau fibrils and/or less nonspecific binding to tissues are urgently needed to complement high-contrast senile plaque imaging agents, including widely studied [11C]Pittsburgh Compound-B ([11C]PIB) (Klunk et al., 2004) and United States Food and Drug Administration-approved [18F]florbetapir (Yang et al., 2012). In addition, [18F]FDDNP and several other candidate tau probes do not bind to tau inclusions in non-AD tauopathy brains without plaque deposition (Okamura et al., 2005), and therefore can be clinically characterized only in AD patients with comingled Aβ and tau amyloids. Hence, compounds that detect diverse tau aggregates, including tau inclusions in non-AD neurodegenerative diseases and tau Tg models, could be used to interrogate in vivo interactions between exogenous ligands and tau pathologies.
Here, we found that the lipophilicity of β-sheet ligands is associated with their selectivity for tau versus Aβ fibrils, and that the core dimensions of these chemicals are major determinants of their reactivity with a broad spectrum of tau aggregates in diverse tauopathies and mouse models of tau pathology. Building on these observations, we developed a novel series of fluorescent compounds capable of detecting diverse tau lesions using optical and PET imaging in living Tg mouse models of tauopathies. Finally, we identified a radiotracer that produced the highest contrast for tau inclusions in animal PET and was then used inexploratory in vivo imaging studies of AD patients, providing the first clear demonstration of signal intensification in tau-rich regions, in sharp distinction from [11C]PIB-PET data reflecting plaque deposition.
RESULTS
Identification of PBBs as ligands for diverse tau inclusions in human tauopathies
We screened an array of fluorescent chemicals capable of binding to β-sheet conformations (see Compounds subsection in Experimental Procedures). Fluorescence labeling with these compounds were examined in sections of AD brains bearing Aβ and tau amyloids (Figs. 1A and 2A) and non-AD tauopathy brains characterized by tau inclusions and few or no Aβ plaques (Fig. 2). Amyloid PET tracers currently used for human PET studies, PIB (Klunk et al., 2004) and BF-227 (Kudo et al., 2007), tightly bound to senile plaques, while they only weakly reacted with AD NFTs (Figs. 1A and S1). PET probes reported to selectively label tau aggregates, BF-158 (Okamura et al., 2005) and THK523 (Fodero-Tavoletti et al., 2011), detected AD NFTs (Fig. 2A and S1), but microscopically detectable fluorescence signals produced by FDDNP, which are presumed to bind to both Aβ and tau fibrils (Small et al., 2006), were consistent with dense cores of classic plaques and distinct from tau lesions (Figs. 2A and S1). While all the above-mentioned PET ligands were not reactive with tau inclusions in non-AD tauopathies, such as Pick bodies in Pick’s disease (Figs. 2A and S1) and neuronal and glial fibrillary lesions in PSP and CBD (data not shown), these pathologies were intensely labeled with a widely used amyloid dye, thioflavin-S, and a derivative of another classic amyloid dye Congo red, (E, E)-1-fluoro-2,5-bis(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB) (Higuchi et al., 2005; Maeda et al., 2007) (Figs. 1, 2A and S1), although these chemicals may not undergo efficient transfer through the blood-brain barrier (BBB) (Zhuang et al., 2001). Since compounds possessing a π-electron-conjugated backbone longer than 13Å exhibited affinities for pathological inclusions in a broad range of tauopathies, we examined binding of additional chemicals with a variety of structural dimensions to tau aggregates, and found that affinity for non-AD tau inclusions could be attributed to a core structure with a specific extent ranging from 13 to 19Å (Fig. S1). Based on this view and the known fact that chemicals with a flat and slender backbone could pass through and attach to channel-like accesses in β-pleated sheets (Krebs et al., 2005), we developed a new class of compounds, phenyl/pyridinyl-butadienyl-benzothiazoles/benzothiazoliums (PBBs), by stretching the core structure of a prototypical fluorescent amyloid dye, thioflavin-T, with two C=C double bond inserts between aniline (or aminopyridine) and benzothiazole (or benzothiazolium) groups (Fig. 1B).
Figure 1. Design and characterization of PBB compounds as potential imaging agents for tauopathies.

(A) Confocal fluorescence images of frontal cortex sections from an AD patient. Following fluorescence labeling (pseudocolors are converted to green) with PIB (top row) and FSB (middle row), the samples were immunostained with an antibody against AβN3(pE) (red in the right column). PIB intensely labeled Aβ plaques (white arrowheads), but did not clearly label NFTs (arrows). By contrast, NFTs and neuropil threads were intensely labeled by FSB, while the staining of diffuse plaques was negligible. A section was also doubly immunolabeled (bottom row) with AT8 (green) and anti-AβN3(pE) antibodies (red in the right panel), to demonstrate the abundance of tau and Aβ amyloids in this area. Yellow arrowheads indicate tau-positive dystrophic neurites associated with senile plaques. (B) Structures of PBBs. Neutral benzothiazoles (PBB1–4) are newly synthesized chemicals, and a charged benzothiazolium, PBB5, is identical to a commercially available near-infrared laser dye. (C) Confocal fluorescence images of PBBs (pseudocolors are converted to green) and AβN3(pE) (red in the right column) staining in sections adjacent to those displayed in A. The intensity of plaque staining (arrowheads) relative to that of NFTs (arrows) was positively associated with the lipophilicity of PBBs. As compared with PBB1 (top row) staining, labeling of diffuse plaques with PBB3 (middle row) was substantially attenuated. PBB5 was nearly unreactive with diffuse plaques (bottom row), and subsequent double immunofluorescence staining of the same section (bottom row in C) illustrated good agreement of PBB5 labeling with the distribution of AT8-positive NFTs. Scale bar: 50 μm (A, C).
Figure 2. Binding of tau ligands to tau lesions in AD and non-AD tauopathy brains.
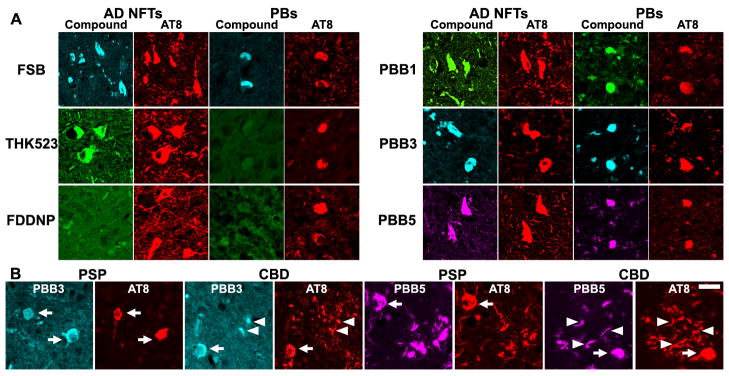
(A) Double fluorescence staining of AD NFTs and Pick bodies (PBs) in Pick’s disease with PBBs, other tau ligands and anti-phospho-tau antibody (AT8). FSB and PBBs sensitively captured AD NFTs and PBs. AD NFTs were labeled with THK523. Meanwhile, PBs were not visualized by these compounds. NFTs and PBs were barely recognizable by using FDDNP. (B) Double fluorescence staining of neuronal tau inclusions (arrows) in PSP and CBD and putative astrocytic plaques (arrowheads) in CBD. A substantial portion of tau fibrils in neurons were captured by PBB3 and PBB5, but a much smaller subset of phosphorylated tau aggregates in astrocytic plaques were labeled with these compounds. Scale bar: 20 μm (A, B).
All PBB compounds intensely labeled NFTs, neuropil threads and plaque neurites in AD brains (Fig. 1C). Interestingly, the affinity of these PBBs for Aβ plaques lacking dense cores was positively correlated with their lipophilicity (Fig. 1C), and thereby three potential probes with relatively low logP (log of the octanol/water partition coefficient) values, including PBB3, 2-[4-(4-methylaminophenyl)-1,3-butadienyl]-benzothiazol-5,6-diol (PBB4) and PBB5 (structurally identical to Styryl 7, CAS registry number 114720-33-1), appeared suitable for visualizing tau pathologies in living organisms with reasonable selectivity. High affinity of PBBs for tau lesionswas further demonstrated by fluorometric analyses using Aβ and tau filaments assembled in a test tube (Table S1; experimental procedures are given as Supplemental Experimental Procedures online), but the most and least lipophilic PBB members displayed similar selectivity for in vitro tau versus Aβ pathologies, implying a methodological limitation in screening chemicals for tau-selective ligands based on binding to synthetic peptides and recombinant proteins. PBBs and FSB were also shown to label tau inclusions in non-AD tauopathies, such as Pick’s disease (Figs. 2A and S1), PSP and CBD (Fig. 2B), all of which were immunodetected by an antibody specific for phosphorylated tau proteins (AT8).
In vitro and ex vivo fluorescence imaging of tau lesions in tau Tg mice by PBBs
To obtain in vivo evidence of direct interaction between PBBs and tau lesions, we employed Tg mice expressing a single human four-repeat tau isoform with the P301S FTDP-17 mutation (PS19 line, see Fig. S2 for neuropathological features of this Tg strain) (Yoshiyama et al., 2007). Similar to the findings in non-AD tauopathy brains, NFT-like inclusions in the brain stem and spinal cord of PS19 mice were clearly recognized by PBBs (Figs. 3A and S1). We then performed ex vivo labeling of tau lesions in PS19 mice with intravenously administered, fluorescently labeled PBBs. Brains and spinal cords were removed 60 min after tracer injection, and fluorescence microscopy revealed an intense accumulation of these compounds in fibrillary tau inclusions abundantly seen throughout the sections by staining with thioflavin-S, FSB and AT8 (Fig. 3B). On the other hand, no overt in vitro (Fig. 3A) or ex vivo (data not shown) fluorescence of these ligands was noted in the corresponding regions of non-Tg wild-type (WT) mice. Consistent with these observations, two-photon laser scanning fluorescence microscopy of ex vivo samples demonstrated somatic and neuritic staining of a subset of tangle-bearing neurons with intravenously injected 2-[4-(4-methylaminophenyl)-1,3-butadienyl]-benzothiazol-6-ol (PBB2) and PBB4 in unsliced spinal cord blocks from PS19 mice (Fig. 3B).
Figure 3. In vitro and ex vivo labeling of NFTs in PS19 mice with PBB compounds.
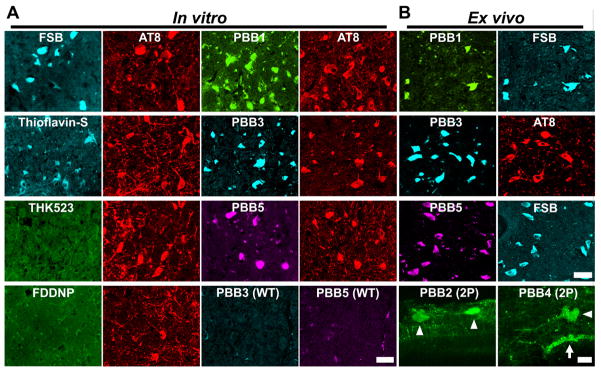
(A) Double fluorescence staining of intraneuronal tau aggregates in postmortem brain stem slices of a 12-month-old PS19 mouse with PBB, other amyloid ligands and anti-phospho-tau antibody (AT8). (B) Binding of intravenously administered PBBs (0.1 mg/kg PBB5 and 1 mg/kg PBB1 and PBB3) to NFTs in PS19 mice at 10–12 months of age. The tissues were sampled at 60 min after tracer administration. The brain stem (top row) and spinal cord (second and third rows from the top) sections abundantly contained neurons showing strong fluorescence (left), and subsequent staining with FSB or AT8 (right) indicated that these cells were laden with tau amyloid fibrils (right). Putative intraneuronal tau inclusions in unsectioned spinal cords (arrowheads in the bottom row) removed from PS19 mice at 60 min after intravenous injection of PBB2 and PBB4 were also clearly visible by using a two-photon (2P) fluorescence microscopic system. Arrow in the bottom row indicates a cluster of autofluorescence signals from blood cells. Scale bars: 25 μm (A); 30 μm (top to third rows in B); 20 μm (bottom row in B).
In vivo macroscopic and mesoscopic optical detection of fibrillar tau pathologies in a mouse model using PBB5
We next characterized PBBs with the use of in vivo fluorescence imaging modalities, which permitted a quick assessment of candidate chemicals without the need for radiolabeling. Because PBB5 is fluorescent, with peak excitation and emission wavelengths in a near-infrared range (Table S1), this compound is applicable to in vivo optical imaging of tau deposits in laboratory animals. To examine this possibility, fluorescence images were obtained from living mice over a time course following intravenous PBB5 injections using a small animal-dedicated system permitting the intravital observation of fluorescence signals at magnifications varying between macroscopic and microscopic levels. Tail vein administration of PBB5 in PS19 mice revealed strong fluorescence relative to non-Tg WT mice in the CNS above the slit between the base of the skull and first vertebra, through the skin and connective tissues overlaying the cisterna magna (Fig. S3A–D), suggesting a concentration of this tracer in the PS19 spinal cord. In line with this in vivo observation, the hindbrain and spinal cord of PS19 mice, which were dissected out at 2 hours after the injection of PBB5, exhibited increased retention of this compound compared to non-Tg WT mice (Fig. S3E–G).
In vivo optical imaging of tau Tg mice was subsequently performed using a device equipped with a pulsed diode laser and a photomultiplier tube to detect deep signals through the skull. Elevated levels of fluorescence intensity were found in homogenized brain stem samples collected from PS19 mice at 20 hours after the intravenous tracer administration (Fig. S4A), indicating a long-lasting in vivo binding of PBB5 to tau fibrils. To support the ex vivo evidence, fluorescence intensity was noninvasively analyzed in living PS19 and non-Tg WT mice treated with PBB5. The mice, with their heads shaved in advance, were prescanned, and autofluorescence signals were detected at a relatively high level in an area corresponding to the frontal forebrain. Using these baseline signals as landmarks, regions of interest (ROIs) were defined in the frontal cortex, brain stem and spinal cord (Fig. 4A). The near-infrared fluorescence was notably increased immediately after the intravenous injection of PBB5 (Fig. S4C), and the fluorescence in the brain stem and spinal cord ROIs of PS19 mice much exceeded that in WT mice at 30 min (Fig. 4B). Fluorescence intensity in the frontal cortex ROI, normalized on the basis of integration time and laser power, was lower in PS19 mice than in WT mice over 120 min after tracer injection (Fig. S4B), which may reflect impaired CNS delivery of the tracer in Tg mice due to degenerative changes (see Fig. S4C–L for details), and thereafter this became almost equivalent between the two genotypes (Fig. S4B). Meanwhile, persistent retention of the signals in the brain stem and spinal cord ROIs of PS19 mice was observed beyond 240 min (Figs. 4B and S4B). A more quantitative index comparable among different mice was determined by calculating the target-to-frontal-cortex ratio of fluorescence intensity, and was shown to increase over time particularly in PS19 mice (Fig. 4C, D). This ratio was significantly greater in PS19 mice than in WT mice at 240 min (Fig. 4E), beyond which the difference between the two lines of mice became nearly constant (Fig. 4C, D). The intensity ratio of the spinal cord ROI to the frontal cortex in PS19 mice at 240 min was also significantly correlated with the abundance of NFTs stained with FSB (Fig. 4F), but such correlations were not statistically significant in the brain stem (Fig. 4F), implying limitations of the intensitometry in some brain regions below the cerebellum and fourth ventricle.
Figure 4. Noninvasive near-infrared imaging of tau pathology in living tau Tg mice using pulsed laser optics and PBB5.
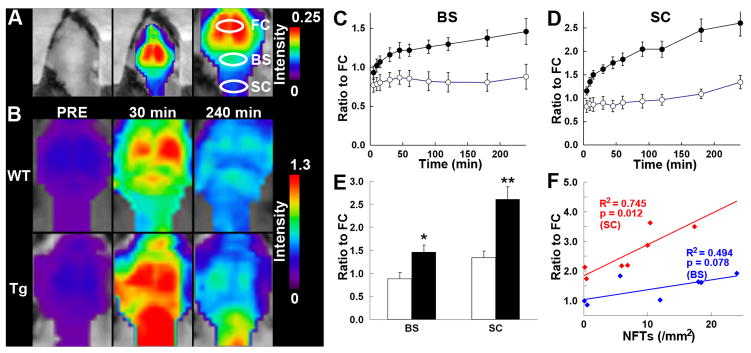
(A) Baseline autofluorescence signals (middle) are overlaid on the visible background image of a shaven non-Tg WT mouse head (left). Ellipsoidal ROIs are defined above the frontal cortex (FC), brain stem (BS) and cervical spinal cord (SC) guided by a relatively intense emission from the FC region (right). (B) Fluorescence intensity maps in 12-month-old WT (top) and PS19 (Tg; bottom) mice before and at 30 and 240 min after the intravenous administration of PBB5 (0.1 mg/kg). The intensity maps (A, B) are normalized by the FC ROI value at 30 min after tracer injection. Long-lasting retention of the tracer was noted in the BS and SC ROIs of the Tg mouse. C, D, Target-to-FC ratios of fluorescence intensity in the BS (C) and SC (D) ROIs over the image acquisition time in the WT (open circles; n = 7) and PS19 (closed circles; n = 7) mice. There were significant main effects of time, region and genotype in 2-way, repeated-measures ANOVA (time, F(11, 132) = 17.6, p < 0.001; region, F(1, 12) = 29.9, p < 0.001; genotype, F(1, 12) = 23.6, p < 0.001). (E) Target-to-FC ratios in the BS and SC ROIs of the WT (open columns) and tau Tg (closed columns) mice at 240 min after tracer injection. *, p < 0.05; **, p < 0.01; 2-way repeated-measures ANOVA with Bonferroni’s post hoc analysis. (F) Scatterplots of target-to-FC ratios at 240 min versus the number of FSB-positive NFTsper unit area of postmortem 20 -μm tissue slices in BS (blue symbols) and SC (red symbols) ROIs of tau Tg mice. Solid lines represent regressions; p values were determined by t-test. Vertical bars in the graphs represent SEs.
Intravital imaging of individual tau inclusions by PBB3 and two-photon laser scanning fluorescence microscopy
Two-photon excitation microscopy, which enables optical sectioning, potentially up to 1 mm deep, in living tissues, could be utilized to visually demonstrate transfer of a fluorescent probe from the plasma compartment into the cytoplasm of CNS neurons and its binding to intraneuronal tau inclusions. We therefore captured fluorescence signals from intravenously administered PBB3 by in vivo two-photon laser scanning microscopic imaging of the spinal cord of laminectomized PS19 mice. Within 3 sec of PBB3 injection, green fluorescence signals emerged in blood vessels pre-labeled with red with intraperitoneal treatment using Sulforhodamine 101, and subsequently diffused from the vasculatures to the spinal cord parenchyma over the next few minutes (Fig. 5A–F). These diffuse signals declined thereafter due to the clearance of PBB3 from the tissue, while intense labeling of putative tau inclusions with green fluorescence appeared in a subpopulation of large cells morphologically identified as neurons at 3–5 min after PBB3 injection (Fig. 5G, H). These intracellular PBB3 fluorescent signals were not found in the spinal cord of WT mice (Fig. 5I). As the BBB of the brain and spinal cord are presumed to be identical, the two-photon microscopic data obtained here provide compelling evidence that PBB3 rapidly transits the BBB and neuronal plasma membranes, where it binds to intraneuronal tau inclusions. Accumulation of injected PBB3 in AT8-positive, NFT-like lesions of Tg mice was postmortemly confirmed by ex vivo microscopy (Fig. 5J, K).
Figure 5. Real-time two-photon laser scanning images of PBB3 diffusing from vessels, binding to intraneuronal tau inclusions, and clearing from spinal cord.

(A–H) A maximum projection of fluorescence in a 3-D volume of the spinal cord of a living PS19 mouse at 12 months of age before (A) and at various time points after (B–H) intravenous administration of PBB3 (1 mg/kg). Blood vessels were labeled with Sulforhodamine 101 (red) intraperitoneally injected at 15 min before PBB3 administration. Green fluorescence indicates a rapid transfer of PBB3 from the plasma to tissue parenchyma (B–E) and subsequent washout from the tissue (F). Background PBB3 signals were further attenuated beyond 300 sec, while somatodendritic labeling by this compound was observed in a subset of neurons (arrowheads in G, H). (I) Fluorescence image of WT spinal cord at 300 sec after PBB3 injection, demonstrating no overt retention of the tracer in the tissue. (J, K) Ex vivo microscopy for a brain stem section of the same Tg mouse. Tissues were obtained at 60 min after PBB3 injection. Signals of intravenously administered PBB3 (J) overlapped with AT8 immunoreactivity (K). Scale bars: 50 μm (A–F); 25 μm (G–I), 25 μm (J, K).
Autoradiographic and PET imaging of tau lesions in PS19 mice by radiolabeled PBBs
We investigated the kinetic properties of PBBs by HPLC analyses of plasma and brain samples collected from non-Tg WT mice treated with these ligands. Following intravenous administration, PBB5 was rapidly converted into a major metabolite, which at 5 min was found at high levels in both plasma and brain extracts. Subsequent liquid chromatography-mass spectrometry (LC-MS) assays suggested that the major metabolite was likely a reduced, electrically neutralized derivative of PBB5 (Fig. S5A, B). Besides trans-ventricular uptake of unmetabolized PBB5 as implied above, this uncharged form incapable of emitting near-infrared light could readily penetrate the BBB as well as cell membranes, and thereafter could be reoxidized into its original form, thereby enabling it to bind to tau fibrils, and particularly at sites exposed to oxidative stress in pathological conditions. In addition, PBB4 was promptly converted to metabolites capable of entering the brain. Finally, studies of PBB2 and PBB3 showed that they exhibited reasonable biostability and sufficient entry into and clearance from the brain. Indeed, HPLC assays demonstrated that fractions of unmetabolized PBB2 and PBB3 in mouse plasma were 23.5% and 16.3%, respectively, at 3 min after intravenous administration, and were 4.6% and 2.8%, respectively, at 30 min. There were also no metabolites of PBB2 and PBB3 detectable in the mouse brain at 3 and 30 min.
We then radiolabeled PBB2 and PBB3 with 11C to conduct autoradiographic and PET assays using PS19 mice. In vitro autoradiography using frozen tissue sections showed binding of these radioligands to the brain stem of PS19 mice and neocortex of AD patients (Fig. 6A). As expected from their lipophilicities, [11C]PBB3 yielded high-contrast signals with less nonspecific labeling of myelin-rich white matter than did [11C]PBB2, and the accumulation of [11C]PBB3 in pathological regions was nearly completely abolished by the addition of nonradioactive compounds. Similarly, ex vivo autoradiographic studies demonstrated that intravenously administered [11C]PBB3 selectively labeled the brain stem and spinal cord of PS19 mice harboring neuronal tau inclusions, while tau-associated [11C]PBB2 radiosignals were less overt due to a considerable level of nonspecific background (Figs. 6B and S6C–F). Finally, in vivo visualization of tau lesions in PS19 mouse brains was enabled by a microPET system using these two tracers (Figs. 6C and S6A, B). Following intravenous injection, [11C]PBB3 rapidly crossed the BBB, and unbound and nonspecifically bound tracers were promptly washed out from the brain with a half life of ~10 min (left panel in Fig. 6E). The retention of [11C]PBB3 signals in the brain stem of 12-month-old PS19 mice lasted over the imaging time (90 min), producing a pronounced difference from that in age-matched non-Tg WT mice (left panel in Fig. 6E). By selecting the striatum as a reference region lacking tau deposits, the target-to-reference ratio was estimated for the brain stem, with the value in PS19 mice peaking at around 70 min, contrasting with its continuous decrease over 60 min in WT mice (right panel in Fig. 6E). The mean ratio at 45–90 min was increased by 40% in 12-month-old PS19 mice as compared with age-matched WT mice (p < 0.01 by t-test). The agreement between localizations of PET signals and tau inclusions in PS19 mice was proven by postmortem FSB staining of brain sections from scanned mice (Fig. 6D). Significantly, the mean target-to-reference ratio in the brain stem quantified by PET correlated closely with the number of FSB-positive inclusions per brain section in the same region of the postmortem sample (p < 0.001 by t-test; data not shown). [11C]PBB2 exhibited slower clearance from the brain and higher nonspecific retention in myelin-rich regions than [11C]PBB3 (Fig. S6G), resulting in insufficient contrast of tau-bound tracers in the brain stem of PS19 mice and a small difference in the target-to-reference ratio of radioactivities between PS19 and WT mice (8% at 45–90 min; p < 0.05 by t-test; Fig. S6H) relative to those achieved with [11C]PBB3.
Figure 6. PET and autoradiographic detection of tau pathologies in PS19 mice using [11C]PBB2 and [11C]PBB3.

(A) In vitro autoradiograms of PS19 and non-Tg WT hindbrains (coronal sections) and AD frontal cortex. Fibrillar aggregates in the mouse brain stem and AD gray matter produced intense radiolabeling with both tracers, but nonspecific background signals were also observed at a considerably high level with the use of [11C]PBB2. Binding of [11C]PBB3 was profoundly abolished by the addition of nonradioactive PBB3 (10 μM). (B) Autoradiographic labeling with intravenously injected [11C]PBB2 and [11C]PBB3 in PS19 (Tg) and WT mice. The brains were removed at 45 min after injection, and were cut into sagittal slices. The autoradiographic section of PS19 brain was also stained with FSB. Arrows indicate the brain stem containing numerous tau inclusions displayed at intermediate and high magnifications. (C) Sagittal and coronal PET images generated by averaging dynamic scan data at 60–90 min after intravenous administration of [11C]PBB3. The images are overlaid on the MRI template (images of the template alone are presented at the bottom). Arrows and asterisks indicate the brain stem and striatum, respectively, and arrowhead denotes intense radiolabeling in the medial brain stem of the PS19 mouse. (D) FSB staining of PS19 mouse brain shown in c. Sagittal (left) and coronal (middle) images and a high-power view of fibrillar inclusions (right) are displayed. Corresponding to high-level retention of [11C]PBB3 in PET scans, abundant FSB-positive lesions were found in the medial brain stem (arrow and arrowhead). (E) Time-radioactivity curves (left) in the striatum (ST) and brain stem (BS) and BS-to-ST ratio of radioactivity (right) over the imaging time in PS19 (Tg; red symbols) and WT (black symbols) mice (n = 5 each). Vertical bars in the graphs denote SEs. Scale bars: 1 cm (A, top, middle and bottom-left panels in B); 1 cm (C, left and middle panels in D); 100 μm (bottom-middle panel in B); 100 μm (bottom-right panel in B, right panel in D).
As radiolabeling at the dimethylamino group in PBB5 with 11C was unsuccessful, 11C-methylation of a hydroxyl derivative of this compound was performed, leading to the production of [11C]methoxy-PBB5 ([11C]mPBB5; Fig. S5C). PET images demonstrated complex pharmacokinetics of [11C]mPBB5 (Fig. S5D, E), and the difference in the specific radioligand binding between Tg and WT mice was small relative to the [11C]PBB3-PET data (Fig. S5F). After taking all of these findings into consideration, [11C]PBB3 was selected as the most suitable ligand for in vivo PET imaging of tau pathology in tau Tg nice and human subjects.
It is noteworthy that the hippocampus of many PS19 mice was devoid of overt [11C]PBB3 retention (Fig. 6C), although a pronounced hippocampal atrophy was noted in these animals. This finding is in agreement with the well-known neuropathological features of PS19 mice in the hippocampus, since the accumulation of AT8-positive phosphorylated tau inclusions results in the degeneration of the affected hippocampal neurons prior to or immediately after NFT formation, followed by the clearance of their preNFTs or NFTs that are externalized into the interstitial CNS compartment (Fig. S2). To explore the feasibility of our imaging agents in studies with other tauopathy model mice, we also performed fluorescence labeling with PBBs for brain sections generated from rTg4510 mice (Santacruz et al., 2005, and Supplemental Experimental Procedures). As reported elsewhere (Santacruz et al., 2005), these mice developed numerous thioflavin-S-positive neuronal tau inclusions in the neocortex and hippocampus, and reactivity of these lesions with PBBs was demonstrated by in vitro and ex vivo fluorescence imaging (Fig. S7).
Detection of tau pathologies in living brains of AD patients by comparative PET imaging with [11C]PBB3 and [11C]PIB
In order to compare the bindings of [11C]PBB3 and [11C]PIB to tau-rich regions in the human brain, in vitro autoradiography was carried out with sections of AD and control hippocampus. A notable difference in labeling between these two radioligands was observed in the CA1 sector and subiculum of the AD hippocampus, where fibrillar tau aggregates predominantly localized to NFTs and neuropil threads (Fig. 7A).
Figure 7. Accumulation of [11C]PBB3 in the hippocampal formation of AD patients revealed by in vitro autoradiography and in vivo PET.

(A) Autoradiographic labeling of adjacent brain sections from an AD patient with 10 nM of [11C]PBB3 (left) and [11C]PIB (middle). The slices contain the hippocampus (Hi), parahippocampal gyrus (PH), fusiform gyrus (FF) and white matter (asterisks). Total binding (top) of [11C]PBB3 and [11C]PIB was markedly abolished (bottom) by addition of nonradioactive PBB5 (100 μM) and thioflavin-S (10 μM), respectively, except for the nonspecific (NS) labeling of white matter with [11C]PIB. The hippocampal CA1 sector and subiculum displayed intense [11C]PBB3 signals without noticeable binding of [11C]PIB, and binding of [11C]PBB3 in cortical areas flanking the collateral sulcus (identified by a red dot) and hippocampal CA2 sector (arrows) was also abundant relative to that of [11C]PIB. FSB staining of amyloid fibrils in the sections used for autoradiography indicated the predominance of NFTs and diffuse plaques in the hippocampal subiculum (Sub) and fusiform gyrus (FF), respectively (right panels), supporting the strong reactivity of [11C]PBB3 with AD NFTs. (B) MRI (left) and PET imaging with [11C]PBB3 (middle) and [11C]PIB (right) performed in the same AD (top) and normal control (NC; bottom) subjects. Coronal images containing the hippocampal formation (arrowheads) are displayed. [11C]PBB3- and [11C]PIB-PET images were generated by estimating SUVRs at 30–70 min and 50–70 min after radiotracer injection, respectively, and were superimposed on individual MRI data. In the hippocampal formation, prominently increased retention of [11C]PBB3 in the AD patient was in sharp contrast to the modest or negligible changes in [11C]PIB binding as compared with NC. Scale ranges for SUVRs were 0.75–1.50 ([11C]PBB3) and 0.75–3.00 ([11C]PIB).
We subsequently conducted an exploratory clinical PET study for patients with probable AD (n = 3) and age-matched cognitively normal control (NC) subjects (n = 3). All AD patients exhibited a marked increase in the retention of [11C]PIB in plaque-rich areas, and all NC were negative for this PET assay. These subjects then received a [11C]PBB3-PET scan, and the [11C]PIB and [11C]PBB3 images were compared in the same individuals. Intravenously injected [11C]PBB3 was delivered to the brain tissue despite its relatively rapid metabolism in humans (Fig. 9A, B). Unlike [11C]PIB, [11C]PBB3 showed minimal nonspecific binding to white matter and other anatomical structures with high myelin content, although it accumulated in dural venous sinuses in control and AD brains (Figs. 7B, 8, 9B). Time courses of regional radioactivity (Fig. 9C, D and S8A, B) and the standardized uptake value ratio (SUVR) to the cerebellum (Fig. S8C, D) demonstrated accumulation of [11C]PBB3 in several brain regions of AD patients as compared to controls (definition of these VOIs is indicated in Fig. S8E). In agreement with autoradiographic findings, binding of [11C]PBB3 to the medial temporal region including the hippocampus contrasted strikingly with the low-level retention of [11C]PIB in this area (Fig. 7B). There was a slight increase in the retention of [11C]PBB3 primarily in the medial temporal region of a control subject with a loss of several points in Mini-Mental State Examination (MMSE) (Subject 3 in Fig. 8), appearing similar to the tau pathology at Braak stage III–IV or earlier (Braak et al., 1991), distinct from the lack of enhanced [11C]PIB signals. Indeed, mild increase of medial temporal SUVR (Fig. 9E) contrasted with unremarkable change in lateral temporal and frontal SUVRs in this subject (Fig. 9G, H). Signals of [11C]PBB3 were also intense mainly in the limbic region of a subject with early AD (Subject 4 in Fig. 8), but profound and moderate increases of SUVRs were also observed in the lateral temporal and frontal cortices, respectively, of this case (Fig. 9G, H), resembling the localization of tau deposits at Braak stage V–VI (Braak et al., 1991). With the further cognitive decline as scored by MMSE (Subjects 5 and 6 in Fig. 8), additional increase in the retention of [11C]PBB3 was found in the medial temporal region, precuneus and frontal cortex (Fig. 9E, F, H). Meanwhile, a substantial decline of [11C]PBB3 binding was noted in the lateral temporal cortex of Subject 6 (Fig. 8, 9G). The SUVRs in the medial temporal region, precuneus and frontal cortex were consequently well correlated with the decline of MMSE scores (Fig. 9E, F, H). In distinction from [11C]PBB3-PET data, there was no overt association between the binding of [11C]PIB and disease severity in AD patients (Fig. 8), consistent with previous observations. These data support the potential utility of [11C]PBB3 for clarifying correlations between the distribution of tau deposition and the symptomatic progression of AD.
Figure 9. Pharmacokinetic profiles of [11C]PBB3 administered to humans and PET images of a patient clinically diagnosed as having corticobasal syndrome.

(A) Time course of unmetabolized [11C]PBB3 fraction in plasma following intravenous radiotracer injection. The plot was generated by averaging data from 6 individuals. (B) Time-radioactivity curves in different brain regions of cognitively normal control subjects over 70 min after intravenous injection of [11C]PBB3. Data were generated by averaging values in two individuals, and are presented as standard uptake values (SUVs). (C, D) Comparisons of time-radioactivity curves in the medial temporal region (C) and precuneus (D) of normal controls (black symbols and lines; n = 3) and AD patients (red symbols and lines; n = 3). (E–H) Scatterplots illustrating correlation of SUVRs with MMSE scores in the medial temporal region (E), precuneus (F) and lateral temporal (G) and frontal (H) cortices. Numbers beside symbols denote subject ID as indicated in Fig. 8. Coefficients of determination (r2) and p values by t-test are displayed in graphs. (I) [11C]PBB3- and [11C]PIB-PET images in a subject with clinical diagnosis of corticobasal syndrome. Images were generated as in Figs. 7 and 8. Accumulation of [11C]PBB3 was noticeable in the basal ganglia (red arrowheads) with right-side dominance and an area containing the thalamus and midbrain (yellow arrowhead).
Figure 8. Orthogonal [11C]PBB3-PET images in all human subjects examined in the present exploratory clinical study.

Data are displayed as parametric maps for SUVR. The [11C]PBB3 binding to the hippocampal formation (arrowheads) was increased consistently in AD patients in contrast to minimum radiotracer retention in normal control (NC) subjects with MMSE score of 29–30 points (Subjects 1 and 2). Another NC subject with MMSE score of 27 points (Subject 3) was negative for [11C]PIB-PET, but exhibited slight accumulation of radiotracer signals primarily around the hippocampus, resembling fibrillar tau deposition at Braak stage III–IV or earlier. Sagittal slices around the midline illustrate that radioligand signals were the most intense in the limbic system but began to expand to the neocortex in a patient with the mildest AD (Subject 4), in agreement with the tau pathology at Braak stage V–VI, and was further intensified in most neocortical areas, corresponding to Braak stage VI, apparently as a function of the disease severity assessed by MMSE (Subjects 5 and 6). The AD patient with the lowest MMSE score (Subject 6) displayed less profound increase of [11C]PBB3 retention in the lateral temporal and parietal cortices than did the other two AD cases, and this is attributable to marked cortical atrophy in this individual and/or toxic loss of tau-bearing neurons in these brain areas at an advanced pathological stage. In contrast to the spatial profiles of [11C]PBB3 binding, the distribution of [11C]PIB signals appeared unchanged among AD subjects.
As in vitro fluorescence staining indicated that PBB3 was reactive with not only tau lesions but also several types of senile plaques, particularly dense core plaques, density of binding sites and affinity of [11C]PBB3 for these sites were quantified by autoradiographic binding assays with hippocampal and neocortical sections of AD brains enriched with NFTs and senile plaques, respectively. These analyses demonstrated that specific radioligand binding sites were primarily constituted by high-affinity, low-capacity binding components in NFT-rich regions and low-affinity, high-capacity binding components in plaque-rich regions (Figs S9A, B). A subsequent simulation for radioligand binding in an area containing these two types of binding sites at a ratio of 1:1 indicated that the selectivity of [11C]PBB3 for NFTs versus plaques may be inversely associated with concentration of free radioligands (Fig. S9C). In a range of free concentration in the brain achievable at a pseudoequilibrium state in human PET imaging (< 0.2 nM), [11C]PBB3 is presumed to preferentially bind to tau lesions relative to in vitro autoradiographic (~1 nM) and fluorescence (> 100 nM) labeling.
We also estimated contribution of [11C]PBB3 bound to dense core plaques to total radiosignals in the neocortical gray matter of AD patients, by conducting autoradiography and FSB histochemistry for the same sections. Radiolabeling associated with dense cored plaques accounted for less than 1% and 3% of total gray matter signals in the temporal cortex and precuneus, respectively (Fig. S9D–H). Moreover, fluorescence labeling of adjacent sections with PBB3 demonstrated that approximately 2% and 5% of total gray matter fluorescence signals were attributable to PBB3 bound to dense core plaques in the temporal cortex and precuneus, respectively. Hence, dense cored plaques were conceived to be rather minor sources of binding sites for [11C]PBB3.
Finally, PET scans with [11C]PBB3 and [11C]PIB were conducted for a subject clinically diagnosed as having corticobasal syndrome. Retention of [11C]PIB stayed at a control level, but notable accumulation of [11C]PBB3 was observed in the neocortex and subcortical structures (Fig. 9I), providing the first evidence for in vivo detection of tau lesions in plaque-negative tauopathies. Interestingly, right-side dominant [11C]PBB3-PET signals in the basal ganglia was consistent with laterality of atrophy in this area (Fig. S8F). These findings may also be associated with right-side dominant decrease in cerebral blood flow and left-side dominant motor signs in this patient.
DISCUSSION
Here, we report our efforts to develop BBB-penetrant ligands that are capable of binding to and visualizing intracellular tau aggregates in AD and non-AD tauopathies. These compounds may accordingly be useful for the differential diagnosis of neurological conditions in elderly subjects on the basis of the distribution of tau lesions, thereby opening up novel avenues for research toward elucidating mechanisms of tau-mediated neurodegeneration as well as tau-focused biomarkers and therapies.
Despite numerous efforts to develop imaging ligands to visualize tau pathologies in the brains of patients with AD and related tauopathies, the urgent need for these tau biomarkers remains largely unmet. To address this significant challenge, we also took advantage of a multimodal imaging system, which facilitates a quick and label-free validation of candidate compounds in terms of their transfer to the brain and retention in tau-rich regions. In addition, subcellular-resolution imaging optics exemplified by two-photon laser scanning microscopy provided proof of the rapid transfer of intravenously administered potential tau pathology imaging agents from plasma to the CNS extracellular matrix and subsequently to the cytoplasm of neurons where they can bind to intracellular tau inclusions. Based on these encouraging preliminary data using non-labeled compounds, a subset of these compounds was radiolabeled for use in PET imaging of Tg mice that model tau pathology, and a radioligand yielding the best visualization of tau lesions in these Tg mice was selected for further testing in human AD patients and NC subjects as well as a patients with probable CBD. This stepwise strategy enabled us to identify and advance the most promising PET probe for the visualization and quantitative assessment of tau pathology in the CNS of living human subjects. It is also of great interest that another research group has recently reported development of 18F-labeled PET ligands for tau lesions mostly through assessments of binding to brain tissues but not recombinant tau assemblies (Zhang et al., 2012; Chien et al., 2013), as in the present approach. These radioligands have been implied to produce considerably high contrasts for tau pathologies in living AD brains, and relatively long radioactive half-life of 18F would enable delivery of radioligands from a radiosynthesis sites to multiple PET facilities. [11C]PBB3 has distinct advantages over these compounds, as exemplified by affinity for diverse tau lesions including Tg mouse tau aggregates, applicability to multimodal imaging and induction of smaller radioactive exposure than 18F-labeled ligands.
In the present work, we clinically validated the performance of [11C]PBB3 as a tau imaging agent by comparing its distribution with that of [11C]PIB in AD brains. Tau deposits in patients with moderate or severe AD are thought to be distributed extensively in the neocortical and limbic regions (classified as Braak stage V – VI) (Braak et al., 1991), thereby resembling localization of senile plaques except for the predominance of tau aggregates in the hippocampal formation. This rationalizes the use of radioactivity in the medial temporal area as an index to validate an imaging probe for tau pathology versus Aβ deposits in AD patients from prodromal to advanced stages. Furthermore, our preliminary data suggest that [11C]PBB3 may be capable of capturing the temporospatial spreading of neurofibrillary tau pathologies from the limbic system (Braak stage III–IV or earlier) to neocortical areas (Braak stage V – VI) with the progression of AD (Fig. 8). A considerable subset of tau lesions at Braak stage I–II are composed of phosphorylated tau deposits barely reactive with thioflavin-S (i. e. pretangles), and NFTs are relatively low in number and are confined to the transentorhinal cortex (Braak et al., 1991; 2011). Therefore, detection of these early tau pathologies would be more difficult. Our next stage clinical study with expanded sample size and wider range of MMSE scores is currently ongoing to pursue tau accumulation in normal controls and subjects with mild cognitive impairments and AD at diverse stages, and will bring more compelling insights into the significance of tau PET imaging in early diagnosis and prediction of AD. In addition, alterations of [11C]PBB3 retention were indicated in the transition from mild to moderate AD. Loss of PET signals in the lateral temporal cortex of a patient with moderate AD (Subject 6 in Fig. 8) might not result from atrophy of this region, as the hippocampus of the same subject exhibited strong [11C]PBB3 binding despite marked atrophy. Possible explanations for this change include formation of extracellular NFTs and their envelopment by astrocytes in the degenerating neocortex, profoundly modifying accessibility of these NFTs to exogenous molecules (Schmidt et al., 1988). This notion would need to be examined by combined autoradiogarphic and immunohistochemical assays of different brain regions.
Being able to visualize tau deposits with [11C]PBB3 in non-AD tauopathies such as PSP, CBD and related disorders is also of major importance, as suggested in the present PET data supporting detectability of tau deposition in living CBD brains. Although abundant tau deposits are largely confined to specific neuroanatomical locations of the CNS in tau-positive, plaque-negative illnesses, as exemplified by PSP and CBD (Dickson et al., 2011), the homogenous and low-level background signals of [11C]PBB3 in brain parenchyma indicate the possibility of detecting tau lesions that are less abundant and/or less widespread in these disorders than NFTs and neuropil threads in AD. Following such in vivo assessments, a postmortem neuropathological evaluation of scanned subjects would be required as a reference standard for PET assays of non-AD tau pathologies.
It has been documented that [11C]PIB-positive plaque formation nearly plateaus prior to the progression of brain atrophy in AD (Engler et al., 2006), but tau abnormalities may bridge the chasm between Aβ fibrillogenesis and neuronal death. Consistent with this notion, our PET/MRI data indicate that the deposition of tau inclusions as visualized by the intense [11C]PBB3 labeling but lacking overt [11C]PIB binding is closely associated with a local volume reduction in the hippocampal formation. Indeed, our pilot clinical PET study demonstrated that localized accumulation of [11C]PBB3 in the medial temporal region of AD patients was accompanied by marked hippocampal atrophy (Fig. 7B). It is noteworthy that [11C]PBB3-PET signals were substantially increased, notwithstanding the atrophy-related partial volume effects on PET images, and this observation may support the contribution of tau fibrils to toxic neuronal death in AD. However, these data do not immediately imply neurotoxicities of [11C]PBB3-reactive tau fibrils, in light of MRI-detectable neurodegeneration uncoupled with [11C]PBB3 retention in the hippocampus of PS19 mice. In the hippocampal formation of AD patients, neurons bearing NFTs resembling those in the PS19 hippocampus may drive neurodegeneration similar to that observed in either of the PS19 hippocampus or brain stem, and this issue could be addressed in future studies using [11C]PBB3-PET and MRI in diverse mouse models, including PS19 and rTg4510 mice, and human subjects.
Our analyses of multiple β-sheet ligands illustrated electrochemical and/or conformational diversities of β-pleated sheets among amyloid aggregates, producing a selectivity of these compounds for a certain spectrum of fibrillar pathologies (Figs. 1 and S1). Lipophilicities of the β-sheet ligands could determine their reactivity with non-cored plaques, as noted among the PBBs studied here (Fig. 1), although the molecular properties underlying this variation are yet to be elucidated. Meanwhile, it should also be noted that all β-sheet ligands tested in the present study were reactive with dense core plaques irrespective of their lipophilicities. This may affect in vivo PET signals particularly in AD brain areas with abundant cored plaques, such as the precuneus. However, our combined autoradiographic and histochemical assessments indicated that [11C]PBB3 bound to dense core plaques accounts for less than 10% of total specific radioligand binding in these areas, and this percentage in fact includes binding to tau fibrils in plaque neurites in addition to Aβ amyloid core. A second possibility to account for the diversity of ligand reactivity to tau lesions may arise from the packing distance between two juxtaposed β-sheets in tau filaments, and is discussed in the supplement (Supplemental Discussion).
It is also noteworthy that selectivity of [11C]PBB3 for tau versus aggregates may depends on free radioligand concentration in the brain. Our autoradiographic binding assays suggested that affinity of [11C]PBB3 for NFTs is 40–50 fold higher than senile plaques, but binding components on tau fibrils may be more readily saturated by this radioligand than those on Aβ fibrils. [11C]PBB3-PET data in humans indicated that uptake of this radioligand into the brain is less than 1/3 of [11C]PIB uptake, and that free radioligand concentration in the brain at a pseudo-equilibrium state is approximately 0.2 nM or lower. In this range of concentration, [11C]PBB3 could preferentially interact with high-affinity binding components formed by tau assemblies. Excessive amount of radioligand in the brain would result in saturation of its binding to tau lesions and increased binding to low-affinity, high-capacity binding components in Aβ plaques, and such overload of free radioligand is more likely in regions with less abundant tau pathologies. This could be even more critical in capturing early tau pathologies originating in the hippocampal formation, and may require technical improvements and methodological refinements, including high-resolution imaging, correction for motions of subjects during scans and robust definition of VOIs on the atrophic hippocampus.
Although nonspecific [11C]PBB3-PET signals in control human subjects were generally low, radioligand retention in dural venous sinuses was noticeable in all scanned individuals. Possible mechanisms underlying this property are discussed in the supplement (Supplemental Discussion).
The present work has also implied the potential utility of multimodal imaging systems for translational development of therapeutic agents counteracting tau fibrillogenesis. Optical imaging with a near-infrared fluorescent probe such as PBB5 could provide the least invasive technique to assess tau accumulation in living mouse models. As demonstrated by our in vitro and ex vivo fluorescence labeling, all PBBs share a similarity in terms of their reactivity with tau aggregates. Hence, PBB5-optics may be applicable to early screening of therapeutic agents suppressing tau deposition, and the data on abundance of tau lesions obtained by this approach may be translatable to advanced stages of assessments using [11C]PBB3-PET in animal models and humans. By contrast, pharmacokinetic properties of PBB5 (Fig. S5) were found to be distinct from those of electrically neutral PBBs, including PBB2 and PBB3. These considerations would be of importance in developing and using fluorescent ligands applicable to optical and PET imaging.
To conclude, our new class of multimodal imaging agents offer the possibility of visual investigations of fibrillary tau pathologies at subcellular, cellular and regional levels. These assay systems are potentially powerful tools for the longitudinal evaluation of anti-tau treatments (Marx, 2007), as a single probe may facilitate a seamless, bidirectional translation between preclinical and clinical insights. PET tracers would also serve a more immediate therapeutic purpose by enabling the assessment of the effects of anti-Aβ as well as anti-tau therapies on tau pathologies in living AD patients.
EXPERIMENTAL PROCEDURES
Compounds and reagents
PBB1 (Wako Pure Chemical Industries), PBB2 (ABX), PBB3 (Nard Institute), PBB4 (ABX), mPBB5 (Nard Institute), desmethyl precursor of [11C]PBB2 (2-[4-(4-aminophenyl)buta-1,3-dienyl]benzothiazol-6-ol; Nard Institute), desmethyl precursor of [11C]PBB3 protected with a silyl group (5-[4-(6-tert-butyldimethylsilyloxy-benzothiazol-2-yl)buta-1,3-dienyl]pyridine-2-amine; Nard Institute), desmethyl precursor of [11C]mPBB5 (2-[4-(4-dimethylaminophenyl)buta-1,3-dienyl]-3-ethyl-6-hydroxybenzothiazol-3-ium; Nard Institute) and 2-[8-(4-dimethylaminophenyl)octa-1,3,5,7-tetraenyl]-3-ethylbenzothiazol-3-ium (DM-POTEB; Nard Institute) were custom-synthesized. Information on other chemicals is provided in the supplement (Supplemental Experimental Procedures). ClogP for each compound was calculated using ACD Chemsketch logP software (Advanced Chemistry Development, Toronto, Canada).
Animal models
Tg mice heterozygous for human T34 (4-repeat tau isoform with 1 N-terminal insert) with FTDP-17 P301S mutation driven by mouse prion protein promoter, also referred to as PS19 mice (Yoshiyama et al., 2007), were bred and kept on a C57BL/6 background. All mice studied here were maintained and handled in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals and our institutional guidelines. Protocols for the present animal experiments were approved by the Animal Ethics Committees of the National Institute of Radiological Sciences.
Postmortem brain tissues
Procedures for preparation of human and mouse brain sections are given as Supplemental Experimental Procedures online.
In vitro and ex vivo fluorescence microscopy
Six-μm paraffin sections generated from patient brains and 20-μm frozen sections of mouse brains were stained with 10−3% β-sheet ligands dissolved in 50% ethanol for 1 hr at room temp. Images of the fluorescence signals from these compounds were captured by non-laser (BZ-9000; Keyence Japan) and confocal laser scanning (FV-1000; Olympus) microscopes. In the confocal imaging, excitation/emission wave lengths (nm) were optimized for each compound as follows: 405/420–520 (PBB3, FSB, PIB, BF-227, BF-158, FDDNP, thioflavin-S), 488/520–580 (PBB2, PBB4), 515/530–630 (PBB1, curcumin) and 635/645–720 (PBB5, BF-189, DM-POTEB). Subsequently, the tested samples and adjacent sections probed serially with each ligand were autoclaved for antigen retrieval, immunostained with the anti-tau monoclonal antibody AT8 that is specific for tau phosphorylated at Ser 202 and Thr 205 (Endogen), as well as a polyclonal antibody against AβN3(pE), and inspected using the microscopes noted above. For ex vivo imaging, PS19 and non-Tg WT at 10–12 months of age were anesthetized with 1.5% (v/v) isoflurane, and were given 1 mg/kg PBB1–4, 0.1 mg/kg PBB5 or 10 mg/kg FSB by syringe via tail vein. The animals were killed by decapitation at 60 min after tracer administration. Brain and spinal cord were harvested and cut into 10-μm-thick sections on a cryostat (HM560). The sections were imaged using microscopes as in the in vitro assays, and were labeled with either FSB or AT8, followed by microscopic re-examination.
Ex vivo and in vivo multiphoton imaging
Experimental procedures are given as Supplemental Experimental Procedures online.
In vivo and ex vivo pulsed laser scanning imaging
Noninvasive scans of isoflurane-anesthetized non-Tg WT and tau Tg mice at 12 months of age were performed using a small animal-dedicated optical imager (eXplore Optix; ART). Scan protocols are given in the supplement (Supplemental Experimental Procedures).
Radiosynthesis of [11C]PBB2
Experimental procedures are given as Supplemental Experimental Procedures online.
Radiosynthesis of [11C]PBB3
[11C]Methyl iodide was produced and transferred into 300 μl of dimethyl sulphoxide (DMSO) containing 1.5–2 mg of tert-butyldimethylsilyl desmethyl precursor and 10 mg of potassium hydroxide at room temperature. The reaction mixture was heated to 125°C and maintained for 5 min. After cooling the reaction vessel, 5 mg of tetra-n-butylammonium fluoride hydrate in 600 μl of water was added to the mixture to delete the protecting group, and then 500 μl of HPLC solvent was added to the reaction vessel. The radioactive mixture was transferred into a reservoir for HPLC purification (CAPCELL PAK C18 column, 10 mm × 250 mm; acetonitrile/50 mM ammonium formate=4/6, 6 ml/min). The fraction corresponding to [11C]PBB3 was collected in a flask containing 100 μl of 25% ascorbic acid solution and 75 μl of Tween 80 in 300 μl of ethanol, and evaporated to dryness under a vacuum. The residue was dissolved in 10 ml of saline (pH 7.4) to obtain [11C]PBB3 (970–1990 GBq at EOS) as an injectable solution. The final formulated product was radiochemically pure (≥95%) as detected byanalytic HPLC (CAPCELL PAK C18 column, 4.6 mm × 250 mm; acetonitrile/50 mM ammonium formate=4/6, 2 ml/min). The specific activity of [11C]PBB3 at EOS was 37–121 GBq/μmol, and [11C]PBB3 maintained its radioactive purity exceeding 90% over 3 hrafter formulation.
Radiosynthesis of [11C]mPBB5
Experimental procedures are given as Supplemental Experimental Procedures online.
Radiosynthesis of [11C]PIB
Radiolabeling of PIB was performed as described elsewhere (Maeda et al., 2011). The specific activity of [11C]PIB at EOS was50–110 GBq/μmol.
In vitro and ex vivo autoradiography
Experimental procedures are given as Supplemental Experimental Procedures online.
In vivo PET imaging of mice
PET scans were performed using a microPET Focus 220 animal scanner (Siemens Medical Solutions) immediately after intravenous injection of [11C]PBB2 (28.3 ± 10.3 MBq), [11C]PBB3 (29.7 ± 9.3 MBq) or [11C]mPBB5 (32.8 ± 5.9 MBq). Detailed procedures are provided in the supplement (Supplemental Experimental Procedures).
In vivo PET imaging of humans
Three cognitively normal control subjects (64, 72 and 75 years of age; mean age, 70.3 years) and 3 AD patients (64, 75 and 77 years of age; mean age, 72 years) were recruited to the present work (Fig. 8). Additional information on these subjects is given in the supplement (Supplemental Experimental Procedures). The current clinical study was approved by the Ethics and Radiation Safety Committees of the National Institute of Radiological Sciences, Chiba, Japan. Written informed consent was obtained from the subjects or their family members. PET assays were conducted with a Siemens ECAT EXACT HR+ scanner (CTI PET Systems). Detailed PET scan protocols are provided in the supplement (Supplemental Experimental Procedures). A fraction of radioactivity corresponding to unmetabolized [11C]PBB3 in plasma at 3, 10, 20, 30 and 60 min was determined by HPLC (Waters mBondapak C18 column, 7.8 mm × 300 mm; acetonitrile/ammonium formate mobile phase with gradient elution = 40/60, 52/48, 80/20, 80/20, 40/60 and 40/60 at 0, 6, 7, 8, 9 and 15 min, respectively; flow rate, 6 ml/min) as described elsewhere (Suzuki et al., 1999). The radiotracer injection and following scans and plasma assays were conducted in a dimly lit condition to avoid photoracemization of the chemicals.
Individual MRI data were coregistered to the PET images using PMOD software (PMOD Technologies). Volumes of interest (VOIs) were drawn on coregistered MR images, and were transferred to the PET images. Procedures of image analyses are provided in the supplement (Supplemental Experimental Procedures).
We additionally carried out PET scans of a patient who was clinically diagnosed as having corticobasal syndrome, as described in Supplemental Experimental Procedures online.
Supplementary Material
Highlights.
Compounds for in vivo imaging of diverse types of tau inclusions were developed.
These compounds enabled optical and PET imaging of tau lesions in model mice.
PET with one of these compounds illuminated tau-rich regions in Alzheimer’s disease.
Our probe produced PET images consistent with spreading tau pathology.
Acknowledgments
The authors thank Mr. T. Minamihisamatsu and Mr. Y. Matsuba for technical assistance; staff of the Molecular Probe Group, National Institute of Radiological Sciences, for support with radiosynthesis; Dr. Y. Yoshiyama at Chiba East National Hospital for supports to clinical PET studies; and Dr. T. Iwatsubo at the University of Tokyo and Dr. H. Inoue at Kyoto University for critical discussion. This work was supported in part by grants from the National Institute on Aging of the National Institutes of Health AG10124 and AG17586 (J. Q. T. and V. M.-Y. L.), Grants-in-Aid for Japan Advanced Molecular Imaging Program, Young Scientists 21791158 (M. M.), Scientific Research (B) 23390235 (M. H.), Core Research for Evolutional Science and Technology (T. S.) and Scientific Research on Innovative Areas (“Brain Environment”) 23111009 (M. H.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, Thomas H. Maren Junior Investigator Fund from College of Medicine, University of Florida (N. S.), and research fund of Belfer Neurodegeneration Consortium (Q. C. and M.-K. J.).
Footnotes
Author Information
M. M., H. Shimada, T. S., M.-R. Z. and M. H. are named as inventors on a patent application 0749006WO1, claiming subject matter related to the results described in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-β ligand in transgenic mice. Proc Natl Acad Sci U S A. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Tredici KD. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J Mol Neurosci. 2011;45:384–389. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, Wall A, Ringheim A, Långström B, Nordberg A. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129:2856–2866. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O’Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Iwata N, Matsuba Y, Sato K, Sasamoto K, Saido TC. 19F and 1H MRI detection of amyloid β plaques in vivo. Nat Neurosci. 2005;8:527–533. doi: 10.1038/nn1422. [DOI] [PubMed] [Google Scholar]
- Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. In vivo detection of amyloid-β deposits by near-infrared imaging using an oxazine-derivative probe. Nat Biotechnol. 2005;23:577–583. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Krebs MRH, Bromley EH, Donald AM. The binding of thioflavin-T to amyloid fibrils: localization and implications. J Struct Biol. 2005;149:30–37. doi: 10.1016/j.jsb.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Okamura N, Furumoto S, Tashiro M, Furukawa K, Maruyama M, Itoh M, Iwata R, Yanai K, Arai H. 2-(2-[2-Dimethylaminothiazol-5-yl]ethenyl)-6- (2-[fluoro]ethoxy)benzoxazole: a novel PET agent for in vivo detection of dense amyloid plaques in Alzheimer’s disease patients. J Nucl Med. 2007;48:553–561. doi: 10.2967/jnumed.106.037556. [DOI] [PubMed] [Google Scholar]
- Maeda J, Ji B, Irie T, Tomiyama T, Maruyama M, Okauchi T, Staufenbiel M, Iwata N, Ono M, Saido TC, Suzuki K, Mori H, Higuchi M, Suhara T. Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer’s disease enabled by positron emission tomography. J Neurosci. 2007;27:10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda J, Zhang MR, Okauchi T, Ji B, Ono M, Hattori S, Kumata K, Iwata N, Saido TC, Trojanowski JQ, Lee VMY, Staufenbiel M, Tomiyama T, Mori H, Fukumura T, Suhara T, Higuchi M. In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. J Neurosci. 2011;31:4720–4730. doi: 10.1523/JNEUROSCI.3076-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx J. Alzheimer’s disease. A new take on tau Science. 2007;316:1416–1417. doi: 10.1126/science.316.5830.1416. [DOI] [PubMed] [Google Scholar]
- Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, Arai H, Yanai K, Sawada T, Kudo Y. Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci. 2005;25:10857–10862. doi: 10.1523/JNEUROSCI.1738-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, A β N3(pE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Gur RE, Gur RC, Trojanowski JQ. Intraneuronal and extracellular neurofibrillary tangles exhibit mutually exclusive cytoskeletal antigens. Ann Neurol. 1988;23:184–189. doi: 10.1002/ana.410230212. [DOI] [PubMed] [Google Scholar]
- Small GW, Kepe V, Ercolim LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Eng J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Takei M, Kida T. Development of an analyzing system for the sensitive measurement of radioactive metabolites on the PET study. J Labelled Cpd Radiopharm. 1999;42:S658–660. [Google Scholar]
- Thompson PW, Ye L, Morgenstern JL, Sue L, Beach TG, Judd DJ, Shipley NJ, Libri V, Lockhart A. Interaction of the amyloid imaging tracer FDDNP with hallmark Alzheimer’s disease pathologies. J Neurochem. 2009;109:623–630. doi: 10.1111/j.1471-4159.2009.05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Rieves D, Ganley C. Brain amyloid imaging – FDA approval of florbetapir F18 injection. N Engl J Med. 2012;367:885–887. doi: 10.1056/NEJMp1208061. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VMY. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Szardenings AK, Wang E, Walsh JC, Xia C, Yu C, Zhao T, Kolb HC. A highly selective and specific PET tracer for imaging of Tau pathologies. J Alzheimers Dis. 2012;31:601–612. doi: 10.3233/JAD-2012-120712. [DOI] [PubMed] [Google Scholar]
- Zhuang ZP, Kung MP, Hou C, Skovronsky DM, Gur TL, Plössl K, Trojanowski JQ, Lee VMY, Kung HF. Radioiodinated styrylbenzenes and thioflavins as probes for amyloid aggregates. J Med Chem. 2001;44:1905–1914. doi: 10.1021/jm010045q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.