

Abstract
Purpose
To investigate the anti-apoptotic mechanism of leptin in non-small cell lung cancer.
Materials and Methods
The influences of leptin on apoptosis were investigated, analyzing the mechanism that triggers growth of A549 cells. The effects of leptin on cell proliferation were examined by XTT analysis. Leptin, C/EBP homologous protein (CHOP), phosphorylated-PKR-like ER kinase (p-Perk), inositol requiring proteins-1, spliced X-box transcription factor-1 (XBP1), cleaved activating transcription factor-6 (ATF6), eukaryotic translation initiation factor-2α, caspase-12 and CHOP protein were detected in four groups by western blot, and endoplasmic reticulum (ER) stress related mRNA were detected by reverse transcription PCR.
Results
The expression of leptin in A549 and leptin transfected cells inhibited cisplatin activated ER stress-associated mRNA transcription and protein activation. Two ER stress unfolded protein response pathways, PERK and ATF6, were involved, and XBP1 and tumor necrosis factor receptor-associated factor 2 (TRAF2) were increased significantly when treated with cisplatin in A549-siRNA against leptin cells. Furthermore, CHOP expression was inhibited upon leptin expression in A549, LPT-PeP and LPT-EX cells.
Conclusion
Leptin serves as an important factor that promotes the growth of A549 cells through blocking ER stress-mediated pathways. This blocking is triggered by p-Perk and ATF6 via inhibition of CHOP expression.
Keywords: Apoptosis, ER stress, cell growth, leptin, TRAF2, XPB1
INTRODUCTION
Lung cancer is a widespread disease, with a high incidence rate, and is a leading cause of mortality worldwide. In particular, non-small cell lung cancer (NSCLC) accounts for more than 80% of all lung cancers.1 In clinical, NSCLC is divided into 3 types, including squamous cell carcinoma, adenocarcinoma and large cell lung cancer.2 NSCLC commonly develops resistance to radiation and chemotherapy, and often presents at stages too late for surgical therapy. Current therapy methods are very limited, so the effective methods are urgent to be involved to decrease the incidence of pulmonary neoplasms.3 Therefore, exploring targets for NSCLC therapy has led to the development of promising methods for potential clinical use.
Endoplasmic reticulum (ER) is a central organelle in cells, which plays an important role in protein folding and maturation, and lipid synthesis. The ER can be affected by a variety of toxic insults.4-6 There is an increasing evidence that ER stress acts a significant function in the apoptosis regulation. Two specific signaling pathways were involving the ER stress process, such as, unfolded protein responses (UPR) and ER-associated protein degradation7,8 UPR pathway participates in the activation of some specific proteins, including activating transcription factor 6 (ATF6), PKR-like ER kinase (PERK), and inositol requiring proteins 1 (IRE1).9 The above three pathways of UPR activate several transcription factors, such as eukaryotic translation initiation factor-2α (eIF-2α) and X-box transcription factor-1 (XBP1). The pro-apoptotic transcription factor C/EBP homologous protein (CHOP)/GADD153, which suppresses the transcription of Bcl-2, can also be induced by a combination of the PERK/ATF4 and ATF6 pathways.
Leptin, originally described as an adipocyte-derived hormone regulating food intake and energy expenditure, is a pleiotropic hormone that plays both a proliferative and an anti-apoptotic role in several conditions, such lung cancer,10 breast cancer,11 and gastric cancer.12 Previously, the long isoform leptin receptor was identified in normal human lung tissue, suggesting that lung is a peripheral site of action for leptin. The circulating levels of leptin and/or overexpression of leptin mRNA are increased in adipose tissue. However, the anti-apoptosis effect and mechanism of leptin in lung cancer remain unknown. Accordingly, the present study attempted to establish an understanding of the anti-apoptotic mechanisms involving leptin in NSCLC.
MATERIALS AND METHODS
Plasmid construction
Leptin gene was amplified by the PCR technique, using cDNA from human adipocyte cells isolated from the subcutaneous fat of patients; written informed consent and approval from an Institutional Review Board were obtained prior to conducting this study. PCR was conducted with the forward primer (5'-GCGAATTCATGGTTCCAATCCAAAAAG TCCAAGAGG-3', at BamH I site) and reverse primer (5'-TATGGATCCTCA GCACCCAGGGCTGA GG-3', at Not I site). The PCR product was ligated to vector pMD18-T and sub-cloned into vector pcDNA3.1(+), yielding recombinant plasmid pcDNA3.1-LPT. The PCR conditions were as follows: 94℃ for 1.5 min, followed by 94℃ for 30 s, 63℃ for 30 s and 72℃ for 30 s for a total of 30 cycles, and a final extension at 72℃ for 10 min.
Cell culture and transfection
Lung adenocarcinoma cell line A549 and non-tumorigenic human bronchial epithelial cell line BEAS2B were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). Cells were cultured in complete culture medium (RPMI 1640 containing 10% FCS and 200 IU/mL penicillin/100 µg/mL streptomycin). All cells were cultured at 37℃ with 5% CO2. A549 and BEAS2B cells were plated into 6-well or 96-well plates (Falcon, Tokyo, Japan) 24 h before transfection (for the BEAS2B cells). Different amounts of plasmids (2 µg DNA per well in a 6-well plate and 0.2 µg DNA per well in a 96-well plate) were transfected into the BEAS2B monolayer cells with Lipofectamine™ 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA). A549 and BEAS2B cells were harvested by trypsin/EDTA in ethylenediaminetetraacetic acid 24 h or 48 h after transfection, and subsequently pelleted by short centrifugation and suspended in the lysis buffer as described by Wang, et al.13 Cells were supplemented with complete proteasomal inhibitor mixture (Merck, Darmstadt, Germany).
Small interfering RNA (siRNA) transfection
siRNA against leptin (siLPT) were purchased from Invitrogen Inc. Transient transfection was carried out with Lipofectamine™ 2000 transfection reagent according to the manufacturer's protocol. A549 cells were seeded on 6-well plates for RNA or protein preparation and 96-well plates for DNA fragmentation or cell growth assays (named A549-siLPT). After 24 hours of incubation, media were replaced with serum-free RPMI 1640 containing siRNA (100 nmol/L) and transfection reagent. Cells were harvested for assays daily for three consecutive days after transfection with the siRNA duplexes. The experiments were repeated on the at least three separate occasions.
Western blots
The cell lysates were separated by 15% SDS-PAGE and electro-transferred onto nitrocellulose membranes. After blocking with 5% defatted milk in phosphate buffered saline overnight at 4℃, the membranes were incubated with 1 : 2000 leptin specific monoclonal antibody (mAb) (Santa Cruz, Dallas, TX, USA), 1 : 1000 goat pAb anti-human CHOP, 1 : 2000 mice mAb anti-human p-Perk, 1 : 3000 mice mAb anti-human IRE1, 1 : 600 mAb anti-human β-actin (Santa Cruz, Dallas, TX, USA), 1 : 1000 polyclonal Ab anti-human caspase 3 (Santa Cruz, Dallas, TX, USA), 1 : 1000 mAb anti-full length and spliced XBP1 (Stressgen, Pennsylvania, USA), 1 : 1000 mAb anti-full length and cleaved ATF6 (Santa Cruz, Dallas, TX, USA), and 1 : 1000 anti-eIF-2α (Santa Cruz, Dallas, TX, USA) for 2 h at room temperature, after which there were incubated with 1 : 4000 horseradish peroxidase-conjugated anti-mouse, 1 : 1000 anti-rabbit or anti-goat IgG (Santa Cruz, Dallas, TX, USA). The reactive signals were visualized by ECL kit (PE Applied Biosystems, Foster City, CA, USA).
Proliferation analysis
To measure the effect of leptin on cell proliferation, the A549 cells were cultured and transiently transfected with individual leptin-expressing plasmids. XTT assay was employed to measure the cell proliferation using cytotoxicity detection kit (Cayman, San Diego, CA, USA). Briefly, 24 h or 48 h after transfection, the XTT activity from each well was terminated by adding 1N HCl to dissolve the formazan product. The multi-well plates were read at 490 nm on an ELISA plate reader (Thermo, Waltham, MA, USA). Every analysis was performed for at least six wells in duplication.
Detection of apoptosis
Cell apoptosis was detected by flow cytometry analysis that monitored Annexin V-FITC binding and propidium iodide uptake simultaneously, according to the manufacturer's instructions (Sigma, Shanghai, China). The samples were analyzed by fluorescence on a FACScan flow cytometer (Beckman, Miami, FL, USA). Potential DNA fragmentation was examined by the TUNEL apoptosis detection kit (Chemicon, Temecula, CA, USA) following the manufacturer's instructions. Apoptosis and death bodies were stained as brilliant blue.
Cisplatin treatment
1×105 BEAS2B cells or A549 cells were grown in 6-well plates and treated with cisplatin for 24 hours or 48 hours at a final concentration of 1 µmol/L. Cell viability was quantified using the XTT analysis as described above. Data were collected from three independent experiments in triplicate.
RNA extraction and hemi-quantitative RT-PCR
To evaluate the transcriptional status of the ER stress-related factors in the tested A549, A549-siLPT and BEAS2B cells, a series of hemi-quantitative RT-PCR assays were performed, including XBP1, eIF-2α and tumor necrosis factor receptor-associated factor 2 (TRAF2). The specific primers for XBP1, eIF-2α and TRAF2 were synthesized according to previous studies (Table 1).14-16 In parallel, individual β-actin was selected as an internal control. With an RNAsimple Total RNA Kit (TIANGEN, Beijing, China), total cellular RNA (including A549, A549-siLPT and leptin transfected BEAS2B cells) was prepared. Reverse transcription was performed using SuperScript™ III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Two µL of RT reaction products were amplified by PCR in a volume of 50 µL under the following conditions: 94℃ for 40 s, 60℃ for 30 s and 72℃ for 30 s. After electrophoresis on 1.5% agarose gel, the gel images of each PCR product were digitally captured with a CCD camera and analyzed with NIH Imager beta version 2. Relative transcriptional values of each factor in hemi-quantitative RT-PCR were presented as a ratio of the signal value of the specific PCR product and that of the individual β-actin.
Table 1.
The Sequences of the Primers for the ER-Stress Related Genes
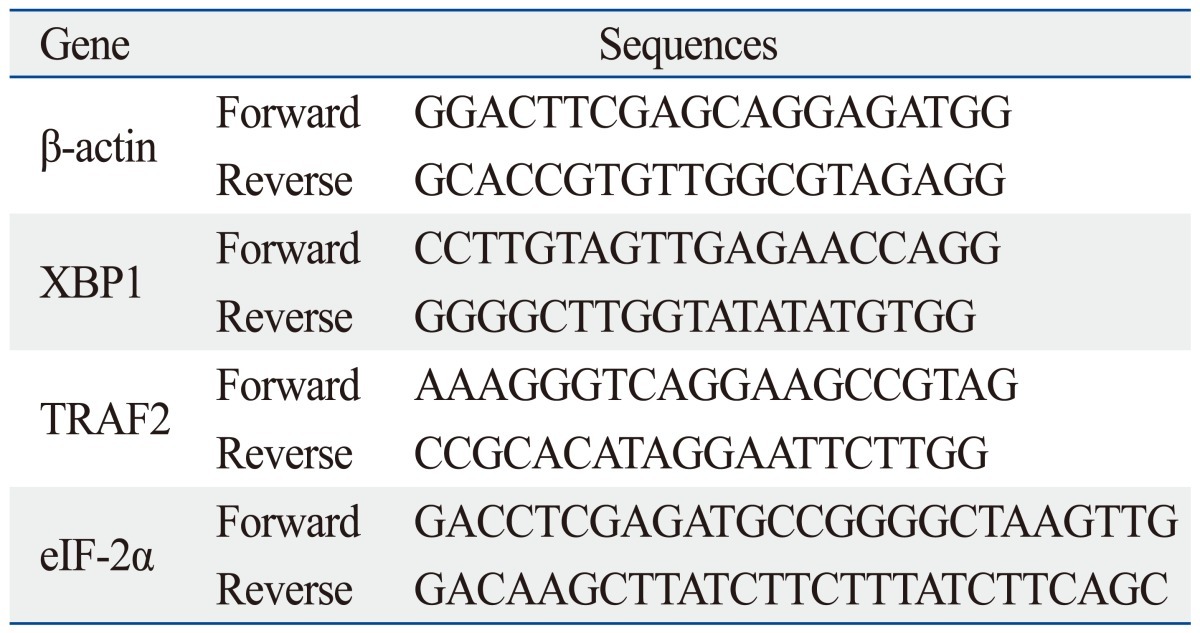
ER, endoplasmic reticulum; XBP1, X-box transcription factor-1; eIF-2α, eukaryotic translation initiation factor-2α.
Leptin peptide treatments
Human leptin peptide (100 nm) (sequence: ASN-VAL-ILE-GLN-ILE-SER-ASN-ASP-LEU-GLU-ASN-LEU-ARG, LPT-PeP) (Bio Vision, Milpitas, CA, USA) was employed into BEAS2B cells 4 hours before treated with cisplatin or without. Later, the ER stress-associated proteins were detected by western blot or XTT analysis.
Statistic analysis
Quantitative analysis of immunoblotting bands was performed using the computer-assisted software Image Total Tech (Pharmacia, St. Paul, MN, USA). The immunoblotting bands were scanned with Typhoon (Pharmacia, St. Paul, MN, USA), digitalized, and saved in TIF format. The gray values of each bands were evaluated detailed. All data were calculated as a mean±SD. Statistical analysis was acted utilizing the t-test. All p-values that less than 0.05 were considered as significant differences.
RESULTS
Leptin expression in cells and interfered leptin in A549-siLPT cells
The levels of leptin in A549 tumor cells, siRNA interfered A549-siLPT cells and leptin-expressing BEAS2B cells were detected to explore the pathogenicity of leptin in NSCLC. In the preparations of A549 cells, high levels of leptin were intrinsically expressed 24 h after culturing. The peptide LPT-PeP also activated high levels of leptin in BEAS2B cells 24 hours post incubation. In the leptin-expressing BEAS2B cells (LPT-EX), the expression levels of leptin were also high. For the siRNA interfered control group (A549-siLPT), vector pcDNA3.1 carrying siRNA was transfected into the cells, and no leptin bands were detected (Fig. 1). Furthermore, intrinsic expressed leptin in the A549 cells was higher than that expressed in the LPT-PeP and LPT-EX groups (p<0.05).
Fig. 1.

Detection of intrinsic leptin in A549, LPT-PeP, LPT-EX, and A549-siLPT cells with western blot assay. (A) Intrinsic or expressed leptin detected with leptin-specific monoclonal antibody. Various leptin proteins are indicated above the western blot bands. Protein molecular weights are shown on the right. (B) Statistical analysis. In the figure, LPT-PeP represents the leptin in peptide incubated BEAS2B cells, LPT-EX represents the expressed leptin in BEAS2B cells, and A549-siLPT represents the leptin in the interfered A549 cells. The relative value of each preparation is calculated by each gray numerical value of the specific product vs. that of β-actin. The average data of each preparation are evaluated based on three independent reactions and represented as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.01, †p<0.001, respectively. siLPT, siRNA against leptin.
Leptin expression promotes the proliferation of human lung cell lines
To observe the influence of leptin expression on the proliferation ability in abnormal and normal lung cell lines, the proliferation activities were measured with XTT analysis 24 hours post transfection (or incubation). XTT analysis results revealed no difference in the proliferation viabilities among A549, LPT-PeP and LPT-EX groups, both treated with and without cisplatin. However, in the inhibited leptin expression preparations of the A549-siLPT group, when treated with cisplatin, proliferation viabilities decreased significantly (p<0.001) 24 hours after transfection (Fig. 2A). Cell death and apoptosis were also detected by the TUNEL method, and we found that cisplatin did not affect cells death and apoptosis of A549, LPT-PeP and LPT-EX cells, which expressed leptin protein (Fig. 2B). The results also indicated that cisplatin could induce cell death and apoptosis in A549-siLPT cells, lacking leptin expression. Accordingly, we determined that leptin expression in BEAS2B cells saved dead cells and promoted cell proliferation.
Fig. 2.
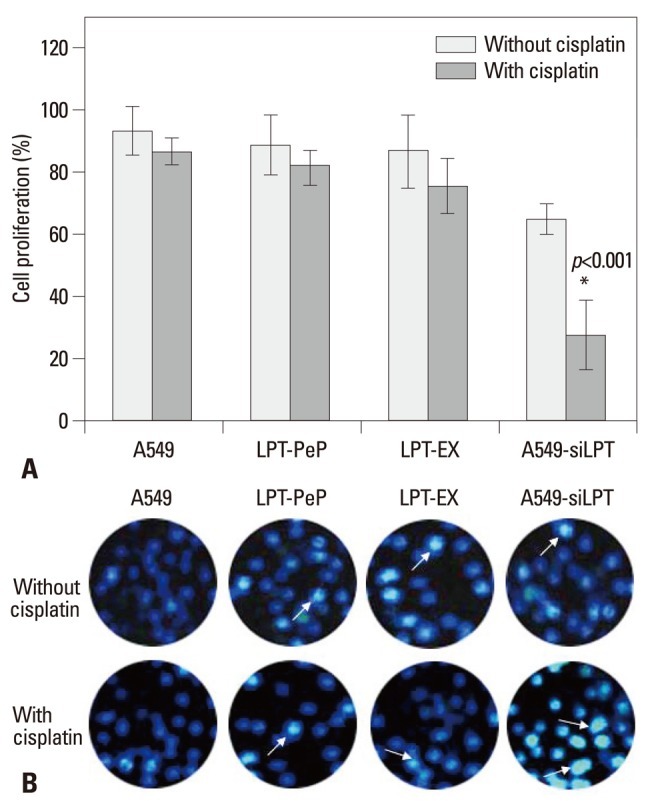
Cell proliferation effects of leptin in the four groups. (A) A549, LPT-PeP, LPT-EX and A549-siLPT cells were incubated with cisplatin, respectively. The cell proliferations were measured by the XTT method. (B) TUNEL analysis of leptin expressing cells treated with or without cisplatin. The arrows show TUNEL positive cells. The average data of each preparation are evaluated based on three independent reactions and represented as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.001. siLPT, siRNA against leptin.
Leptin inhibits the early and late apoptosis of lung cells
In order to investigate the mechanism of leptin-induced cell proliferation, we employed flow cytometry analysis to detect early and late apoptosis in all cell groups. For the A549, LPT-PeP, LPT-EX groups, there were no significant differences in early or late apoptosis between cells treated with cisplatin and those that were not (p>0.05). However, when treated with cisplatin, early or late apoptosis in A549-siLPT cells was significantly increased compared with cells not treated with cisplatin (p<0.001) (Fig. 3). Accordingly, we discerned cell apoptosis might be blocked by the expression of leptin.
Fig. 3.

Early and late apoptosis phenomena were observed in cisplatin treatment and non-treatment groups. (A) Annexin V/PI double staining assays of cells incubated with or without cisplatin. (B) Statistical analysis. Y axis indicates the numbers of PI stained cells. X axis indicates the numbers of Annexin V-FITC strained cells. Results for three independent experiments are shown. The mean data of each preparation were the results of three independent western blots scan and calculation, and indicated as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.01, †p<0.001, respectively. PI, propidium iodide; siLPT, siRNA against leptin.
Two ER stress UPR pathways are involved in the inhibition of apoptosis by leptin
In order to evaluate changes in UPR pathway factors, three main UPR factors, p-Perk, IRE1 and ATF6, were detected by western blot assay. As shown in Fig. 3, the expression of leptin blocked the phosphorylation of Perk and cleaving of ATF6 protein. Therefore, the amounts of p-Perk and cleaved ATF6 were not triggered by the treatment of cisplatin in the A549, PLT-PeP and PLT-EX groups (Fig. 4). However, in the A549-siPLT group, for the blocking of leptin expression, the phosphorylation of Perk and cleaving of ATF6 were significantly increased upon treatment with cisplatin (p<0.05) (Fig. 4).
Fig. 4.

Detection of three UPR pathway proteins. (A) Detection of IRE1, p-Perk and cleaved ATF6. Statistical analysis of p-Perk (B), IRE1 (C) and Cleaved ATF6 (D). The average gray value of each preparation was calculated by the gray numerical value of each blot vs. that of β-actin. The mean data of each preparation were the results of three independent western blots scan and calculation, and indicated as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.001. p-Perk, phosphorylated-PKR-like ER kinase; ER, endoplasmic reticulum; ATF6, activating transcription factor-6; UPR, unfolded protein response; IRE1, inositol requiring proteins-1; siLPT, siRNA against leptin.
UPR downstream mRNA and proteins were highly inhibited in leptin expressed cells
In order to further investigate the pathways of apoptosis, the mRNA and proteins associated with ER stress (UPR pathway), including spliced XBP1, TRAF2, and eIF-2α, were analyzed with hemi-quantitative RT-PCR 12 h after transfection. As shown in Fig. 5, when treated with cisplatin, the mRNA levels of XBP1 and TRAF2 were increased significantly in A549-siLPT cells, compared with cells not treated with cisplatin (p<0.05). Additionally, the levels of eIF-2α remained comparable among the preparations for all three groups (Fig. 5). Interestingly, there were no significant differences in spliced XBP1 and TRAF2 between cells treated with cisplatin and those that were not, among the A549, LPT-PeP and LPT-EX groups (Fig. 5).
Fig. 5.

Hemi-quantification PCR detects the mRNA levels of ER stress (UPR) associated genes. (A) Detection of spliced XBP1 mRNA. (B) Detection of TRAF2 mRNA. (C) Detection of the eIF-2α mRNA. The products were separated in 1.5% agarose gels. The relative values of spliced XBP1, TRAF2 and eIF-2α were evaluated by the calculation of gray scan value of each preparation vs. that of β-actin protein. The mean data of each preparation were the results of three independent western blots scan and calculation, and indicated as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.001, respectively. siLPT, siRNA against leptin; ER, endoplasmic reticulum; UPR, unfolded protein response; XBP1, X-box transcription factor-1; eIF-2α, eukaryotic translation initiation factor-2α; TRAF2, tumor necrosis factor receptor-associated factor 2.
Meanwhile, downstream ER stress associated proteins (spliced XBP1, TRAF2, and eIF-2α proteins) were also detected. The results were similar to those above for mRNA transcription. In the A549-siLPT group, the levels of spliced XBP1 and TRAF2 proteins increased significantly when treated with cisplatin (p<0.05) (Fig. 6). However, cisplatin treatment did not affect the protein levels of XBP1 and TRAF2 in the A549, LPT-PeP and LPT-EX groups.
Fig. 6.

Detection of ER stress (UPR) associated genes. (A) Detection of spliced XBP1 protein, TRAF2 protein, e-IF-2α protein. (B) Detection of spliced XBP1 protein. (C) Detection of TRAF2 protein. (D) Detection of eIF-2α protein. The relative values of spliced XBP1, TRAF2 and eIF-2α proteins were evaluated by the calculation of gray scan value of each preparation vs. that of β-actin protein. The mean data of each preparation were the results of three independent western blots scan and calculation, and indicated as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.001 respectively. ER, endoplasmic reticulum; UPR, unfolded protein response; XBP1, X-box transcription factor-1; eIF-2α, eukaryotic translation initiation factor-2α; siLPT, siRNA against leptin; TRAF2, tumor necrosis factor receptor-associated factor 2.
Leptin expression blocked CHOP triggered apoptosis
To detect potential changes in the ER stress induced apoptosis related markers after expression or incubation with leptin, the cellular levels of cleaved caspase 12 and CHOP protein were evaluated by individual western blot assay (Fig. 7A). Absent cisplatin treatment, there were no changes in caspase 12 and CHOP for all groups (Fig. 7B and C). Additionally, we observed no significant differences in cleaved caspase 12 between the cisplatin treated cells compared with non-treated cells for all three groups (Fig. 7B). When treated with cisplatin, CHOP levels in A549-siLPT cells were significantly higher than that of those untreated with cisplatin (Fig. 7C). However, no changes in CHOP were discovered between the cisplatin treated cells compared with the non-treated cells for the A549, LPT-PeP and LPT-EX groups (Fig. 7C). CHOP was shown to be activated in A549-siLPT cells, which triggered apoptosis. Accordingly, we determined the expression of leptin blocked cisplatin-induced apoptosis via CHOP pathways in human normal lung cells (such as LPT-PeP and LPT-EX cells).
Fig. 7.

Changes of ER stress-associated events in the cells expressing leptin. (A) The levels of CHOP and caspase-12 in A549, A549-siLPT, LPT-PeP, and LPT-EX cells were evaluated by individual western blots. (B) Statistical analysis of Cleaved caspase-12. (C) Statistical analysis of CHOP. The average gray value of each preparation was evaluated by the calculation of gray scan value of each preparation vs. that of β-actin protein. The mean data of each preparation were the results of three independent western blots scan and calculation, and indicated as mean±SD. Statistical differences of the data of cisplatin treatment compared with that of non-treatment are illustrated as *p<0.001. ER, endoplasmic reticulum; siLPT, siRNA against leptin.
DISCUSSION
This study is the first to indicate that leptin inhibits cisplatin-triggered apoptosis of human NSCLC cell lines, such as A549. In the present study, normal human lung cell lines, BEAS2B, treated with cisplatin resisted cisplatin-induced apoptotic effects. Additionally, our study indicated that leptin inhibits apoptosis through the PERK and ATF6 pathways, and blocks apoptosis by inactivating CHOP protein.
Our results revealed that all cell groups expressed leptin protein, but these levels in A549 cells and leptin expressing cells were significantly higher than that in A549-siLPT cells (Fig. 1). It has been shown that the malignant transformation of cancer requires cells to keep growing and block the apoptosis of transformed cells. Leptin expression patterns in the present study implied that the expression of leptin might inhibit the death of cells.
In the present study, the cell proliferation of A549, LPT-PeP, LPT-EX and A549-siLPT group cells were also detected. We hypothesized that higher proliferation in these three cells was due to the blocking of apoptosis. Accordingly, the cells were treated with cisplatin to induce ER stress, and the resulting cell viability changes were observed. Upon adding cisplatin to the experimental cells, cell viability in the leptin-expressing cells or those incubated in leptin underwent no significant changes between cisplatin treated and untreated cells. Cell viability in the cisplatin treated A549-siLPT cells was obviously lower than that in the untreated A549-siLPT cells. These changes indicated that the expression of leptin inhibits cisplatin mediated apoptosis and cell death.
In regards to ER stress (UPR pathway) associated protein detection, we found that p-Perk and cleaved ATF6 proteins were activated in A549-siLPT cells when treated with cisplatin. Therefore, the UPR pathway may involve in leptin-blocked apoptosis. Such leptin-induced apoptosis inhibition may help to further illuminate the role of leptin in cancer progression. Furthermore, downstream factors of spliced XBP1, TRAF2, and eIF-2α were also detected at the mRNA and protein levels, and the data strongly indicated the emergence of ER stress (or UPR pathway) after treatment with cisplatin in A549-siLPT cells. More importantly, the expression of leptin inhibited cisplatin induced ER stress in LPT-PeP, LPT-EX and A549 cells. Accordingly, we surmised that in human NSCLC cell lines leptin may participate in the pathogenic processes of lung cancer.
In order to identify the specific apoptosis factors that leptin inhibited, ER stress-associated factors (cleaved caspase 12 and CHOP protein) were also detected in the A549, A549-siLPT and leptin expressing (incubating) cells. According to Wang, et al.17 study, cleaved caspase 12 activates caspase 3 to trigger apoptosis, and CHOP can directly induce ER stress-associated apoptosis. In this study, we found that there were no significant changes in cleaved caspase 12 (activated) among all three cell groups (p>0.05) when treated with cisplatin. Interestingly, upon cisplatin treatment, CHOP levels in the A549-siLPT group were significantly increased, compared with untreated cells (p<0.05); however, there were no changes in the A549, LPT-PeP and LPT-EX cells. As our results were consistent with other research teams,18,19 we concluded that the expression or the existence of leptin in lung cells may indirectly inhibit CHOP protein mediated apoptosis. Actually, as the induction of the transcription factor CHOP/GADD153 can kill cells by an apoptotic mechanism,20,21 we discovered a novel result that leptin may block CHOP induced ER stress.
In conclusion, leptin may serve as an important factor to promote the growth of A549 cells via blocking of ER stress-mediated pathways. This blocking may be triggered by p-Perk and ATF6 pathways through inhibition of CHOP expression.
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584–594. doi: 10.4065/83.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen I, Petersen S. Towards a genetic-based classification of human lung cancer. Anal Cell Pathol. 2001;22:111–121. doi: 10.1155/2001/374304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al Husaini H, Wheatley-Price P, Clemons M, Shepherd FA. Prevention and management of bone metastases in lung cancer: a review. J Thorac Oncol. 2009;4:251–259. doi: 10.1097/JTO.0b013e31819518fc. [DOI] [PubMed] [Google Scholar]
- 4.Yung HW, Korolchuk S, Tolkovsky AM, Charnock-Jones DS, Burton GJ. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J. 2007;21:872–884. doi: 10.1096/fj.06-6054com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007;67:10631–10634. doi: 10.1158/0008-5472.CAN-07-1705. [DOI] [PubMed] [Google Scholar]
- 6.Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res. 2005;3:597–605. doi: 10.1158/1541-7786.MCR-05-0221. [DOI] [PubMed] [Google Scholar]
- 7.Joung KH, Cho SC. Stress responses of neonates related to maternal characteristics. Yonsei Med J. 2011;52:98–103. doi: 10.3349/ymj.2011.52.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Momoi T, Fujita E, Senoo H, Momoi M. Genetic factors and epigenetic factors for autism: endoplasmic reticulum stress and impaired synaptic function. Cell Biol Int. 2009;34:13–19. doi: 10.1042/CBI20090250. [DOI] [PubMed] [Google Scholar]
- 9.Hung JY, Hsu YL, Ni WC, Tsai YM, Yang CJ, Kuo PL, et al. Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lung cancer cells. Lung Cancer. 2010;68:355–365. doi: 10.1016/j.lungcan.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Terzidis A, Sergentanis TN, Antonopoulos G, Syrigos C, Efremidis A, Polyzos A, et al. Elevated serum leptin levels: a risk factor for non-small-cell lung cancer? Oncology. 2009;76:19–25. doi: 10.1159/000177952. [DOI] [PubMed] [Google Scholar]
- 11.Caldefie-Chézet F, Damez M, de Latour M, Konska G, Mishellani F, Fusillier C, et al. Leptin: a proliferative factor for breast cancer? Study on human ductal carcinoma. Biochem Biophys Res Commun. 2005;334:737–741. doi: 10.1016/j.bbrc.2005.06.077. [DOI] [PubMed] [Google Scholar]
- 12.Geng Y, Wang J, Wang R, Wang K, Xu Y, Song G, et al. Leptin and HER-2 are associated with gastric cancer progression and prognosis of patients. Biomed Pharmacother. 2012;66:419–424. doi: 10.1016/j.biopha.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Dong CF, Shi Q, Shi S, Wang GR, Lei YJ, et al. Cytosolic prion protein induces apoptosis in human neuronal cell SH-SY5Y via mitochondrial disruption pathway. BMB Rep. 2009;42:444–449. doi: 10.5483/bmbrep.2009.42.7.444. [DOI] [PubMed] [Google Scholar]
- 14.Xuan B, Qian Z, Torigoi E, Yu D. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol. 2009;83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S, et al. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect Prev. 2005;29:544–551. doi: 10.1016/j.cdp.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Chérasse Y, Maurin AC, Chaveroux C, Jousse C, Carraro V, Parry L, et al. The p300/CBP-associated factor (PCAF) is a cofactor of ATF4 for amino acid-regulated transcription of CHOP. Nucleic Acids Res. 2007;35:5954–5965. doi: 10.1093/nar/gkm642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Shi Q, Xu K, Gao C, Chen C, Li XL, et al. Familial CJD associated PrP mutants within transmembrane region induced Ctm-PrP retention in ER and triggered apoptosis by ER stress in SH-SY5Y cells. PLoS One. 2011;6:e14602. doi: 10.1371/journal.pone.0014602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moon DO, Park SY, Choi YH, Ahn JS, Kim GY. Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5: involvement of ROS-dependent ER-stress. Biochem Pharmacol. 2011;82:1641–1650. doi: 10.1016/j.bcp.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 19.Tagawa Y, Hiramatsu N, Kasai A, Hayakawa K, Okamura M, Yao J, et al. Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP) Free Radic Biol Med. 2008;45:50–59. doi: 10.1016/j.freeradbiomed.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Kim KM, Kim HC, Jeon KN, Kim HG, Kang JH, Hahm JR, et al. Rituximab-CHOP induced interstitial pneumonitis in patients with disseminated extranodal marginal zone B cell lymphoma. Yonsei Med J. 2008;49:155–158. doi: 10.3349/ymj.2008.49.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang SC, Lu MC, Chen HL, Tseng HI, Ke YY, Wu YC, et al. Cytotoxicity of calotropin is through caspase activation and downregulation of anti-apoptotic proteins in K562 cells. Cell Biol Int. 2009;33:1230–1236. doi: 10.1016/j.cellbi.2009.08.013. [DOI] [PubMed] [Google Scholar]