

Abstract
Various types of stress, such as disruption of calcium homeostasis, inhibition of protein glycosylation and reduction of disulfide bonds, result in accumulation of misfolded proteins in the endoplasmic reticulum (ER). The initial cellular response involves removal of such proteins by the ER, but excessive and/or long-term stress results in apoptosis. In this study, we used a randomized ribozyme library and ER stress-mediated apoptosis (tunicamycin-induced apoptosis) in SK-N-SH human neuroblastoma cells as a selective phenotype to identify factors involved in this process. We identified a double-stranded RNA-dependent protein kinase (PKR) as one of the participants in this process. The level of nuclear PKR was elevated, but the level of cytoplasmic PKR barely changed in tunicamycin-treated SK-N-SH cells. Furthermore, tunicamycin also raised levels of phosphorylated PKR in the nucleus. We also detected the accumulation of phosphorylated PKR in the nuclei of autopsied brain tissues in Alzheimer's disease. Thus, PKR might play a role in ER stress-induced apoptosis and in Alzheimer's disease.
Keywords: Alzheimer's disease; ER stress; neuronal degenerative diseases; PKR; ribozyme; Abbreviations:; ER, endoplasmic reticulum; AD, Alzheimer's disease; Rz, hammerhead ribozyme; Tm, tunicamycin; GRP, glucose-regulated protein; PKR, double-stranded RNA-dependent protein kinase; PACT, PKR-activating protein; eIF2α, eucaryotic translation initiation factor subunit 2α; EBV, Epstein–Barr virus; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TUNEL, TdT-mediated dUTP nick end-labeling; Aβ, amyloid β peptide; PBS, phosphate-buffered saline; WT, wild type; DN, dominant negative
Introduction
Approximately one-third of newly synthesized proteins, in particular, transmembrane and secretory proteins, are transported to the lumen of the endoplasmic reticulum (ER). Many post-translational modifications, including folding and oligomerization of such proteins, which are destined for distal compartments in the secretory pathway, occur in the ER. The ER also functions as an efficient quality-control system, and one of its roles is to prevent incompletely folded molecules from moving along secretory pathways. However, some forms of cellular stress (known collectively as ER stress), such as depletion of calcium ions from the ER lumen, inhibition of protein glycosylation, reduction of disulfide bonds, expression of mutant proteins and ischemic insults, lead to the accumulation of unfolded proteins in the ER (Kozutsumi et al, 1988; Kaufman, 1999).
Misfolding of proteins triggers three compensatory responses: the unfolded protein response, which is mediated by increased expression of molecular chaperones, such as GRP94 (GRP: glucose-regulated protein) and GRP78/Bip, which promote the proper folding of proteins (Mori, 2000; Urano et al, 2000); the generalized suppression of translation (Harding et al, 1999); and ER-associated degradation (Ng et al, 2000). These three protective responses act transiently to control the accumulation of misfolded proteins within the ER, but sustained ER stress leads to apoptosis, with the characteristic fragmentation of nuclei, condensation of chromatin and shrinkage of cell bodies (Imaizumi et al, 2001).
It appears that ER stress is associated with certain genetic and/or neuronal degenerative diseases, such as Alzheimer's disease (AD). Several possible pathogenic mechanisms have been proposed for AD, including mutations in or expression of alternatively spliced variants of presenilin, impairment of the signaling of ER stress and increased sensitivity to stress-induced apoptosis (Katayama et al, 1999, 2001; Sato et al, 1999, 2001).
Hammerhead ribozymes (Rzs) are catalytic RNA molecules that bind to and cleave defined complementary target RNAs (Uhlenbeck, 1987; Haseloff and Gerlach, 1988; Symons, 1992; Zhou and Taira, 1998). Rz-mediated cleavage requires a NUX triplet in the target RNA (where N can be any ribonucleotide and X can be A, U or C; Zhou and Taira, 1998). This strict specificity allows the use of Rzs for suppression of the expression of specific genes, and a reverse functional genomic approach to gene discovery has been established that exploits randomized Rz libraries (Kruger et al, 2000; Welch et al, 2000; Kawasaki et al, 2002; Kawasaki and Taira, 2002; Chatterton et al, 2004; Nelson et al, 2003).
An Rz-mediated change in the phenotype of cells allows rapid identification of genes responsible for that particular phenotype. Using this technology, we have identified genes that are related to specific diseases (Kawasaki et al, 2004; Kawasaki and Taira, 2002; Onuki et al, 2002; Suyama et al, 2003a, 2003b) and some functional microRNAs (Kuwabara et al, unpublished data; Kawasaki et al, 2004). However, the isolation of active Rzs requires the design of a suitable selection system in each case.
To identify genes involved in AD, we focused on genes that facilitate tunicamycin-induced (Tm-induced) ER stress-mediated apoptosis. We constructed an Rz-screening system for the isolation of genes that function in Tm-mediated apoptosis and identified the gene for a double-stranded RNA (dsRNA)-dependent protein kinase (PKR). We found that Tm-mediated ER stress promoted the activation of PKR in the human neuroblastoma cell line SK-N-SH. PKR, an interferon-induced protein kinase, was identified initially in a study of responses to viral infection. It is activated by the extensive secondary structure of the viral RNA (Gale and Katze, 1998). Upon binding to dsRNA, PKR is autophosphorylated and increases cellular sensitivity to apoptotic and/or proinflammatory stimuli through a number of putative pathways, which include the phosphorylation of eucaryotic translation initiation factor subunit 2α (eIF2α) which, in turn, blocks protein synthesis (Wu and Kaufman, 1997; Srivastava et al, 1998). PKR is involved in apoptosis that is induced not only by viral infection but also by, for example, the addition or removal of specific growth factors, lipopolysaccharide or Ca2+ ions (Prostko et al, 1995; Srivastava et al, 1995, 1998).
We examined the localization of PKR in Tm-treated SK-N-SH cells and found that levels of PKR were significantly elevated in the nuclei, where PKR formed aggregates. Furthermore, the level of phosphorylated PKR, an activated form, was also elevated in a nuclear fraction. We found PKR in the cell nuclei of autopsied brain tissue from AD patients, and the level of phosphorylated PKR was significantly higher than in the nuclear fractions from disease-free controls. Our results suggest a relationship between the activation of PKR and neuronal cell death in AD.
Results
Identification of genes involved in Tm-induced apoptosis using a randomized ribozyme library
To identify genes involved in ER stress, we used a simple apoptotic phenotype-based selection system, designed to select Rzs that suppress the expression of proapoptotic genes (Kawasaki et al, 2002; Kawasaki and Taira, 2002). As shown schematically in Figure 1A, human neuroblastoma SK-N-SH cells were transiently transfected with a randomized Rz library. After 24 h, cells were treated with Tm (2 μg/ml) for another 24 h as the first round of selection. This treatment caused apoptosis in most of the empty vector-transfected cells, and we isolated Rz-expressing plasmids from cells that survived the first round of transfection with the randomized ribozyme library. We amplified the isolated plasmids in bacteria and sequenced them. In the second round, cells were transfected with the recovered Rzs and exposed to Tm for a longer period (48 h) to eliminate ‘weak positives'. The Rzs isolated after the second round of exposure to Tm were also amplified in bacteria. Escherichia coli transformants that contained the individual Rz expression plasmids appeared about five times more frequently than empty vector-transfected ‘mock' cells after the first round (Figure 1B). Furthermore, the ratio of surviving Rz-containing cells and empty vector-transfected cells was about seven to one after the second round (Figure 1B). These results suggested that the frequency of false positives was reduced after the second round.
Figure 1.
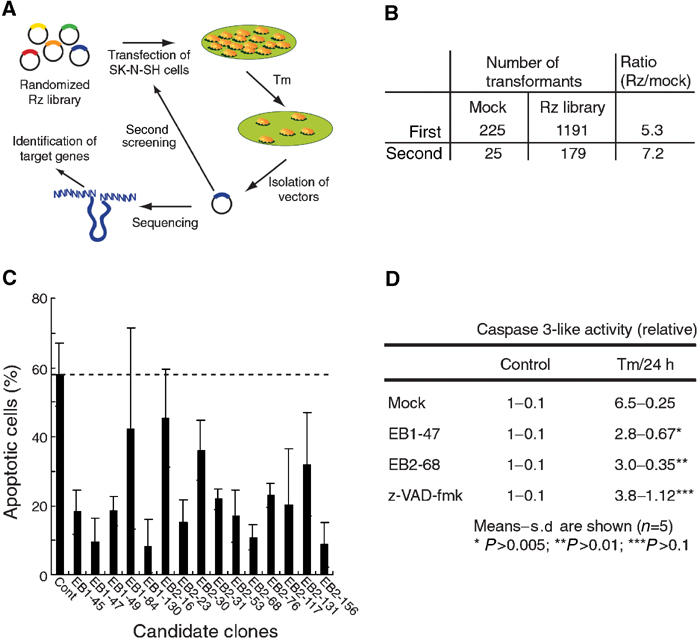
Selection of Rzs that suppress Tm-induced cell death from a randomized library. (A) Schematic representation of the selection system. (B) Number of transformants after each round of selection. Vectors isolated from surviving cells were introduced into DH5α. (C) Individual Rzs, selected from the randomized library, were examined for their ability to protect cells against Tm-mediated apoptosis. SK-N-SH cells that expressed selected Rzs were exposed to 2 μg/ml Tm for 24 h. Apoptosis was assessed by TUNEL staining. (D) The activation of caspase-3 depended on the activation of PKR. Cells were treated with Tm for 24 h or untreated (control), and then caspase-3 activity was measured. Cells treated with z-VAD-fmk were examined as controls.
As we selected cells that transiently expressed Rzs, we could not exclude the possibility that these cells might contain multiple Rzs. Multiple ribozymes might inhibit cell death cooperatively or only one might suppress cell death. To examine the individual effects of each selected Rz, we tried to construct Epstein–Barr virus-based (EBV-based) expression vectors to generate semistable cell lines. EBV-based vectors can replicate autonomously in mammalian cells and we established 15 semistable lines of cells (Figure 1C; lines EB1-45, EB1-47, EB1-49, EB1-84 and EB1-130 were obtained after the first round of screening; lines EB2-16, EB2-23, EB2-30, EB2-31, EB2-53, EB2-68, EB2-76, EB2-117, EB2-131 and EB2-156 were obtained after the second round of screening). To confirm the effects of each Rz, we treated each cell line with Tm for 24 h and then examined apoptosis by TdT-mediated dUTP nick end-labeling (TUNEL) staining. In addition, we used mock-transfected cells as a control. We counted positive cells after TUNEL staining and calculated the proportion of apoptotic cells (Figure 1C). The EB1-84, EB2-16 EB2-30, EB2-31, EB2-76 and EB2-131 clones were slightly resistant to Tm-induced apoptosis. Moreover, the Rzs expressed by clones EB1-45, EB1-47, EB1-49, EB1-130, EB2-23, EB2-53, EB2-68, EB2-117 and EB2-156 effectively suppressed Tm-induced cell death, as compared to cell death in Tm-treated control cells. These results indicated that our screening system had allowed efficient and easy selection of Rzs that suppressed the activities of proapoptotic mRNAs in Tm-induced apoptosis.
As caspase-3 is activated in Tm-treated apoptotic cells, we also confirmed the viability of EB1-47 and EB2-68 by measuring the activity of caspase-3 (Siman et al, 2001). In both EB1-47 and EB2-68 cells, caspase-3 activity was significantly lower than that in mock-transfected cells and it was similar to that in z-VAD-fmk-treated cells (Figure 1D). It appeared that expression of each Rz completely inhibited activation of caspase-3. These two clones were selected for further analysis.
Tm-mediated ER stress promotes the expression of PKR in SK-N-SH cells
We determined the sequences of the selected Rzs and identified their target genes using the BLAST search program. We focused on Rz 47 and Rz 68 from clones EB1-47 and EB2-68, and the sequences of the binding arms of these Rzs are shown in Figure 2A. The BLAST search suggested that these Rzs might be able to cleave PKR mRNA, with Rz 47 cleaving the first intron of the PKR transcript and Rz 68 cleaving PKR mRNA between nucleotides 700 and 714 (Figure 2A, arrows).
Figure 2.
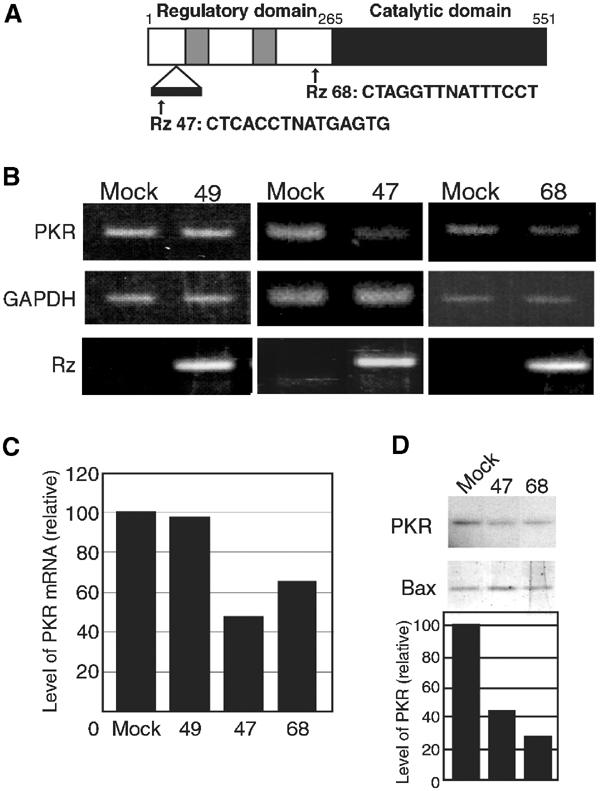
Rz 47 and Rz 68 suppress the expression of PKR. (A) Schematic illustration of candidate Rzs (Rz 47 and Rz 68) with binding sites and cleavage sites in the mRNA for PKR. Two gray boxes indicate dsRNA-binding motifs; white and black boxes indicate regulatory and catalytic domains, respectively. (B) Expression of PKR mRNA in cells transfected with the empty vector (mock) or the Rz 49, Rz 47 or Rz 68 expression vector. PKR mRNA was detected by RT–PCR with primers specific for PKR (upper). GAPDH mRNA served as an internal control (middle). Expression of Rzs was confirmed by RT–PCR with specific primers (lower). (C) Quantitative analysis of PKR mRNA. Levels of PKR mRNA were quantified and normalized by reference to levels of GAPDH mRNA. (D) Levels of PKR in three lines of cells as indicated. Extracts were subjected to Western blotting with anti-PKR (PKR) and anti-Bax antibodies (Bax). Results of quantitative analysis are also shown.
We examined levels of Rz 47 and 68 in EB1-47 and EB2-68 cells and performed an analysis by RT–PCR with Rz-specific primers. Figure 2B shows that expression of each Rz was detectable in Rz-transfected cells but not in mock-transfected cells. To confirm the activities of Rz 47 and Rz 68 against the PKR transcript, we also performed analysis by RT–PCR using primers specific for genes for PKR and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The level of PKR rises and PKR is activated by dsRNA of more than 30 bp and these phenomena hindered the initial application of RNAi technology to mammalian cells (Elbashir et al, 2001; Miyagishi and Taira, 2002). As PKR might have been activated by expression of a ribozyme itself, we used Rz 49, which does not recognize PKR mRNA, as a control. After electrophoresis, we quantified levels of PKR mRNA and normalized them by reference to levels of GAPDH mRNA. As shown in Figure 2B, the expression of Rz 49 did not change the level of PKR mRNA from that in mock-transfected cells. However, the level of PKR mRNA in EB1-47 cells was 53% lower than that in empty vector-transfected cells (Figures 2B and C). Similar results were obtained in the case of Rz 68 (37% decrease).
We performed Western blotting analysis with anti-PKR or anti-Bax antibodies to determine levels of PKR in these cells (Figure 2D). We normalized levels of PKR by reference to levels of Bax. In Rz 47-expressing cells, 58% less PKR was expressed than in mock-transfected cells. Similar results were observed in Rz 68-expressing cells (Figure 2D). Thus, Rz 47 and Rz 68 specifically suppressed the expression of PKR at the post-transcriptional level.
Role of PKR in ER stress-induced apoptosis
Srivastava et al (1995) provided evidence that depletion of Ca2+ ions in the ER triggers stress signaling that might activate eIF2α kinase, a downstream effector of PKR. Thus, elucidation of the relationship between PKR and Tm-induced ER stress in SK-N-SH cells is clearly important. PKR is localized in both the cytoplasm and the nucleus (Jeffrey et al, 1995; Besse et al, 1998). To identify the role of PKR in vulnerability to ER stress, we prepared cytoplasmic and nuclear fractions from Tm-treated and nontreated cells and subjected them to Western blotting with anti-actin and anti-KDEL antibodies. In this experiment, we employed the level of actin (rather than the level of histone) as a loading control, because it was reported that degradation of histone was caused by apoptosis (Zhang et al, 2001). Anti-KDEL antibody interacts with molecular chaperones GRP94 and GRP78/Bip in the ER. Normal cells respond to ER stress by increasing the transcription of genes for chaperones, such as GRP94 and GRP78/Bip, in the ER. At first, we performed Western blotting using anti-histone or anti-actin antibodies to check whether the fractionation had worked well. The expression of histone was observed only in the nuclear fraction using wild type (WT) of cells; therefore, we used this method for further analysis (Supplementary Material 1).
Levels of actin remained unchanged but levels of GRP94 and GRP78/Bip in the cytoplasmic fraction increased after an 8-h incubation with Tm (Figure 3A), as described previously (Kozutsumi et al, 1988; Kaufman, 1999). Neither of these chaperones was detected in the nuclear fraction, suggesting that the nuclear fraction was not contaminated by cytoplasmic material, in agreement with the results shown in Supplementary Material 1. Western blotting analysis with anti-PKR and anti-phospho-PKR antibodies revealed that, when cells were stimulated with Tm for 8 h, the level of PKR rose in the nuclei (Figure 3A and left histogram in Figure 3B) while the level of cytoplasmic PKR barely changed.
Figure 3.
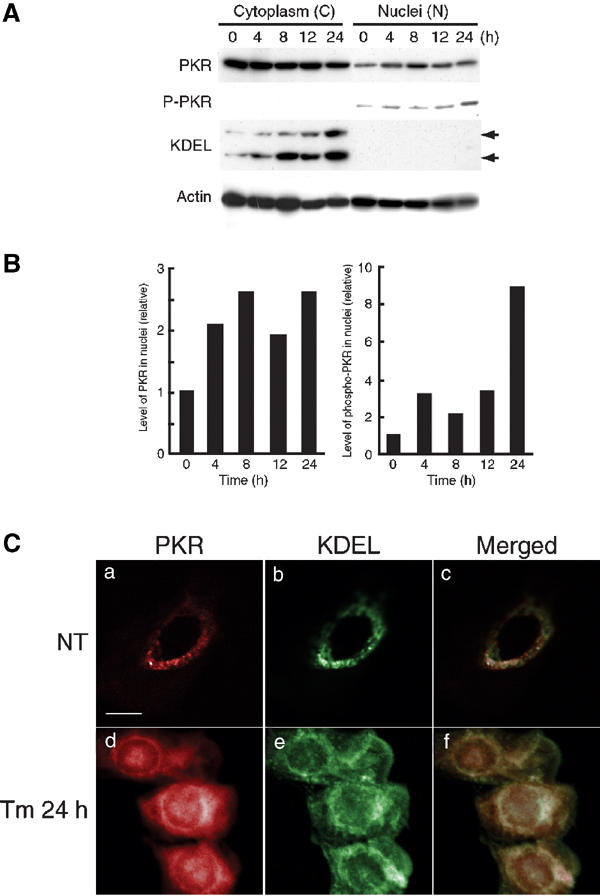
Subcellular localization of PKR and phosphorylated PKR in Tm-treated SK-N-SH cells. (A) Levels of nuclear PKR and phosphorylated PKR were elevated in SK-N-SH cells after exposure to ER stress (Tm: 2 μg/ml) for the indicated times. Cytoplasmic and nuclear fractions were subjected to Western blotting with anti-PKR, anti-phospho-PKR, anti-KDEL (upper arrow, GRP94; lower arrow, GRP78) and anti-actin antibodies as described in the text. (B) Relative intensities of bands (PKR and phosphorylated PKR) were normalized by reference to bands of β-actin. (C) Translocation of PKR to the nuclei in Tm-treated cells. Untreated (NT) and Tm-treated (2 μg/ml) cells were stained with anti-PKR and anti-KDEL antibodies (panels a and d, PKR, red; panels b and e, KDEL, green; panels c and f, overlapping images, yellow). Scale bar, 10 μm.
Phosphorylation or cleavage of PKR is required for kinase activity. Using an anti-phospho-PKR antibody (which recognizes phosphorylation of Thr446), we detected phosphorylated PKR in the nuclear fraction, and the level of this form of PKR depended on the incubation time. The maximum level of phosphorylated PKR was detected 24 h after the start of treatment (Figure 3A and right histogram in Figure 3B).
When we examined the subcellular localization of PKR by immunofluorescence microscopy after SK-N-SH cells had been treated with Tm, we found most PKR (red) in untreated control in the cytoplasm (Figure 3C, upper left). After cells had been treated with Tm, levels of nuclear PKR increased and PKR was clearly localized in the nuclei (Figure 3C, lower left), while levels of GRP94 and GRP78/Bip (green) increased in the ER of treated cells (Figure 3C, middle). These results suggested that Tm-mediated ER stress promoted the expression of PKR in SK-N-SH cells.
Alanine-mutated PKR confers resistance to Tm
We investigated whether phosphorylated PKR might affect Tm-induced cell death. We constructed mutated PKR in which threonine at residues 446 and 451 was changed to alanine. This mutant lacks an important phosphorylation site and acts as a dominant-negative (DN) protein (Jagus et al, 1999). SK-N-SH cells were transfected with the expression vector for DN-PKR or the empty vector. We also tried to establish cells expressing the WT PKR; however, overexpression of WT PKR caused apoptosis as described previously (Srivastava et al, 1995). Therefore, we employed mock-treated cells as a control. Selection yielded clones with the empty vector and the vector that encoded DN-PKR, namely, AlaC1 and AlaC2. We confirmed the expression of DN-PKR using anti-GFP antibody, since GFP was fused to PKR as a tag (Figure 4A).
Figure 4.
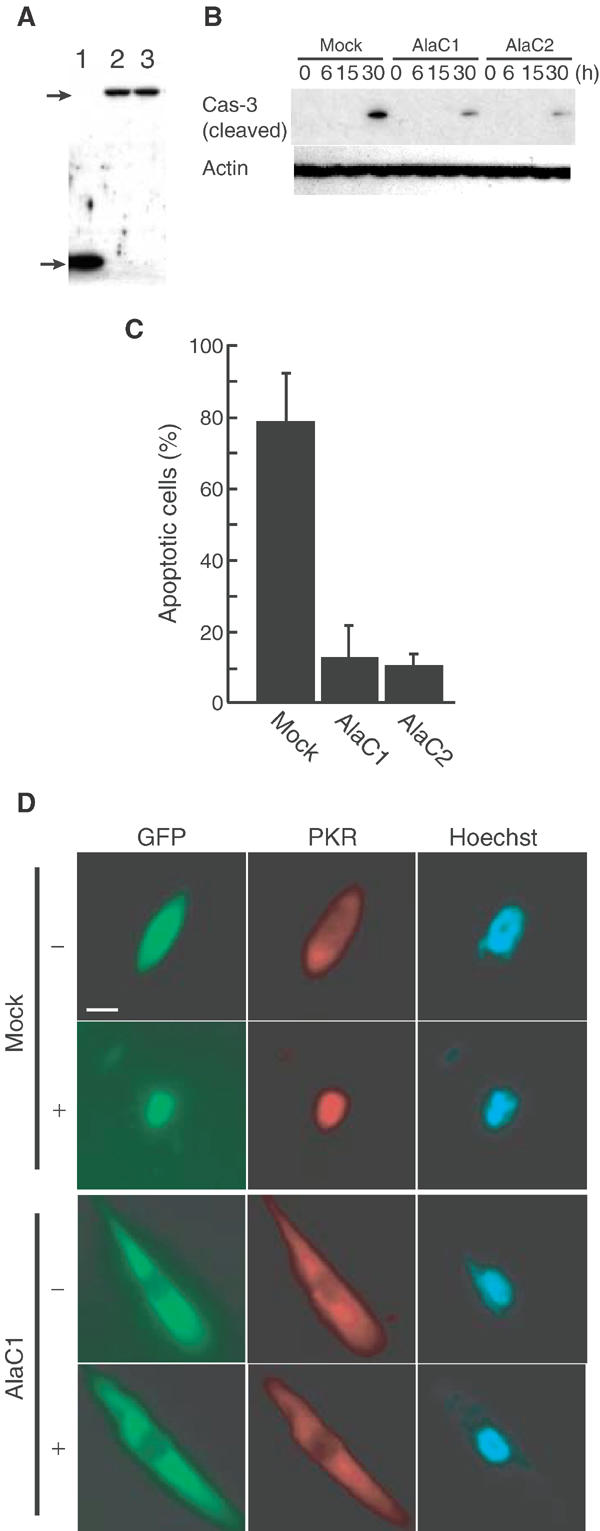
Expression of DN-PKR inhibits Tm-induced apoptosis. (A) Immunoblotting analysis of the expression of DN-PKR. Extracts of GFP-expressing cells (lane 1) and two independent clones (lanes 2 and 3) expressing DN-PKR fused to GFP were examined with anti-GFP antibody. The upper arrow indicates DN-PKR fused to GFP and the lower arrow indicates GFP. (B) Levels of activated caspase-3 in Tm-treated cells. Cells expressing GFP or GFP-DN-PKR were incubated with Tm (2 μg/ml), and activation of caspase-3 was examined by Western blotting with anti-cleaved caspase-3 antibody. Levels of β-actin were analyzed as an internal control. (C) Tm-mediated apoptosis is inhibited by expression of DN-PKR. Cells were treated with 2 μg/ml Tm for 24 h and apoptosis was assessed by TUNEL staining. (D) Localization of PKR and DN-PKR in Tm-treated cells. Untreated cells (−) and Tm-treated cells (+) were stained with anti-PKR antibody (PKR; red). Green fluorescence indicates DN-PKR; the nuclei were stained with Hoechst 33258 (blue). Scale bar, 10 μm.
We incubated AlaC1, AlaC2 and the control cells with Tm for 30 h and examined levels of cleaved caspase-3 (activated caspase-3). As shown in Figure 4B, small amounts of cleaved caspase-3 were detected in AlaC1 and AlaC2 clones, but a large amount of cleaved caspase-3 was detected in the mock-transfected clone. Levels of actin were used as internal controls that remained unchanged. Next, we incubated AlaC1, AlaC2 and mock cells with Tm for 24 h, and then performed TUNEL staining. As shown in Figure 4C, about 75% of cells that expressed DN-PKR survived, while only 20% of mock-transfected cells survived (Figure 4C), suggesting that expression of DN-PKR inhibited apoptosis.
Next, we examined the localization of PKR in cells that expressed DN-PKR (Figure 4D). Control and DN-PKR-expressing cells were incubated with Tm. Before treatment, most PKR was localized in the cytoplasm in both cell lines. After incubation, PKR was localized in the nuclei, while DN-PKR remained in the cytoplasm, suggesting that expression of mutated PKR changed the localization of PKR and inhibited apoptosis.
Levels of nuclear PKR in AD brain tissue
Considerable evidence suggests that ER stress is closely related to genetic and/or neuronal degenerative diseases, such as AD (Katayama et al, 1999, 2001; Sato et al, 1999, 2001; Imaizumi et al, 2001). Therefore, we examined levels of PKR in the autopsied brains of sporadic AD patients. We prepared crude nuclear and cytoplasmic fractions (for seven disease-free controls and 17 patients with AD) for Western blotting with anti-phospho-PKR and anti-actin antibodies. Phosphorylated PKR was barely detectable in the cytoplasmic fractions from AD tissues and tissues from age-matched controls (data not shown). Levels of phosphorylated PKR were significantly higher in crude nuclear fractions of AD tissues than of controls (Figure 5A; actin was used as a loading control). Levels of phosphorylated PKR were quantified and normalized by reference to levels of actin (Figure 5B). In most AD samples, the level of phosphorylated PKR was significantly higher than in age-matched control samples.
Figure 5.
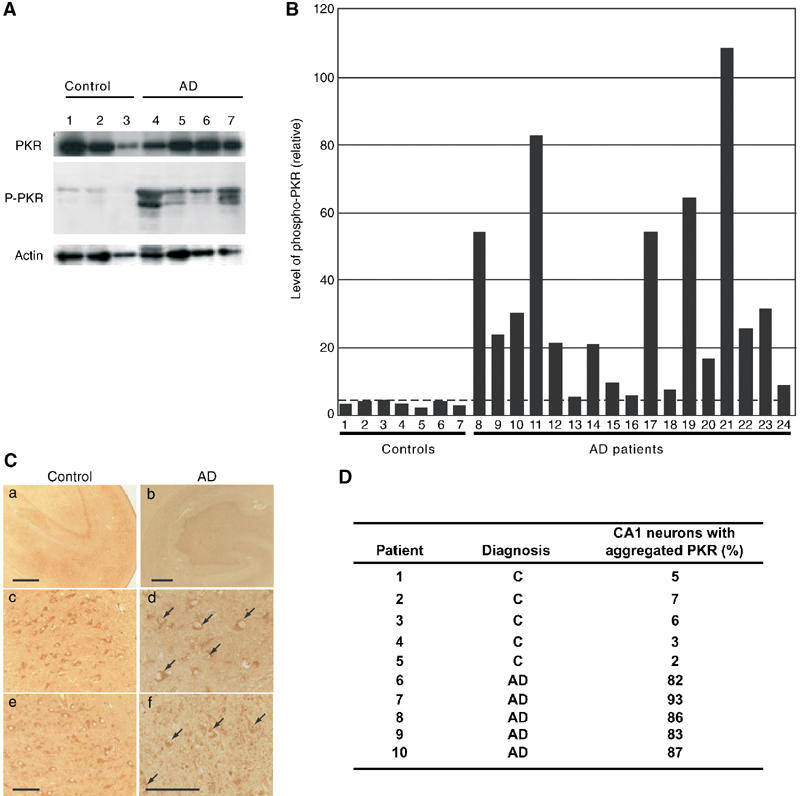
PKR is concentrated in the nuclei and is phosphorylated in autopsied AD brains. (A) Levels of phosphorylated PKR in autopsied AD brains. Nuclear proteins in the extracts of temporal cortex from patients with AD (lanes 4–7) were subjected to Western blotting with anti-PKR (PKR), anti-phospho-PKR (P-PKR) and anti-actin (actin) antibodies. Controls were age-matched and are described in the text (lanes 1–3). (B) Quantitative analysis of levels of phosphorylated PKR, normalized by reference to levels of β-actin, in the temporal cortex. Numbers 1–7, disease-free controls; numbers 8–24, sporadic AD patients (AD). Numbers 1–3 correspond to lanes 1–3 and numbers 8–11 correspond to lanes 4–7 in (A). (C) Expression of PKR in AD autopsy tissue. Panels a, c and e show tissues from an age-matched control and panels b, d and f show tissues from an AD autopsy. Panels a and b, hippocampus; panels c and d, CA1 region; panels e and f, CA3 region. Arrows indicate nuclear aggregates of PKR. Scale bar: panels a and b, 100 μm; panels e and f, 10 μm. (D) Aggregated PKR in AD brains. Aggregated PKR was significantly more common in sporadic AD brains than in controls. AD, sporadic AD patient; C, age-matched control.
To study the localization of PKR, we examined the autopsied hippocampus of an AD patient. The hippocampus is affected early in the course of AD and has been studied extensively (Selkoe, 1994). Immunohistochemical analysis suggested that PKR immunoreactivity was slightly higher in the hippocampus of the AD patient than in the control (Figure 5C, panel a versus b). Under high magnification, PKR was observed specifically in the nuclei, in particular in the CA1 and CA3 regions (Figure 5C, panels d and f, arrows). Similar observations were made in all the three samples of AD tissue examined. In contrast, immunoreactive PKR in age-matched controls was detected only in the cytoplasm of cells (Figure 5C, panels c and e).
To determine the proportion of neurons with aggregated PKR in sporadic AD (five cases) and age-matched controls (non-neurological diseases; five cases), we performed an immunohistochemical analysis of sections of the hippocampal CA1 region in each case (Figure 5D). Aggregation of PKR was observed in all specimens from AD brains, and the proportion of positive neurons in AD brains was above 80%. In contrast, positive neurons accounted for only 2–7% of the total in age-matched controls. These results suggest that phosphorylation and aggregation of PKR in the nucleus of neurons might be associated with the pathology of AD.
Discussion
In this study, we developed a system for selection of Rzs that suppress the expression of proapoptotic genes that are involved in Tm-induced apoptosis (Figures 1A and B). To confirm the activity of 10 selected Rzs, we performed TUNEL staining after treatment of Rz-expressing cells with Tm. As shown in Figure 1C, most of the selected Rzs conferred resistance against Tm-induced apoptosis. However, we could not eliminate contamination by false-positive clones after the second selection. This observation suggested that Tm failed to cause apoptosis uniformly and that a small fraction of cells was able to avoid apoptosis in the absence of active Rzs. Thus, to confirm the reproducibility of the effect of each selected Rz on Tm-induced apoptosis, we used EBV-based vectors to express each individual Rz for a third round of selection and further analysis.
EBV-based vectors are extrachromosomal vectors that replicate autonomously in mammalian cells, but not in rodent cells, and they are distributed to both daughter cells after cytokinesis (Tanaka et al, 1999). Such vectors are never inserted into the host genome and, thus, the level of Rz expression remains constant in each transfected cell and it is not necessary to select multiple clones for analysis of a specific Rz.
Using semistable EBV-transfected clones, we performed TUNEL staining as a third selection to eliminate weak positives (Figure 1C). Our established protocol allowed the efficient and easy selection of active Rzs from the randomized library, and the selected Rzs suppressed ER stress-mediated apoptosis. As each Rz binds to and cleaves a defined RNA that has a complementary target sequence, the sequences of the substrate-binding arms of the selected Rzs allowed us to identify ER stress-related genes using a computerized search program.
The results of the third selection and the assay of caspase-3 activity focused our attention on Rz 47 and Rz 68, which appeared to target PKR mRNA (Figures 1C and D). dsRNAs of more than 30 bp bind to and activate PKR. To examine whether expression of Rzs could, itself, activate PKR, we performed RT–PCR using Rz 49-expressing cells (Rz 49 does not recognize PKR mRNA). As shown in Figures 2B and C, levels of PKR mRNA were the same in mock-transfected cells and Rz 49-transfected cells. Thus, a nonspecific Rz did not activate PKR. Cells expressing Rz 47 and Rz 68 efficiently suppressed the expression of PKR mRNA, as indicated by RT–PCR (Figures 2B and C) and so we selected these two clones for further analysis.
We examined the role of PKR in Tm-mediated ER stress using WT SK-N-SH cells and their Rz-expressing derivatives. In general, PKR is expressed in an inactive state. However, when cells are subjected to certain stresses, two independent pathways activate PKR. One involves the autophosphorylation of PKR after the binding of dsRNA (Gale and Katze, 1998) or a dsRNA-independent activator, PKR-activating protein (PACT) (Prostko et al, 1995; Patel and Sen, 1998; Patel et al, 2000; Peters et al, 2001). The other involves caspase-catalyzed cleavage of PKR between its regulatory and kinase domains (Satoh et al, 1999; Saelens et al, 2001), without a requirement for phosphorylation.
As shown in Figure 3, we analyzed the relationship between activation of PKR and Tm-induced ER stress in WT SK-N-SH cells. When ER stress was induced by Tm, the level of PKR or of phosphorylated PKR increased in the nuclei (Figures 3A and B), but no caspase-dependent cleavage of PKR was detected (data not shown). We also examined levels of PACT, which remained unchanged during Tm-induced cell death (data not shown).
We examined whether phosphorylation of PKR is necessary for apoptosis. We constructed a DN form of PKR (DN-PKR) in which Thr446 and Thr451 were changed to alanine, since phosphorylation of these two residues is critical for activation of PKR (Jagus et al, 1999). Two independent clones expressing DN-PKR were resistant to Tm-induced cell death (Figures 4A–C). Therefore, activation of PKR seemed to depend on its phosphorylation, and only phosphorylated PKR acted as a proapoptotic factor in Tm-induced apoptosis. We did not, however, identify any specific activator that binds to PKR. We also established a cell line that overexpressed PKR fused to GFP as a tag. We monitored the localization of PKR during Tm-induced apoptosis in these cells, as shown in Figure 4D. We failed to establish cell lines that overexpressed intact PKR because overexpression of PKR leads to apoptosis, as described previously (Srivastava et al, 1998). As shown in ‘Supplementary Material 2', we observed the localization of WT PKR fused with GFP transiently. In control cells (expressing GFP protein), fluorescence of GFP diffused both in the nucleus and the cytoplasm. WT PKR fused with GFP also localized in the nucleus and the cytoplasm; however, the nuclear PKR seemed to aggregate like those in Tm-induced WT cells (Figures 3C, panel d). Furthermore, this localization is clearly different from that of DN-PKR-expressing cells. These results suggest that GFP fusing does not inactivate PKR activity. In addition, transient expression of WT PKR fused with GFP also caused apoptosis (data not shown).
In a recent report, we suggested that a relationship might exist between stress, at the level of the ER, and neuronal cell death in AD (Katayama et al, 1999). It has been reported that caspase-12 is localized in the ER and is activated by ER stress. In addition, caspase-12-deficient cortical neurons are defective in apoptosis that is induced by amyloid-β peptide (Aβ; a major constituent of senile plaques). Therefore, caspase-12 appears to mediate an ER-specific pathway to apoptosis and might contribute to the neurotoxicity of Aβ (Nakagawa et al, 2000; Nakagawa and Yuan, 2000). Thus, the apoptotic cascade that originates in the ER might play a critical role in neuronal cell death. For this reason, we compared PKR in the brains of AD patients with that in age-matched disease-free controls. As shown in Figures 5A and B, levels of phosphorylated PKR were significantly elevated in the nuclear fractions of AD brains, as they are in the nuclear fractions of Tm-treated SK-N-SH cells. Furthermore, we found aggregated PKR in the nuclei of samples from AD brains (Figures 5C and D). Although many cells showed aggregations of PKR in their nuclei, these cells had not, apparently, undergone apoptosis, as judged histochemically. It is possible that the aggregation of PKR in the nuclei occurs at an early phase in the pathogenesis of AD. We also detected elevated levels of PKR in the brains of patients, at autopsy, who had died of Huntington's disease or Parkinson's disease (data not shown; manuscript in preparation). It is possible that PKR might be widely involved in neuronal degenerative diseases.
In summary, we have identified PKR as a proapoptotic protein in Tm-induced apoptosis. During apoptosis, phosphorylation of PKR was elevated and phosphorylated PKR was localized in the nuclei of SK-N-SH cells. In the nuclei in AD brains, similar results underscore the potential importance of phosphorylated PKR.
Our libraries of randomized Rz and/or genome-wide specific siRNAs (Miyagishi and Taira, 2003), some of them targeting micro RNAs (Kawasaki et al, 2004) should serve as powerful tools for the development of gene-suppressing reagents of both therapeutic and general importance and for the rapid identification of genes whose functions are of specific interest.
Materials and methods
Chemicals and antibodies
Tm was obtained from Sigma (Missouri). Anti-PKR (sc-9479; Santa Cruz Biotech., California), anti-Bax (sc-493; Santa Cruz Biotech.), anti-phospho-PKR (07-148; Upstate Biotech., New York), anti-KDEL (SPA-827; Stressgen Biotech., California), anti-cleaved caspase-3 antibody (Cell Signaling Technology, Massachusetts) and anti-actin (Chemicon International, California) antibodies were purchased for this study. Anti-GFP antibody was kindly provided by Dr Nagasaki (AIST, Ibaraki, Japan).
Culture and transfection of cells
SK-N-SH cells were grown in MEM-α (Invitrogen, The Netherlands) supplemented with 10% fetal calf serum (Invitrogen). Cells were transiently, semistably or stably transfected with plasmids using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's protocol. After transfection, cells were selected with Geneticin® (Sigma).
The randomized Rz expression library and construction of Rz expression vectors
The randomized Rz expression library was prepared in the vector pUC-dt, as described previously (Suyama et al, 2003a). The pEB6-CAG vector (EBV-based vector) was kindly provided by Dr Miwa (Tsukuba University, Ibaraki, Japan; Tanaka et al, 1999). It was digested with EcoRI and BamHI and self-ligated. The Rz expression cassette that included the promoter of a human gene for tRNAval was inserted at the AflIII site of pEB6-CAG to generate pEBRz. The 10 recovered plasmids were digested with EcoRI and KpnI, and fragments were inserted at the EcoRI/KpnI site of pEBRz to generate vectors for semistable expression.
Screening for active ribozymes
Lines of SK-N-SH cells that had been transiently transfected with the randomized Rz library were treated with Tm (2 μg/ml). For the first screening, transfected cells were exposed to Tm for 24 h. For the second screening, cells were transfected with candidate plasmids from the first screening and collected after incubation with Tm for 48 h. Expression vectors harboring Rzs were isolated from surviving cells and used to transform DH5α (TOYOBO, Tokyo, Japan).
Detection of cell death or apoptosis
Lines of SK-N-SH cells that had been semistably transfected with Rz expression vectors were treated with Tm (2 μg/ml) for 24 h. Apoptotic cells were stained by TUNEL or propidium iodide. For TUNEL staining, SK-N-SH cells were fixed in 4% formaldehyde for 25 min at 4°C and then permeabilized by treatment with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 5 min. After three washes with PBS, cells were subjected to TUNEL staining with the DeadEND™ Fluorometric TUNEL System (Promega, Wisconsin) according to the manufacturer's protocol. Cells were observed under a conventional fluorescence microscope (LSM-510; Carl Zeiss, Germany). To monitor chromatin condensation, we incubated cells with Hoechst 33258 for 10 min, and examined them under the fluorescence microscope.
Measurement of caspase-3-like activity
Caspase-3-like activity was measured spectrophotometrically as described previously (Tamatani et al, 2000). In brief, cells that had been exposed to 2 μg/ml Tm for 24 h were suspended in ice-cold ICE buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 1 mM PMSF, 1 mM EDTA and 10 mM EGTA) with subsequent centrifugation. The concentration of protein in the resulting supernatant was measured. Then, an aliquot of the supernatant, containing 40 μg of protein, was incubated with 50 μM substrate for caspase-3 (Ac-DEVD-MCA; Peptide Institute, Osaka, Japan) for 30 min at 37°C. The enzymatic activity was monitored by measuring absorbance (excitation, 380 nm; emission, 460 nm) in a spectrophotometer (F-3000; Hitachi, Tokyo, Japan). For experiments with an inhibitor of caspase-3, we used 1 μM z-VAD-fmk (Peptide Institute, Osaka, Japan) and exposed SK-N-SH cells to this reagent for 2 h prior to exposure to Tm.
Analysis by RT–PCR
Total RNA was isolated from cells transfected with an EBV-based vector (mock, Rz 47, Rz 49 and Rz 68 expression vectors) with Isogen™ (Nippon Gene, Tokyo, Japan), according to the manufacturer's protocol. For confirmation of expression of Rzs, total RNA was reverse transcribed in the presence of Rz down primer (5′-TTCGGCCTTTCGGCCTCATCAG-3′) after incubation with DNase (Roche Diagnostics, Basel, Switzerland). Products were amplified by PCR with Rz up primer (5′-TCCCCGGTTCGAAACCGGGCA-3′) and Rz down primer and reverse transcribed DNA as template, and products of PCR were analyzed by electrophoresis on a 2% agarose gel. We examined the expression of PKR using PKR RT up primer (5′-GGCTGGTGATCTTTCAGCAG-3′) and PKR RT down primer (5′-CCTTCTGGAAATTCTCTTCC-3′) and cDNA as template after reverse transcription in the presence of a poly-dT primer.
Immunocytochemical analysis
We examined the cellular distribution of PKR immunohistochemically as described previously (Bando et al, 2000). In brief, SK-N-SH cells were plated on chamber slides and then fixed in PBS that contained 0.1% Triton X-100 and 4% paraformaldehyde. After washing with ice-cold PBS, cells were incubated with normal goat serum for 2 h at room temperature and then with anti-PKR or anti-KDEL antibodies, overnight, at 4°C. Binding of the primary antibody was examined, after reaction with second antibody conjugated with Alexa fluor™ 568 or Alexa fluor™ 488 (Molecular Probes, Oregon), under a confocal laser microscope (LSM510; Carl Zeiss).
Cloning of PKR and generation of alanine-mutated PKR
We amplified the human gene for PKR from SK-N-SH cells using three sets of primers: PKR-F1 f (5′-ATG TCGGACG GCG GCA TGG CTG GTG ATC TTT-3′) and PKR-F1 r (5′-GGA AGG TCA AAT CTG GGT GCC-3′); PKR-F2 f (5′-CAA AAA GAT CTT TGG CAC CC-3′) and PKR-F2 r (5′-GTT ACA AGT CCA AAG TCT CC-3′); PKR-F3 f (5′-GGG GTG GAT TAT ATA CAT TC-3′) and PKR-F3 r (5′-GAG GAT CCC TAA CAT GTG TGT CGT T-3′). Each of the products of PCR was subcloned into pGEM-T (Promega) and its sequence confirmed. A full-length gene for PKR was constructed using the BglII site in the PKR gene and changing the vector from pGEM-T to pEGFPN3 vector.
Threonine residues 446 and 451 were changed to alanine with a QuickChange™ Site-Directed Mutagenesis Kit (Stratagene, La Jolla, California). To amplify the mutated gene for PKR, we used the following primers: alanine mutant f (5′-GGA AAG GCC AGG AGT AAG GGA GCC TTG CGA TAC-3′) and alanine mutant r (5′-GTA TCG CAA GGC TCC CTT ACT CCT GGC TCG CTT TCC-3′).
Post-mortem specimens and immunohistochemical analysis
Prior to autopsies, we obtained consent from patients and their families to use samples for research only. Some human tissues were obtained from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, Maryland, USA). Autopsies of AD patients (three; mean age, 71.7±3.3 years) and of age-matched controls who had died of cerebral ischemia or had been clinically and pathologically free of neurological disease (three; mean age, 75.3±4.7 years) were performed between 4 and 8 h after death, and then autopsy specimens were prepared for this study. In each case, diagnosis was confirmed histopathologically. Formalin-fixed samples were used in some experiments.
Preparation of cell extracts and immunoblotting
SK-N-SH cells were collected and lysed in PBS buffer that contained 5 mM EDTA, 1% NP-40, 1 mM DTT (Sigma), 10 μg/ml leupeptin (Roche Diagnostics) and 1 mM Pephabloc SC (Roche Diagnostics). After 5 min on ice, the lysate was centrifuged at 15 000 rpm for 5 min. The supernatant was designated the cytoplasmic fraction. The pelleted nuclei were sonicated in nuclear extraction buffer (20 mM Tris–HCl, pH 7.5, 1% SDS, 5 mM EGTA, 0.5% Triton X-100, 150 mM NaCl, 1 mM DTT, 10 μg/ml leupeptin and 1 mM Pephabloc SC) and the lysate was centrifuged at 15 000 rpm for 5 min. The supernatant was designated the nuclear fraction.
Gray matter was dissected from the temporal cerebral cortex of autopsied brains of four patients with AD and three age-matched controls. Tissues were homogenized in 50 volumes of homogenization buffer (50 mM Tris–HCl, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% NP-40 and 1 mM Pephabloc SC) and incubated for 5 min on ice. Each homogenate was centrifuged at 15 000 rpm for 5 min. The supernatant or the cytoplasmic fraction was subjected to Western blotting. The pellet was resuspended in nuclear extraction buffer and sonicated. The lysate was centrifuged at 15 000 rpm for 5 min and the supernatant was subjected to Western blotting as the nuclear fraction.
Concentrations of protein were determined with a Protein Assay kit (Bio Rad, California) following the manufacturer's protocol. Cell extracts were separated by SDS–PAGE and bands of protein were transferred to a Clear Blot™ Membrane-P (ATTO, Tokyo, Japan). The membrane was probed with various antibodies, as mentioned above, and then with peroxidase-conjugated second antibodies (Amersham Bioscience, New Jersey). Immunoreactions were detected with the ECL plus™ Western blotting system (Amersham Bioscience).
Supplementary Material
Supplementary Material 1 Control immunoblot for the fractionation of a nuclear-residents. Nuclear and cytoplasmic fractions were subjected to Western blotting with anti-histone and anti-actin antibodies as described in the text.
Supplementary Material 2 Localization of WT PKR fused with GFP protein. GFP or WT PKR fused with GFP was transiently transfected into the SK-N-SH cells. Localization of GFP (A), GFP fused to N-terminus of PKR (B) and GFP fused to C-terminus of PKR was observed. Bar, 20 μm.
Acknowledgments
This research was supported by grants from the Ministry of Economy, Trade and Industry (METI) of Japan, by a grant from the New Energy and Industrial Technology Development Organization (NEDO) of Japan, by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Culture (MEXT) of Japan, and by Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology. We thank Dr Yoshihiro Miwa at the University of Tsukuba and Drs Akira Nagasaki, Renu Wadhwa and Laura Nelson at AIST for helpful comments on the original manuscript.
References
- Bando Y, Ogawa S, Yamauchi A, Kuwabara K, Ozawa K, Hori O, Yanagi H, Tamatani M, Tohyama M (2000) 150-kDa oxygen-regulated protein (ORP150) functions as a novel molecular chaperone in MDCK cells. Am J Physiol Cell Physiol 278: C1172–C1182 [DOI] [PubMed] [Google Scholar]
- Besse S, Rebouillat D, Marie I, Puvion-Dutilleul F, Hovanessian AG (1998) Ultrastructural localization of interferon-inducible double-stranded RNA-activated enzymes in human cells. Exp Cell Res 239: 379–392 [DOI] [PubMed] [Google Scholar]
- Chatterton J, Hu X, Wong-Staal F (2004) Ribozymes in gene identification, target validation, and drug discovery. Targets 3: 10–17 [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Gale M Jr, Katze MG (1998) Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther 78: 29–46 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397: 271–274 [DOI] [PubMed] [Google Scholar]
- Haseloff J, Gerlach WL (1988) Simple RNA enzymes with new and highly specific endoribonuclease activities. Nature 334: 585–591 [DOI] [PubMed] [Google Scholar]
- Imaizumi K, Miyoshi K, Katayama T, Yoneda T, Taniguchi M, Kudo T, Tohyama M (2001) The unfolded protein response and Alzheimer's disease. Biochim Biophys Acta 1536: 85–96 [DOI] [PubMed] [Google Scholar]
- Jagus R, Joshi B, Barber GN (1999) PKR, apoptosis and cancer. Int J Biochem Cell Biol 31: 123–138 [DOI] [PubMed] [Google Scholar]
- Jeffrey IW, Kadereit S, Meurs EF, Metzger T, Bachmann M, Schwemmle M, Hovanessian AG, Clemens MJ (1995) Nuclear localization of the interferon-inducible protein kinase PKR in human cells and transfected mouse cells. Exp Cell Res 218: 17–27 [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, Tohyama M (2001) Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer's disease-linked presenilin-1 mutations. J Biol Chem 276: 43446–43454 [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M (1999) Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol 1: 479–485 [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13: 1211–1233 [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Onuki R, Suyama E, Taira K (2002) Identification of genes that function in the TNF-alpha-mediated apoptotic pathway using randomized hybrid ribozyme libraries. Nat Biotechnol 20: 376–380 [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Taira K (2002) Identification of genes by hybrid ribozymes that couple cleavage activity with the unwinding activity of an endogenous RNA helicase. EMBO Rep 3: 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kawasaki H, Wadhwa R, Taira K (2004) World of small RNAs: from ribozymes to siRNA and miRNA. Differentiation, in press [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332: 462–464 [DOI] [PubMed] [Google Scholar]
- Kruger M, Beger C, Li QX, Welch PJ, Tritz R, Leavitt M, Barber JR, Wong-Staal F (2000) Identification of eIF2Bgamma and eIF2gamma as cofactors of hepatitis C virus internal ribosome entry site-mediated translation using a functional genomics approach. Proc Natl Acad Sci USA 97: 8566–8571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagishi M, Taira K (2002) U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol 20: 497–500 [DOI] [PubMed] [Google Scholar]
- Miyagishi M, Taira K (2003) Strategies for generation of an siRNA-expression library directed against the human genome. Oligonucleotides 13: 325–333 [DOI] [PubMed] [Google Scholar]
- Mori K (2000) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101: 451–454 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 150: 887–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Nelson L, Suyama E, Kawasaki H, Taira K (2003) Use of random ribozyme libraries for the rapid screening of apopsotis- and metastasis-related genes. Targets 2: 191–200 [Google Scholar]
- Ng DT, Spear ED, Walter P (2000) The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol 150: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuki R, Nagasaki A, Kawasaki H, Baba T, Uyeda TQ, Taira K (2002) Confirmation by FRET in individual living cells of the absence of significant amyloid beta-mediated caspase 8 activation. Proc Natl Acad Sci USA 99: 14716–14721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CV, Handy I, Goldsmith T, Patel RC (2000) PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 275: 37993–37998 [DOI] [PubMed] [Google Scholar]
- Patel RC, Sen GC (1998) PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17: 4379–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Hartmann R, Qin J, Sen GC (2001) Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol 21: 1908–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prostko CR, Dholakia JN, Brostrom MA, Brostrom CO (1995) Activation of the double-stranded RNA-regulated protein kinase by depletion of endoplasmic reticular calcium stores. J Biol Chem 270: 6211–6215 [DOI] [PubMed] [Google Scholar]
- Saelens X, Kalai M, Vandenabeele P (2001) Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem 276: 41620–41628 [DOI] [PubMed] [Google Scholar]
- Sato N, Hori O, Yamaguchi A, Lambert JC, Chartier-Harlin MC, Robinson PA, Delacourte A, Schmidt AM, Furuyama T, Imaizumi K, Tohyama M, Takagi T (1999) A novel presenilin-2 splice variant in human Alzheimer's disease brain tissue. J Neurochem 72: 2498–2505 [DOI] [PubMed] [Google Scholar]
- Sato N, Imaizumi K, Manabe T, Taniguchi M, Hitomi J, Katayama T, Yoneda T, Morihara T, Yasuda Y, Takagi T, Kudo T, Tsuda T, Itoyama Y, Makifuchi T, Fraser PE, St George-Hyslop P, Tohyama M (2001) Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J Biol Chem 276: 2108–2114 [DOI] [PubMed] [Google Scholar]
- Satoh S, Hijikata M, Handa H, Shimotohno K (1999) Caspase-mediated cleavage of eukaryotic translation initiation factor subunit 2alpha. Biochem J 342 (Part 1): 65–70 [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1994) Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci 17: 489–517 [DOI] [PubMed] [Google Scholar]
- Siman R, Flood DG, Thinakaran G, Neumar RW (2001) Endoplasmic reticulum stress-induced cysteine protease activation in cortical neurons: effect of an Alzheimer's disease-linked presenilin-1 knock-in mutation. J Biol Chem 276: 44736–44743 [DOI] [PubMed] [Google Scholar]
- Srivastava SP, Davies MV, Kaufman RJ (1995) Calcium depletion from the endoplasmic reticulum activates the double-stranded RNA-dependent protein kinase (PKR) to inhibit protein synthesis. J Biol Chem 270: 16619–16624 [DOI] [PubMed] [Google Scholar]
- Srivastava SP, Kumar KU, Kaufman RJ (1998) Phosphorylation of eukaryotic translation initiation factor 2 mediates apoptosis in response to activation of the double-stranded RNA-dependent protein kinase. J Biol Chem 273: 2416–2423 [DOI] [PubMed] [Google Scholar]
- Suyama E, Kawasaki H, Kasaoka T, Taira K (2003a) Identification of genes responsible for cell migration by a library of randomized ribozymes. Cancer Res 63: 119–124 [PubMed] [Google Scholar]
- Suyama E, Kawasaki H, Nakajima M, Taira K (2003b) Identification of genes involved in cell invasion by using a library of randomized hybrid ribozymes. Proc Natl Acad Sci USA 100: 5616–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons RH (1992) Small catalytic RNAs. Annu Rev Biochem 61: 641–671 [DOI] [PubMed] [Google Scholar]
- Tamatani M, Mitsuda N, Matsuzaki H, Okado H, Miyake S, Vitek MP, Yamaguchi A, Tohyama M (2000) A pathway of neuronal apoptosis induced by hypoxia/reoxygenation: roles of nuclear factor-kappaB and Bcl-2. J Neurochem 75: 683–693 [DOI] [PubMed] [Google Scholar]
- Tanaka J, Miwa Y, Miyoshi K, Ueno A, Inoue H (1999) Construction of Epstein–Barr virus-based expression vector containing mini-oriP. Biochem Biophys Res Commun 264: 938–943 [DOI] [PubMed] [Google Scholar]
- Uhlenbeck OC (1987) A small catalytic oligoribonucleotide. Nature 328: 596–600 [DOI] [PubMed] [Google Scholar]
- Urano F, Bertolotti A, Ron D (2000) IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci 113 (Part 21): 3697–3702 [DOI] [PubMed] [Google Scholar]
- Welch PJ, Marcusson EG, Li QX, Beger C, Kruger M, Zhou C, Leavitt M, Wong-Staal F, Barber JR (2000) Identification and validation of a gene involved in anchorage-independent cell growth control using a library of randomized hairpin ribozymes. Genomics 66: 274–283 [DOI] [PubMed] [Google Scholar]
- Wu S, Kaufman RJ (1997) A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem 272: 1291–1296 [DOI] [PubMed] [Google Scholar]
- Zhang D, Pasternack MS, Beresford PJ, Wagner L, Greenberg AH, Lieberman J (2001) Induction of rapid histone degradation by the cytotoxic T lymphocyte protease Granzyme A. J Biol Chem 276: 3683–3690 [DOI] [PubMed] [Google Scholar]
- Zhou DM, Taira K (1998) The hydrolysis of RNA: from theoretical calculations to the hammerhead ribozyme-mediated cleavage of RNA. Chem Rev 98: 991–1026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1 Control immunoblot for the fractionation of a nuclear-residents. Nuclear and cytoplasmic fractions were subjected to Western blotting with anti-histone and anti-actin antibodies as described in the text.
Supplementary Material 2 Localization of WT PKR fused with GFP protein. GFP or WT PKR fused with GFP was transiently transfected into the SK-N-SH cells. Localization of GFP (A), GFP fused to N-terminus of PKR (B) and GFP fused to C-terminus of PKR was observed. Bar, 20 μm.