

Abstract
Current study demonstrates the glioma tumor antigen podoplanin to be present at very high levels (>90%) in both glioblastoma (D2159MG, D08-0308MG, and D08-0493MG) and medulloblastoma (D283MED, D425MED, and DAOY) xenografts and cell line. We constructed a novel recombinant single-chain antibody variable region fragment (scFv), NZ-1, specific for podoplanin from the NZ-1 hybridoma. NZ-1-scFv was then fused to Pseudomonas exotoxin A, carrying a C-terminal KDEL peptide (NZ-1-PE38KDEL). The immunotoxin was further stabilized by a disulfide (ds) bond between the heavy-chain and light-chain variable regions as the construct NZ-1-(scdsFv)-PE38KDEL. NZ-1-(scdsFv)-PE38KDEL exhibited significant reactivity to glioblastoma and medulloblastoma cells. The affinity of NZ-1-(scdsFv), NZ-1-(scdsFv)-PE38KDEL and NZ-1 antibody, for podoplanin peptide was 2.1×10−8 M, 8.0×10−8 M, and 3.9×10−10 M, respectively. In a protein stability assay, NZ-1-(scdsFv)-PE38KDEL retained 33-98% of its activity while that of NZ-1-PE38KDEL declined to 13% of its initial levels after incubation at 37°C for 3 days. In vitro cytotoxicity of the NZ-1-(scdsFv)-PE38KDEL was measured in cells isolated from glioblastoma xenografts, D2159MG, D08-0308MG, D08-0493MG, and in the medulloblastoma D283MED, D425MED, and DOAY xenografts and cell line. The NZ-1-(scdsFv)-PE38KDEL immunotoxin was highly cytotoxic, with an IC50 in the range of 1.6–29 ng/mL. Significantly, NZ-1-(scdsFv)-PE38KDEL demonstrated tumor-growth delay, averaging 24 days (P<0.001) and 21 days (P<0.001) in D2159MG and D283MED in vivo tumor models, respectively. Crucially, in the D425MED intracranial tumor model, NZ-1-(scdsFv)-PE38KDEL caused a 41% increase in survival (P≤0.001). In preclinical studies, NZ-1-(scdsFv)-PE38KDEL exhibited significant potential as a targeting agent for malignant brain tumors.
Keywords: podoplanin, glioblastoma multiforme, medulloblastoma, recombinant immunotoxin, single-chain disulfide Fv
Introduction
Glioblastoma is the most frequent and most malignant primary brain tumor in adults and accounts for 17% of all primary brain tumors, while medulloblastoma an invasive, malignant tumor, accounts for about 13% of all childhood brain tumors in children of ages 0–14 years.1 Despite aggressive multimodality therapy, the median survival of glioblastoma patients until the late 1990s was less than a year.2 Current clinical trials incorporating temozolomide or the monoclonal antibody (mAb) bevacizumab have been shown to be effective. However, they produced only a modest increase in glioblastoma patient survival, with less than 5% of patients surviving five years post-diagnosis.1,3,4 Similarly, recent advances in surgery, radiotherapy, and chemotherapy have led to an increase in five-year survival rate of medulloblastoma patients, from 30% to upwards of 60–70%.5 However, the aggressive nature of these therapies results in long-term adverse effects, including cerebellar mutism, cognitive and endocrine impairments, hearing loss, infertility, and neuropathies.6 To overcome these difficulties, there is a need for the development of novel and effective therapies specifically targeting the tumor cells while preserving the surrounding normal central nervous system (CNS).
Current advances in the understanding of brain tumor biology has identified unique molecular abnormalities that are specifically associated with brain tumors but which are absent in normal brain cells.7 Molecular characterization of brain tumors has led to the identification of tumor-specific proteins that are being exploited for the development of targeted therapies for brain tumor treatment.8 Several mAbs against brain tumor antigens have been generated.9 Over the past two decades, recombinant single-chain variable-region antibody fragment (scFv)-based immunotoxins (ITs), composed of the heavy-chain and light-chain variable regions (VH and VL) of a mAb targeting tumor antigen fused to a toxin molecule, have been widely investigated for the treatment of several cancers.10 The smaller size of a scFv-IT fusion protein should provide greater tumor penetration and therefore enhanced therapeutic efficacy.
Podoplanin is a 162-amino acid type-I transmembrane sialomucin-like glycoprotein consisting of a serine- and threonine-rich extracellular domain, a single transmembrane domain, and a short cytoplasmic domain containing cAMP and protein kinase C phosphorylation sites.11 It is highly expressed by cultured human lymphatic endothelial cells, and podoplanin−/− mice die at birth due to impaired lymphatic vessel pattern formation and respiratory failure.12 Podoplanin expression has been described in several tumors, including brain tumors11,13,14 and is associated with malignant progression involving epithelial-mesenchymal transition, metastasis, and invasion.15–18 Among the CNS tumors, podoplanin expression is observed in 83% and 27% of glioblastoma and medulloblastoma cases, respectively,14 making it an attractive target for brain tumor therapy. Several anti-podoplanin antibodies (NZ-1, D2-40, AB3, 18H5) have been developed and well characterized.19,20 A functional immunotherapeutic agent should be able to bind to the target tumor cells with high affinity and be internalized. The radiolabeled NZ-1 mAb has been previously demonstrated to bind avidly by D397MG cells and rapidly internalized by D2159MG xenograft cells.21 Further, the [131I]SGMIB-NZ-1 mAb exhibited significant tumor accumulation in D2159MG tumor-bearing mice, which thereby established the potential utility of NZ-1 as an immunotherapeutic agent against malignant brain tumors.21
Herein, we demonstrate the expression of the tumor antigen podoplanin in several glioblastoma and medulloblastoma xenograft cells. Further, we report the construction of a recombinant Pseudomonas exotoxin based IT, NZ-1-(scdsFv)-PE38KDEL, from the NZ-1 mAb. The NZ-1-(scdsFv)-PE38KDEL IT inhibited protein synthesis in brain tumor xenograft cells in vitro and exhibited significant delay in tumor growth of brain tumor xenografts in vivo, establishing this immunotoxin as a promising candidate for the therapy of brain tumors.
Material and Methods
Cell line
The medulloblastoma cell line DAOY (ATCC Number: HTB-186) was cultured in complete zinc option (ZO)-10% fetal bovine serum (FBS; Improved Modified Eagle Medium ZO [Richter’s Modification, Cat.No.10373-017] liquid; Invitrogen, San Diego, CA) and passed at confluence with 0.05% Trypsin-EDTA (Invitrogen).
Dissociation of xenografts
Human-biopsy-derived xenograft tissue from malignant glioma lines D2159MG, D08-0308MG, D08-0493MG, D08-0695MG, and D08-0537MG, and medulloblastoma lines D283MED, and D425MED, obtained under sterile conditions from the Duke animal facility, were prepared for cell culture in a laminar flow hood with a sterile technique. Tumor material was finely minced and digested with 100 μg Liberase (Roche, Indianapolis, IN). This mixture was stirred at 37°C for 10 min, and a cell-rich supernatant was obtained. The cells were washed with complete ZO medium, further treated with Ficoll-Hypaque to remove any red blood cells, and then washed once in ZO medium.
Cloning of variable heavy (VH) and variable light (VL) domains of the NZ-1 and P588 mAb
P588 is a mouse monoclonal antibody (IgG1κ) purified from the mouse myeloma cell line P3X63Ag8, which is a subclone of the myeloma cell line MOPC-21.22 The antigen specificity of MOPC-21 is unknown and is widely used as an isotype control antibody (Cell Signaling Technology, Inc., Danvers, MA, Cat.No.4878). Total cellular mRNA from 106 NZ-1 and P3X63Ag8 hybridoma cells was isolated by using a Dynabeads, mRNA direct kit (Invitrogen). A SMART RACE cDNA amplification kit (Clontech, Palo Alto, CA) was used to obtain VH and VL cDNAs of the NZ-1 and P588 mAb. The VH domain was fused to the VL domain by a 15-amino-acid peptide (Gly4Ser)3 linker by PCR. The NZ-1 and P588 scFv fragment was cloned into pRB199 vector using a T4 DNA ligase kit (Roche) to obtain NZ-1-(scFv)-PE38KDEL and P588-(scFv)-PE38KDEL. The NZ-1-(scdsFv)-PE38KDEL and P588-(scdsFv)-PE38KDEL constructs were obtained by mutating residues 44 of VH and 103 of VL, and 44 of VH and 100 of VL respectively, by site-directed mutagenesis using a QuickChange Multi-Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA), and the sequence was verified.
Preparation of recombinant immunotoxins
The different NZ-1 and P588 ITs were generated by fusing the specific scFv or scdsFv with the sequences for domains II and III of Pseudomonas exotoxin A (PE38KDEL; KDEL: Lysine-aspartic acid-glutamic acid-leucine) according to a previously described protocol.23 NZ-1-(scdsFv), NZ-1-(scFv)-PE38KDEL, NZ-1-(scdsFv)-PE38KDEL, P588-(scFv)-PE38KDEL, and P588-(scdsFv)-PE38KDEL were expressed under control of the T7 promoter in E. coli BL21 (λDE3) (Stratagene, La Jolla, CA). All recombinant proteins accumulated in the inclusion bodies. The inclusion bodies were purified and solubilized in 8 M guanidine (Thermo Scientific) and reduced with 0.3 M DTE (Sigma) for 5 h at room temperature, as described previously.23 The reduced protein was rapidly diluted 100 fold in the refolding buffer consisting of 0.1 M Tris pH 8.0 (Invitrogen), 0.5 M L-Arginine (Sigma), 9 mM GSSG (Sigma), and 2 mM EDTA (Invitrogen). Protein samples in refolding buffer at a concentration of 100 μg/ml were incubated at 10°C for 72 h. After renaturation the samples were dialyzed against 20 mM Tris pH 7.5 (Invitrogen) and 100 mM urea (MP Biomedicals), overnight at 4°C. The dialyzed protein sample was filtered and loaded on to a Q-Sepharose fast flow column (GE Healthcare) and the IT was eluted over a linear salt gradient with 1 M NaCl (Sigma) in 20 mM Tris pH 7.5. The IT was further purified as a monomer (64 kDa) by size exclusion chromatography on a TSKgel SuperSW3000 column (TOSOH Bioscience) to greater than 95% purity.
In vitro cytotoxicity assay
The cytotoxicity of the NZ-1-(scdsFv)-PE38KDEL on cells isolated from D2159MG, D08-0308MG, D08-0493MG, D08-0695MG, D08-0537MG, D283MED, and D425MED xenografts and DAOY cells was assayed by inhibition of protein synthesis as described previously.24 P588-(scdsFv)-PE38KDEL was used as negative control. Cells were seeded in 96-well plates at a density of 2×104 cells per well in 200 μl of complete ZO medium 24 h before the assay. Immunotoxins were serially diluted to achieve a final concentration of 0.01 to 1000 ng/mL in phosphate buffered saline containing 0.2% bovine serum albumin (BSA; 0.2% BSA/PBS), and 10 μl of diluted toxin was added to each well. Plates were incubated for 20 h at 37°C and then pulsed with 1 μCi/well of L-[4,5-3H]leucine (PerkinElmer, Shelton, CT) in 25 μl of 0.2% BSA/PBS for 3 h at 37°C. Radiolabeled cells were captured on filter-mats and counted in a MicroBeta scintillation counter (PerkinElmer). The cytotoxic activity of NZ-1-(scdsFv)-PE38KDEL was defined by IC50, which was the toxin concentration that inhibited radioactivity incorporation by 50% as compared to the radioactivity measured in untreated cells. All the assays were performed in triplicate for each cell line. Results are presented as mean ± standard deviation where n ≥ 3.
In vivo xenograft model
Female athymic nude mice (≈20 g; 4–6 weeks) were injected subcutaneously (s.c.) in the right flank with 3×106 D2159MG or D283MED cells suspended in 50 μl of PBS, pH 7.4. When the implanted tumors reached 150–300 mm3, 10 mice per arm were randomly selected for inoculation. The test mice were treated by intratumoral (i.t.) injection every other day with a total of three doses of 0.3 mg/kg of NZ-1-(scdsFv)-PE38KDEL diluted in 200 μl of PBS containing 0.2% human serum albumin (PBS-HSA). The control mice were handled in the same manner, but treated with either 0.2% PBS-HSA or with 0.3 mg/kg of the negative control IT, P588-(scFv)-PE38KDEL. Tumors were measured twice weekly with a handheld vernier caliper, and the tumor volumes were calculated in cubic millimeters by using the formula: ([length]×[width2])/2. When tumor volume met both of the following criteria: (1) >1000 mm3 and (2) 5 times its original treatment size, animals were tested out of the study.
Assessment of response
The s.c. xenograft response was assessed by delay in tumor growth in test mice (T) versus control mice (C). The growth delay (T-C) was the difference between the median times required for tumors in treated and control mice to reach a volume that was 5 times the size at the initiation of therapy and >1000 mm3. Tumor regression was the decrease in tumor volume over two successive measurements.
Statistical analysis was performed as previously described,25 using the Wilcoxon rank-sum test for growth delay and Fisher’s exact test for tumor regressions.
Intracranial implantation of tumor cells
Male NOD scid gamma (NSG) mice (≈20–30 g; 12 weeks) were anesthetized by an intraperitoneal injection of a solution of ketamine 50 mg/kg (Fort Dodge Animal Health, Iowa), and xylazine 10 mg/kg (Akorn, Inc., Illinois). The anterior cranial region was shaved under sterile conditions and an incision ≈1 cm in length was made in the skin over the skull, and a small burr hole was drilled at coordinates 2.5 mm left lateral of the sagittal and 1 mm anterior to the bregma. A 25-gauge 10-μl Hamilton needle was inserted vertically to a depth of 2.5 mm from the dura mater. All mice were injected with 105 D425MED cells in 5 μl of 1X PBS into the cortex.
Intracranial convection-enhanced delivery (CED)
Four days post tumor implantation, a three day Alzet pump (Durect Corporation, California) containing 0.3 μg of the P588-(scdsFv)-PE38KDEL or NZ-1-(scdsFv)-PE38KDEL IT diluted in 100 μl of 0.2% PBS-HSA was implanted subcutaneously on the back of the anesthetized mice slightly posterior to the scapulas. The pump was connected to the cannula through a catheter passing through a tunnel created under the skin of mice and the IT was infused through the cannula into the tumor site over a three day period. The control animals were handled in the same manner, but were either left untreated (sham) or treated with 0.2% PBS-HSA alone. The mice were followed to assess tumor development and death.
Evaluation of intracranial xenograft response to treatment
The response of intracranial (ic) xenografts to treatment was assessed by percentage increase in time to a specific neurologic endpoint (seizure activity, repetitive circling, or other subtle changes such as grooming or decrease in appetite) or to death. Statistical analysis was performed using the Wilcoxon rank order test as previously described.26,27 Animals were observed twice daily for signs of distress or development of neurologic symptoms, at which time, the mice were euthanized.
Results
Expression of podoplanin on glioblastoma and medulloblastoma xenografts and cell line
Prior to the construction of a recombinant IT from a mAb, it is essential to demonstrate the reactivity of the mAb to brain tumor cells. Hence, the expression of podoplanin on brain tumor xenograft cells was analyzed by flow cytometry with NZ-1 mAb (Fig. 1, left panel). FACS data revealed significant levels of podoplanin to be present on both glioblastoma (D2159MG, D08-0308MG, and D08-0493MG) and medulloblastoma (D283MED, D425MED, and DAOY) tumors (Fig. 1, left panel). The specificity of NZ-1 mAb was demonstrated by testing its reactivity on two additional glioblastoma xenograft cells (D08-0695MG and D08-0537MG), exhibiting almost complete absence of podoplanin (Supplementary Fig. S1). The levels of podoplanin receptor on both podoplanin positive and negative brain tumor cells was quantified using NZ-1 mAb by QFACS and the data is presented in Table 2.
Figure 1.

Flow cytometric analysis of brain tumor xenografts to determine reactivity of the NZ-1 mAb and NZ-1-(scdsFv)-PE38KDEL. Cells isolated from brain tumor xenografts (a) D2159MG, (b) D08-0308MG, (c) D08-0493MG, (d) D283MED, (e) D425MED, and (f) DAOY cell line, were stained with NZ-1 mAb/ NZ-1-(scdsFv)-PE38KDEL (grey open peaks) or an isotype control IgG2a antibody/ P588-(scdsFv)-PE38KDEL (filled black peaks).
Table 2.
Cytotoxicity, receptor number, and internalization of NZ-1-(scdsFv)-PE38KDEL on brain tumor cells*
| Xenograft | NZ-1-(scdsFv)-PE38KDEL IC50 ng/mL | Number of podoplanin molecules per cell | Percent Internalized |
|---|---|---|---|
| D08-0695MG | >1000 | 1.0 × 103 | ND |
| D08-0537MG | >1000 | 6.9 × 102 | ND |
| D2159MG | 1.6 ± 1.2 | 3.4 × 106 | 80% |
| D08-0308MG | 3.3 ± 2.0 | 1.1 × 105 | 87% |
| D08-0493MG | 2.7 ± 1.7 | 6.1 × 107 | 42% |
| D283MED | 9.7 ± 4.0 | 7.3 × 103 | ND |
| D425MED | 26.7 ± 4.0 | 6.5 × 104 | 93% |
| DAOY | 29.0 ± 14.4 | 1.3 × 106 | 21% |
Cytotoxicity data are given as an IC50 value, the concentration of immunotoxin that causes a 50% inhibition of protein synthesis after a 20-h incubation with immunotoxin.
ND: Not determined.
Cloning of the variable domains of NZ-1 scFv and generation of NZ-1-(scdsFv)-PE38KDEL immunotoxin
Once the cell surface reactivity of NZ-1 was established, the VH and VL cDNAs were cloned and sequenced. The cloned VH and VL fragments were approximately 351 and 342 bp, respectively. The NZ-1 VH and VL domain amino acid sequences are shown in Supplementary Fig. S2.
The NZ-1 (scFv) was then fused with the sequences for domains II and III of Pseudomonas exotoxin A, PE38KDEL, carrying a modified C-terminus which increases its intracellular retention, in turn enhancing its cytotoxicity. The stability of an IT can be enhanced by mutating a single, key residue in the VH and VL domains to cysteine, which forms a stabilizing disulfide bond.28 Thus, we constructed a scdsFv containing both a disulfide bond, generated by cysteine residues that replace Gly44 in the framework region 2 (FR2) of VH and Gly103 in the FR4 of VL, and a peptide linker. The NZ-1 (scdsFv) PCR fragment was then fused to PE38KDEL. The NZ-1-(scFv)-PE38KDEL and NZ-1-(scdsFv)-PE38KDEL immunotoxin constructs were expressed in E. coli under the control of T7 promoter and collected as inclusion bodies. Both NZ-1-(scFv)-PE38KDEL and NZ-1-(scdsFv)-PE38KDEL were refolded and purified as described in “Materials and Methods” to >95% purity. Size exclusion chromatography fraction 68 of both NZ-1-(scFv)-PE38KDEL and NZ-1-(scdsFv)-PE38KDEL were concentrated (Supplementary Fig. S3). The protein concentration was determined and 2.0 μg of the purified proteins were run in a SDS-PAGE gel (Supplementary Fig. S3). The final yield of the purified protein was ≈2 mg/L of bacterial culture.
Antigen binding characteristics of NZ-1-(scdsFv)-PE38KDEL
To determine whether the NZ-1-(scdsFv)-PE38KDEL IT binds to podoplanin expressed on the cell surface, indirect flow cytometric analysis was performed (Fig. 1, right panel). Similar to the NZ-1 mAb, FACS analysis revealed significant NZ-1-(scdsFv)-PE38KDEL binding to both glioblastoma (D2159MG, D08-0308MG, and D08-0493MG) and medulloblastoma (D283MED, D425MED, and DAOY) tumors (Fig. 1, right panel).
The antigen-binding kinetics of NZ-1-(scdsFv), NZ-1-(scdsFv)-PE38KDEL and NZ-1 mAb was assessed by surface plasmon resonance (Fig. 2). The purified NZ-1-(scdsFv), NZ-1-(scdsFv)-PE38KDEL and NZ-1 mAb was applied to sensor chips coated with a biotinylated podoplanin peptide (hpp38-51, corresponding to amino acids 38–51 of human podoplanin) which was used as an immunogen for the generation of NZ-1 mAb.19,20 All three proteins bound to the podoplanin peptide coated chips, as expected NZ-1 mAb (KD = 3.9×10−10 M) displayed a much higher affinity as opposed to both NZ-1-(scdsFv) (KD = 2.1×10−8 M) and NZ-1-(scdsFv)-PE38KDEL (KD = 8.0×10−8 M) (Fig. 2). These results demonstrate that the NZ-1-(scdsFv)-PE38KDEL IT binds to both podoplanin peptide and to native protein molecules expressed on brain tumor cells.
Figure 2.

Surface plasmon resonance analysis of NZ-1-(scdsFv), NZ-1-(scdsFv)-PE38KDEL and NZ-1 mAb. Binding kinetics and affinity constants of NZ-1-(scdsFv) (a), NZ-1-(scdsFv)-PE38KDEL (b), and NZ-1 mAb (c), were determined by surface plasmon resonance against biotinylated podoplanin peptide (hpp38-51). The association and dissociation rates from the sensogram were KA = 4.8×107 M−1 and KD = 2.1×10−8 M for NZ-1-(scdsFv) (a), KA = 1.2×107 M−1 and KD = 8×10−8 M for NZ-1-(scdsFv)-PE38KDEL (b), and KA = 2.6×109 M−1 and KD = 3.9×10−10 M for NZ-1 mAb (c).
Stability of NZ-1 ITs
The stability of an IT plays a significant role in determining its therapeutic efficacy. Earlier studies indicated scdsFvs might be more stable than scFvs.28 Hence, the stability of NZ-1-(scFv)-PE38KDEL was compared to that of NZ-1-(scdsFv)-PE38KDEL over a six or seven day period at 37°C by measuring its cytotoxic activity against D2159MG xenograft cells (Table 1a) and DAOY cells (Table 1b). While the IC50 value of NZ-1-(scdsFv)-PE38KDEL increased 8-fold from day 0 to day seven, the IC50 value of NZ-1-(scFv)-PE38KDEL increased 33.3-fold over the same time period on D2159MG xenograft cells (Table 1a). Similarly on the medulloblastoma cells DAOY, the IC50 value of NZ-1-(scdsFv)-PE38KDEL increased 1.3-fold from day 0 to day six; however there was a much higher increase in the IC50 value of NZ-1-(scFv)-PE38KDEL, 21.4-fold over the same time period (Table 1b), demonstrating that NZ-1-(scdsFv)-PE38KDEL was more stable than NZ-1-(scFv)-PE38KDEL.
Table 1a.
Stability of NZ-1-(scFv)-PE38KDEL vs NZ-1-(scdsFv)-PE38KDEL* on D2159MG
| Sample | IC50 ng/ml
|
||||||
|---|---|---|---|---|---|---|---|
| 0 | 2 | 3 | 4 | 5 | 6 | 7 (Days) | |
| NZ-1-(scFv)-PE38KDEL | 0.6 | 3 | 4.5 | 5.5 | 6 | 7 | 20 |
| NZ-1-(scdsFv)-PE38KDEL | 0.5 | 1 | 1.5 | 1.5 | 3.5 | 2.5 | 4 |
Table 1b.
Stability of NZ-1-(scFv)-PE38KDEL vs NZ-1-(scdsFv)-PE38KDEL* on DAOY
| Sample | IC50 ng/ml
|
||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 (Days) | |
| NZ-1-(scFv)-PE38KDEL | 28 | 81 | 220 | 220 | 380 | 400 | 600 |
| NZ-1-(scdsFv)-PE38KDEL | 40 | 42 | 41 | 41 | 48 | 48 | 51 |
NZ-1-(scFv)-PE38KDEL and NZ-1-(scdsFv)-PE38KDEL was incubated with 0.2% BSA/PBS at 7.5 μg/ml, over a six or seven day period at 37°C and then assayed for cytotoxic activity on D2159MG xenograft cells or on DAOY medulloblastoma cell line. Cytotoxicity data are given as an IC50 value, the concentration of immunotoxin that causes a 50% inhibition of protein synthesis after a 20-h incubation with immunotoxin.
Cytotoxicity of NZ-1-(scdsFv)-PE38KDEL toward podoplanin expressing brain tumor cells
The cytotoxic activity of NZ-1-(scdsFv)-PE38KDEL was assessed on three podoplanin positive (D2159MG, D08-0308MG, and D08-0493MG) and two podoplanin negative glioblastoma xenograft cells (D08-0695MG and D08-0537MG), and two medulloblastoma xenograft cells (D283MED and D425MED) and a medulloblastoma cell line (DAOY) (Table 2 and Supplementary Fig. S4). Inhibition of protein synthesis by NZ-1-(scdsFv)-PE38KDEL was used as a measure of its cytotoxic effect. The NZ-1-(scdsFv)-PE38KDEL was highly cytotoxic, with IC50 in the range of 1.6–3.3 ng/mL and 9.7–29 ng/mL on the glioblastoma and medulloblastoma cells, respectively (Table 2 and Supplementary Fig. S4). An IgG1κ isotype control derived IT P588-(scdsFv)-PE38KDEL, exhibited a nonspecific cytotoxic effect only at the highest concentration tested (1000 ng/ml) on some of the xenograft cells (D2159MG, D08-0308MG, D08-0493MG and D283MED) (Supplementary Fig. S4). Both NZ-1-(scdsFv)-PE38KDEL and P588-(scdsFv)-PE38KDEL had a nonspecific cytotoxic effect at the highest concentration tested (1000 ng/ml) on the podoplanin negative glioblastoma xenograft cells (D08-0695MG) and no cytotoxic effect on the D08-0537MG xenograft cells (Supplementary Fig. S4a and S4b).
Determination of podoplanin receptor number on brain tumor cells using NZ-1 mAb
Under conditions assuming 1:1 stoichiometry between antibody and antigen, Alexa Fluor 488 (AF488) labeled NZ-1 mAb was used to estimate the number of podoplanin molecules expressed on each brain tumor cell surface by QFACS.29 The results of QFACS analysis of glioblastoma and medulloblastoma cells with NZ-1-AF488 are summarized in Table 2. The levels of surface podoplanin receptors by QFACS on glioblastoma and medulloblastoma xenograft cells varied widely with 3.4×106 on D2159MG, 1.1×105 on D08-0308MG, 6.1×107 on D08-0493MG, 7.3×103 on D283MED, 6.5×104 on D425MED, and 1.3×106 on DAOY. The control glioblastoma xenografts, D08-0695MG (1.0×103) and D08-0537MG (6.9×102), expressed comparatively lower levels of surface podoplanin molecules. QFACS analysis revealed that the number of podoplanin receptors expressed on glioblastoma cells (D2159MG, D08-0308MG, and D08-0493MG) to be significantly higher compared to the levels on medulloblastoma cells (D283MED, D425MED, and DAOY).
NZ-1-(scdsFv)-PE38KDEL internalization rates on brain tumor cells
Upon binding to its cognate receptor, NZ-1-(scdsFv)-PE38KDEL is internalized by endocytosis and kills the target cells. Internalization rates of NZ-1-(scdsFv)-PE38KDEL-AF488 on different brain tumor cells was measured at various time points (1–6 h) and was compared with the total amount of IT bound to the cell surface in the absence of internalization. This value was set as 100%. As shown in Table 2, the percent of internal NZ-1-(scdsFv)-PE38KDEL-AF488 in D2159MG (80%) and D08-0308MG (87%), was 2-fold greater than that of D08-0493MG (42%). On the D425MED xenograft cells, NZ-1-(scdsFv)-PE38KDEL-AF488 was internalized at a much faster rate. At a concentration of 10 μg/mL, 93% of NZ-1-(scdsFv)-PE38KDEL-AF488 on D425MED cells were internalized while only 21% of the IT was internalized on DAOY cells (Table 2). Hence, different brain tumor cells show variation in their rates of NZ-1-(scdsFv)-PE38KDEL internalization.
Anti-tumor activity of NZ-1-(scdsFv)-PE38KDEL in subcutaneous tumor model
The antitumor activity of NZ-1-(scdsFv)-PE38KDEL was investigated in female athymic nude mice bearing D2159MG or D283MED subcutaneous tumors. D2159MG or D283MED tumors were treated intratumorally with three doses of the NZ-1-(scdsFv)-PE38KDEL at a 0.3-mg/kg concentration. The control groups were treated with either 0.2% PBS-HSA or with 0.3-mg/kg of the nonspecific control IT, P588-(scFv)-PE38KDEL. The D2159MG xenograft tumors regressed in 8/10 mice treated with NZ-1-(scdsFv)-PE38KDEL. Compared to the control IT-treated animals, a growth delay of 19 days, with a P-value of <0.001, was observed in NZ-1-(scdsFv)-PE38KDEL treated group (Fig. 3a). Similarly, with the D283MED model a tumor-growth delay of 10 days, with a P-value of <0.001, along with two tumor regressions was observed in the NZ-1-(scdsFv)-PE38KDEL group as opposed to the control group (Fig. 3b). No toxicity-related deaths or adverse effects were observed in any of the treated animals.
Figure 3.
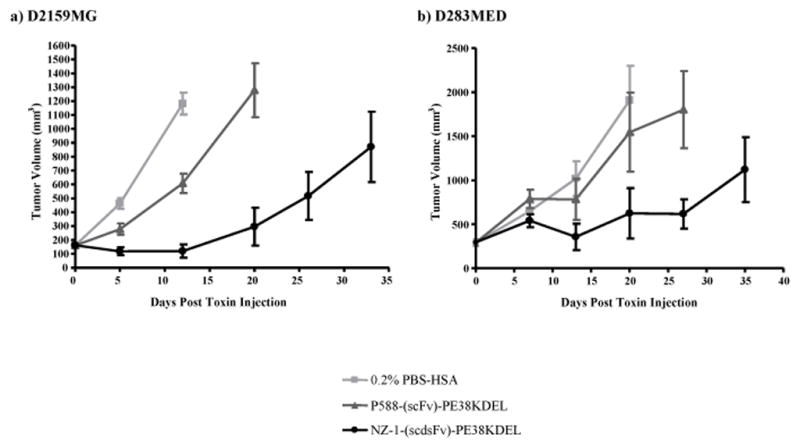
Effect of NZ-1-(scdsFv)-PE38KDEL on D2159MG and D283MED subcutaneous tumors in athymic nude mice. Female athymic nude mice (≈20 g; 4–6 weeks) bearing D2159MG tumors (a) or D283MED tumors (b) were randomized into three groups, 0.2% PBS-HSA (■), P588-(scFv)-PE38KDEL (▲), and NZ-1-(scdsFv)-PE38KDEL (●). Once the tumors reached an average volume of 150–300 mm3, the mice were treated by i.t. injections every other day with a total of three 0.3-mg/kg doses of P588-(scFv)-PE38KDEL or NZ-1-(scdsFv)-PE38KDEL diluted in 0.2% PBS-HSA or with 0.2% PBS-HSA alone.
Intracranial efficacy of NZ-1-(scdsFv)-PE38KDEL on D425MED tumor model
To determine the time course of D425MED intracranial tumor growth in the xenograft model, 1×105 cells/5 μl was injected into male NSG mice (11 mice) and a survival curve was plotted (Supplementary Fig. S5). The D425MED survival curve demonstrated that 100% death occurred at day 14 post tumor implantation; hence, post tumor implantation day 4 was chosen as the optimal day to study the efficacy of NZ-1-(scdsFv)-PE38KDEL. Further, to determine whether NZ-1-(scdsFv)-PE38KDEL would be tolerated intracranially, NSG mice were given different concentrations of the NZ-1-(scdsFv)-PE38KDEL IT (0.1 μg–3.0 μg/100 μl) through an Alzet pump over a three day period (5 mice/group). Supplementary figure S6 depicts that both 0.1 μg and 0.3 μg of NZ-1-(scdsFv)-PE38KDEL had no toxicity associated mortality. Therefore, 0.3 μg of NZ-1-(scdsFv)-PE38KDEL, was selected to be the ideal concentration for intracranial therapy.
Based on the survival and toxicity studies, the efficacy study was performed on day 4 post D425MED tumor implantation with 0.3 μg dose of the NZ-1-(scdsFv)-PE38KDEL. The control groups were either left untreated (sham) or treated with 0.2% PBS-HSA or with 0.3 μg of the nonspecific control IT, P588-(scdsFv)-PE38KDEL. Treatment of the tumor-bearing mice with NZ-1-(scdsFv)-PE38KDEL increased survival by 41% (Fig. 4) and the Kaplan–Meyer survival curve demonstrated a statistically significant (P≤0.001) increase in survival time in the 0.3 μg NZ-1-(scdsFv)-PE38KDEL-treated mice as compared to the controls (Fig. 4).
Figure 4.
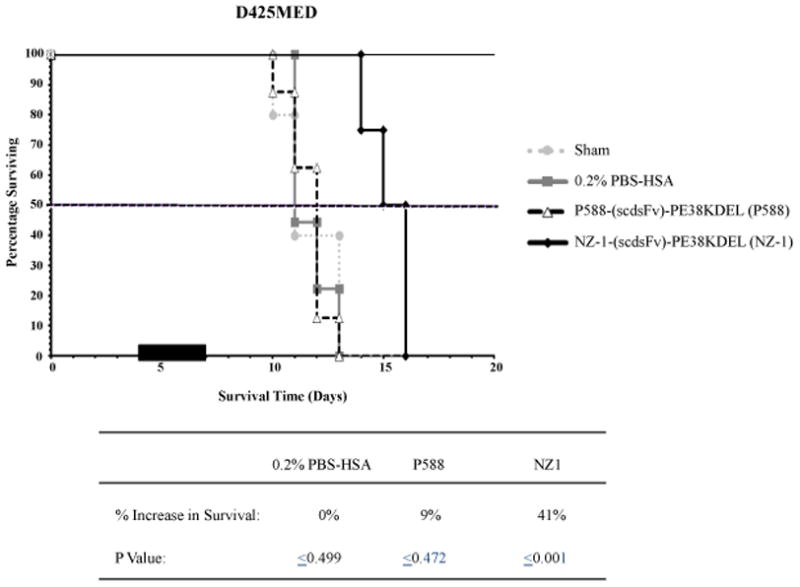
Effect of NZ-1-(scdsFv)-PE38KDEL on D425MED intracranial tumors in NSG mice. Male NSG mice (≈20–30 g; 12 weeks) bearing D425MED tumors were randomized into four groups, sham (●), 0.2% PBS-HSA (■), P588-(scdsFv)-PE38KDEL (3), and NZ-1-(scdsFv)-PE38KDEL (◆). Four days post tumor implantation, the mice were either left untreated (sham) or treated with 0.3 μg of P588-(scdsFv)-PE38KDEL or NZ-1-(scdsFv)-PE38KDEL diluted in 100 μl of 0.2% PBS-HSA or with 0.2% PBS-HSA alone, delivered intracranially through an Alzet pump over a three day period.
Discussion
The clinical success of mAbs and recombinant IT-based targeted therapies against solid intracranial tumors has led investigators to identify novel brain-tumor-specific antigen targets.30 Podoplanin is one such antigen that is highly expressed on several CNS tumors.13,14 NZ-1 is a mAb exhibiting high reactivity to podoplanin (KD = 1.2×10−10 M) and when bound by glioblastoma cells expressing podoplanin, NZ-1 mAb is efficiently internalized,21 thereby demonstrating its potential as an important immunotherapeutic agent. In our current study, we demonstrate the expression of podoplanin on both glioblastoma and medulloblastoma xenografts by flow cytometry using NZ-1 mAb. Further, we constructed a recombinant IT, NZ-1-(scdsFv)-PE38KDEL, by fusing the scFv segment of the NZ-1 mAb with the PE38KDEL molecule. Similar to the NZ-1 mAb, the NZ-1-(scdsFv)-PE38KDEL IT exhibited considerable binding to both glioblastoma and medulloblastoma cells by flow cytometry. The affinity of NZ-1-(scdsFv)-PE38KDEL was determined to be KD = 8.0×10−8 M by Biacore. NZ-1-(scdsFv)-PE38KDEL displayed considerable stability when incubated at 37°C for long time periods. NZ-1-(scdsFv)-PE38KDEL demonstrated significant cytotoxicity against both glioblastoma and medulloblastoma cells. Further, in subcutaneous and intracranial animal models, with glioblastoma or medulloblastoma xenografts, NZ-1-(scdsFv)-PE38KDEL demonstrated significant tumor growth delays (P<0.001), which makes it a novel and efficacious immunotherapeutic agent against malignant brain tumors.
Consistent with earlier studies, we demonstrate podoplanin to be present in both glioblastoma and medulloblastoma xenografts.14 Flow cytometry analysis with cells isolated from glioblastoma (D2159MG, D08-0308MG, and D08-0493MG) and medulloblastoma xenografts (D283MED and D425MED) and a medulloblastoma cell line (DAOY) demonstrate different levels of reactivity to both the NZ-1 mAb and NZ-1-(scdsFv)-PE38KDEL, which directly correlates with podoplanin expression (Fig. 1). Further, the QFACS data demonstrates a higher level of podoplanin to be present on glioblastoma xenografts than on the medulloblastoma xenografts. This difference in cell surface levels of podoplanin could contribute to the lower cytotoxicity values obtained with NZ-1-(scdsFv)-PE38KDEL against glioblastoma xenografts as opposed to medulloblastoma cells (Table 2). Moreover, despite having similar cytotoxicity values within themselves, the levels of podoplanin expression and the rate of podoplanin receptor internalization varied considerably among both glioblastoma and medulloblastoma xenografts (Table 2). Thus, both the cell surface levels of podoplanin and the internalization rate determines the cytotoxic activity of NZ-1-(scdsFv)-PE38KDEL.
The affinity of NZ-1-(scdsFv)-PE38KDEL was determined by surface plasmon resonance with podoplanin peptide (hpp38-51). The association rate, ka = 4.9 × 104 (Ms) −1 and the dissociation rate, kd = 3.9 × 10−3 s−1, of NZ-1-(scdsFv)-PE38KDEL was 2.6-fold lower and 80-fold higher than that of the NZ-1 mAb, respectively. This difference in the association and dissociation rates observed between the NZ-1-(scdsFv)-PE38KDEL and NZ-1 mAb could be explained by the monovalent and bivalent nature of the IT and mAb, respectively. Despite having lower affinity, the cytotoxic activity (IC50 values) of NZ-1-(scdsFv)-PE38KDEL was comparable to the activity obtained with a mutant epidermal growth factor receptor-specific IT currently being evaluated in Phase I clinical trial for glioblastoma patients.24,31.
Additionally, an IT has to be significantly stable over long periods of time to demonstrate therapeutic efficacy when administered to brain tumor patients. The VH and VL domain of NZ-1 scFv are held together by a 15-amino-acid peptide linker, and it is well established that the noncovalent, interdomain interactions provided by the peptide linker are not strong enough to keep them together.32 The linker is rather flexible, and this could promote the interaction between the VH and VL domains of NZ-1-(scFv)-PE38KDEL and a nearby IT molecule, which could thereby affect the stability of the IT. We hypothesized that this problem could be overcome by introducing an interchain disulfide bond between the structurally conserved framework regions of the NZ-1 VH and VL domains.28 The new disulfide-stabilized IT is termed NZ-1-(scdsFv)-PE38KDEL. As hypothesized, the incorporation of a disulfide bond, in addition to the peptide linker, offered higher stability to NZ-1-(scdsFv)-PE38KDEL, which was evident from its lower IC50 values compared to NZ-1-(scFv)-PE38KDEL on both D2159MG xenograft cells and DOAY cells (Table 1a and 1b).
The in vivo results were compatible with the in vitro flow cytometry and cytotoxic activity data. Our in vitro results demonstrated higher reactivity and cytotoxicity of NZ-1-(scdsFv)-PE38KDEL against glioblastoma xenografts. Accordingly, NZ-1-(scdsFv)-PE38KDEL caused greater tumor regression (8/10 mice) and a significant tumor growth delay (P-value of <0.001) against the glioblastoma xenograft D2159MG. Moreover, it was also effective in producing a tumor growth delay of 10 days (P-value of <0.001) against the medulloblastoma model D283MED. In this regard, NZ-1-(scdsFv)-PE38KDEL is a truly novel IT that can target two different CNS malignancies effectively. In our in vivo animal models, the NZ-1-(scdsFv)-PE38KDEL IT was administered by intratumoral injections. In the treatment of malignant brain tumors the NZ-1-(scdsFv)-PE38KDEL IT will be administered directly into the tumor site and thus bypass the blood-brain barrier by CED.31 To replicate an actual brain tumor model, the D425MED xenograft cells were implanted intracranially and were treated by CED with NZ-1-(scdsFv)-PE38KDEL. Similar to the subcutaneous brain tumor models NZ-1-(scdsFv)-PE38KDEL was extremely efficacious in the intracranial model and increased the survival of tumor bearing mice by 41% (P-value of ≤0.001).
In conclusion, our results clearly demonstrate that the disulfide-stabilized recombinant IT NZ-1-(scdsFv)-PE38KDEL is highly efficacious against CNS tumors expressing podoplanin in a preclinical setting and hence is a suitable candidate for targeted therapy of malignant brain tumors.
Supplementary Material
Novelty and Impact.
Podoplanin is an antigen that is overexpressed in various neoplasms, such as brain tumors, mesothelioma, squamous cell carcinoma, and germ cell tumors. NZ-1-(scdsFv)-PE38KDEL is a novel recombinant immunotoxin targeting podoplanin. The specificity and high binding affinity of NZ-1-(scdsFv)-PE38KDEL allows for specific targeting of tumor cells while avoiding adjacent normal tissue thereby having the potential to improve brain tumor patient survival.
Acknowledgments
Grant sponsor: The National Cancer Institute; Grant numbers: NINDS 5P50 NS20023-25, and NCI Merit Award R37 CA 011898-38 (D.D. Bigner); Southeastern Brain Tumor Foundation; Silvian Foundation; Grant sponsor: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (I.H. Pastan).
Abbreviations
- MG
malignant glioma
- MED
medulloblastoma
- BSA
bovine serum albumin
- CDR
complementarity-determining region
- FACS
fluorescence-activated cell sorter
- QFACS
quantitative FACS
- FBS
fetal bovine serum
- FR
framework
- HC
heavy chain
- HSA
human serum albumin
- IC50
50% inhibitory concentration
- i.t
intratumoral
- i.c
intracranial
- IT
immunotoxin
- LC
light chain
- mAb
monoclonal antibody
- s.c
subcutaneous
- scFv
single-chain antibody fragment
- KDEL
Lysine-aspartic acid-glutamic acid-leucine
References
- 1.http://www.cbtrus.org/2011-NPCR-SEER/WEB-0407-Report-3-3-2011.pdf
- 2.Scott JN, Rewcastle NB, Brasher PM, Fulton D, MacKinnon JA, Hamilton M, Cairncross JG, Forsyth P. Which glioblastoma multiforme patient will become a long-term survivor? A population-based study. Ann Neurol. 1999;46:183–8. [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Wagner M, Bigner DD, Friedman AH, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 5.Giangaspero FEC, Haapasalo H, Pietsch T, Wiestler OD, Ellison DW. Medulloblastoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. 4. Lyon, France: International Agency for Research on Cancer; 2007. pp. 132–40. [Google Scholar]
- 6.Roussel MF, Robinson G. Medulloblastoma: advances and challenges. F1000 Biol Rep. 2011;3:5. doi: 10.3410/B3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 8.Thaker NG, Pollack IF. Molecularly targeted therapies for malignant glioma: rationale for combinatorial strategies. Expert Rev Neurother. 2009;9:1815–36. doi: 10.1586/ern.09.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bullard DE, Bigner DD. Applications of monoclonal antibodies in the diagnosis and treatment of primary brain tumors. J Neurosurg. 1985;63:2–16. doi: 10.3171/jns.1985.63.1.0002. [DOI] [PubMed] [Google Scholar]
- 10.Weldon JE, Pastan I. A guide to taming a toxin - recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278:4683–700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raica M, Cimpean AM, Ribatti D. The role of podoplanin in tumor progression and metastasis. Anticancer Res. 2008;28:2997–3006. [PubMed] [Google Scholar]
- 12.Schacht V, Ramirez MI, Hong YK, Hirakawa S, Feng D, Harvey N, Williams M, Dvorak AM, Dvorak HF, Oliver G, Detmar M. T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J. 2003;22:3546–56. doi: 10.1093/emboj/cdg342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishima K, Kato Y, Kaneko MK, Nishikawa R, Hirose T, Matsutani M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol. 2006;111:483–8. doi: 10.1007/s00401-006-0063-y. [DOI] [PubMed] [Google Scholar]
- 14.Shibahara J, Kashima T, Kikuchi Y, Kunita A, Fukayama M. Podoplanin is expressed in subsets of tumors of the central nervous system. Virchows Arch. 2006;448:493–9. doi: 10.1007/s00428-005-0133-x. [DOI] [PubMed] [Google Scholar]
- 15.Kato Y, Kaneko MK, Kunita A, Ito H, Kameyama A, Ogasawara S, Matsuura N, Hasegawa Y, Suzuki-Inoue K, Inoue O, Ozaki Y, Narimatsu H. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci. 2008;99:54–61. doi: 10.1111/j.1349-7006.2007.00634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunita A, Kashima TG, Morishita Y, Fukayama M, Kato Y, Tsuruo T, Fujita N. The platelet aggregation-inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am J Pathol. 2007;170:1337–47. doi: 10.2353/ajpath.2007.060790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin-Villar E, Megias D, Castel S, Yurrita MM, Vilaro S, Quintanilla M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J Cell Sci. 2006;119:4541–53. doi: 10.1242/jcs.03218. [DOI] [PubMed] [Google Scholar]
- 18.Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D, Christofori G. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell. 2006;9:261–72. doi: 10.1016/j.ccr.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Ogasawara S, Kaneko MK, Price JE, Kato Y. Characterization of anti-podoplanin monoclonal antibodies: critical epitopes for neutralizing the interaction between podoplanin and CLEC-2. Hybridoma (Larchmt) 2008;27:259–67. doi: 10.1089/hyb.2008.0017. [DOI] [PubMed] [Google Scholar]
- 20.Kato Y, Kaneko MK, Kuno A, Uchiyama N, Amano K, Chiba Y, Hasegawa Y, Hirabayashi J, Narimatsu H, Mishima K, Osawa M. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem Biophys Res Commun. 2006;349:1301–7. doi: 10.1016/j.bbrc.2006.08.171. [DOI] [PubMed] [Google Scholar]
- 21.Kato Y, Vaidyanathan G, Kaneko MK, Mishima K, Srivastava N, Chandramohan V, Pegram C, Keir ST, Kuan CT, Bigner DD, Zalutsky MR. Evaluation of anti-podoplanin rat monoclonal antibody NZ-1 for targeting malignant gliomas. Nucl Med Biol. 2010;37:785–94. doi: 10.1016/j.nucmedbio.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambson BE, Bernstein R, Mendelow BV. Chromosomal evolution in secretory and nonsecretory subline of MOPC 21. Somat Cell Mol Genet. 1990;16:91–5. doi: 10.1007/BF01650484. [DOI] [PubMed] [Google Scholar]
- 23.Buchner J, Pastan I, Brinkmann U. A method for increasing the yield of properly folded recombinant fusion proteins: single-chain immunotoxins from renaturation of bacterial inclusion bodies. Anal Biochem. 1992;205:263–70. doi: 10.1016/0003-2697(92)90433-8. [DOI] [PubMed] [Google Scholar]
- 24.Beers R, Chowdhury P, Bigner D, Pastan I. Immunotoxins with increased activity against epidermal growth factor receptor vIII-expressing cells produced by antibody phage display. Clin Cancer Res. 2000;6:2835–43. [PubMed] [Google Scholar]
- 25.Friedman HS, Houghton PJ, Schold SC, Keir S, Bigner DD. Activity of 9-dimethylaminomethyl-10-hydroxycamptothecin against pediatric and adult central nervous system tumor xenografts. Cancer Chemother Pharmacol. 1994;34:171–4. doi: 10.1007/BF00685936. [DOI] [PubMed] [Google Scholar]
- 26.Friedman HS, Colvin OM, Skapek SX, Ludeman SM, Elion GB, Schold SC, Jr, Jacobsen PF, Muhlbaier LH, Bigner DD. Experimental chemotherapy of human medulloblastoma cell lines and transplantable xenografts with bifunctional alkylating agents. Cancer Res. 1988;48:4189–95. [PubMed] [Google Scholar]
- 27.Gehan EA. A Generalized Wilcoxon Test for Comparing Arbitrarily Singly-Censored Samples. Biometrika. 1965;52:203–23. [PubMed] [Google Scholar]
- 28.Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nat Biotechnol. 1996;14:1239–45. doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- 29.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57:4130–40. [PubMed] [Google Scholar]
- 30.Li YM, Hall WA. Targeted toxins in brain tumor therapy. Toxins (Basel) 2010;2:2645–62. doi: 10.3390/toxins2112645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampson JH, Brady M, Raghavan R, Mehta AI, Friedman AH, Reardon DA, Petry NA, Barboriak DP, Wong TZ, Zalutsky MR, Lally-Goss D, Bigner DD. Colocalization of gadolinium-diethylene triamine pentaacetic acid with high-molecular-weight molecules after intracerebral convection-enhanced delivery in humans. Neurosurgery. 2011;69:668–76. doi: 10.1227/NEU.0b013e3182181ba8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiter Y, Pastan I. Antibody engineering of recombinant Fv immunotoxins for improved targeting of cancer: disulfide-stabilized Fv immunotoxins. Clin Cancer Res. 1996;2:245–52. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.