

Abstract
Mutations in the X-linked gene DCX result in lissencephaly in males, and abnormal neuronal positioning in females, suggesting a role for this gene product during neuronal migration. In spite of several known protein interactions, the involvement of DCX in a signaling pathway is still elusive. Here we demonstrate that DCX is a substrate of JNK and interacts with both c-Jun N-terminal kinase (JNK) and JNK interacting protein (JIP). The localization of this signaling module in the developing brain suggests its functionality in migrating neurons. The localization of DCX at neurite tips is determined by its interaction with JIP and by the interaction of the latter with kinesin. DCX is phosphorylated by JNK in growth cones. DCX mutated in sites phosphorylated by JNK affected neurite outgrowth, and the velocity and relative pause time of migrating neurons. We hypothesize that during neuronal migration, there is a need to regulate molecular motors that are working in the cell in opposite directions: kinesin (a plus-end directed molecular motor) versus dynein (a minus-end directed molecular motor).
Keywords: brain development, DCX, JIP, JNK, lissencephaly
Introduction
The lissencephaly syndromes in humans involve abnormal cortical lamination and are categorized as neuronal migration defects (Barth, 1987; Aicardi, 1989). Mutations in LIS1 (Reiner et al, 1993) or mutations in X-linked DCX (des Portes et al, 1998; Gleeson et al, 1998) result in lissencephaly. In the mouse, DCX mutants exhibit a lamination defect only in the hippocampus (Corbo et al, 2002) identical to that described in Lis1−/+ mice (Hirotsune et al, 1998), suggesting that the two gene products participate in the same pathway. In addition to these possible genetic interactions, we have shown a physical interaction between LIS1 and DCX (Caspi et al, 2000).
Both these gene products are involved in MT regulation. LIS1 is part of a pathway conserved from Aspergillus nidulans involved in dynein/dynactin regulation (reviewed by Morris et al, 1998; Reiner, 2000). DCX has been shown to be a microtubule-associated protein (MAP) that stabilizes microtubules (MTs) (Francis et al, 1999; Gleeson et al, 1999; Horesh et al, 1999). The interaction with MTs is via an evolutionarily conserved Doublecortin (DC) domain (Sapir et al, 2000; Taylor et al, 2000; Kim et al, 2003), where most missense mutations cluster. The expression and phosphorylation of DCX is regulated during brain development (Francis et al, 1999; Gleeson et al, 1999). In young neuronal culture, DCX is detected in the distal regions of neurites (Francis et al, 1999; Friocourt et al, 2003). Indeed, DCX has been shown to interact with additional proteins: clathrin adaptor proteins, the μ subunits of AP-1/-2 suggesting a potential involvement of DCX in protein sorting or vesicular trafficking (Friocourt et al, 2001), neurabin II, an actin-binding protein (Tsukada et al, 2003), and a phospho-specific interaction with neurofascin (Kizhatil et al, 2002). Neurofascin is a transmembrane protein of the Ig superfamily that engages in protein interactions as well as signaling pathways (Brummendorf et al, 1998; Hortsch, 2000). A possible involvement of DCX in signaling pathways was observed in PC12 cells overexpressing DCX where nerve growth factor (NGF)-dependent neurite outgrowth was inhibited (Shmueli et al, 2001). Several mutations in DCX phosphorylation sites exhibited diminished activity (Shmueli et al, 2001).
DCX contains multiple putative phosphorylation sites, among them sites for c-Jun N-terminal kinase (JNK). This group of mitogen-activated protein kinases (MAPKs) caught our attention for four main reasons: (1) Mice devoid of both Jnk1 and Jnk2 suffer from multiple abnormalities during development of the CNS (Kuan et al, 1999; Sabapathy et al, 1999). (2) In radially migrating neurons of the cerebral cortex, the protein levels of a JNK activator kinase, MUK/DLK/ZPK (Hirai et al, 2002), and JNK activity are specifically increased (Hirai et al, 2002; Kawauchi et al, 2003). Ectopic expression of MUK in neural precursor cells in utero reduced radial migration. (3) Inhibition of JNK activity or overexpression of dominant-negative JNK reduced radial migration, and the effect was mediated through MTs (Kawauchi et al, 2003). (4) The possibility that the JNK pathway links the reelin signal with the cell soma has been suggested (Verhey et al, 2001; Herz and Bock, 2002). The most complete neuronal migration related signaling pathway involves reelin (reviewed by Gupta et al, 2002). Mutations in either reelin (D'Arcangelo et al, 1995; Hirotsune et al, 1995; Ogawa et al, 1995) or in its receptors—the very low-density lipoprotein receptor (VLDLR) and the apoE receptor 2 (ApoER2) (D'Arcangelo et al, 1999; Hiesberger et al, 1999)- or in the intracellular signaling molecule Dab1 result in abnormal lamination in mice and humans (Hong et al, 2000). In addition, a recent link between LIS1 and the reelin pathway has been suggested (Assadi et al, 2003).
Here, we detected DCX as a substrate of JNK; it interacts and co-immunoprecipitates with JNK and the JNK interacting protein (JIP-1). The localization of DCX at neurite tips is determined by its interaction with JIP-1 and the interaction of the latter with conventional kinesin that is a plus-end directed molecular motor. The signaling module is present in migrating neurons and in the marginal zone of the developing cerebral cortex. DCX phosphorylated by JNK is enriched in the actin-rich region of growth cones. Furthermore, DCX phosphorylated by JNK affects neurite outgrowth and neuronal motility.
Results
DCX is a substrate of JNK
It has been previously demonstrated that DCX (Francis et al, 1999) is a phosphoprotein. DCX C-terminus contains multiple S/T-P residues that fit consensus JNK phosphorylation sites. Indeed, in vitro phosphorylation using the constitutive active fusion protein JNK2–MKK7 (Otto et al, 2000) indicated that recombinant GST–DCX (Figure 1A, Supplementary Figure S1b) is a potential substrate. The specificity of this phosphorylation was verified by using a different MAPK, ERK2–MEK1 (Robinson et al, 1998), and its activity resulted in negligible GST–DCX phosphorylation, although ERK2–MEK1 autophosphorylation was higher than that of the positive control JNK2–MKK7 in this experiment (Figure 1B). Ha-tagged JNK2 activated by arsenate phosphorylated GST–DCX, verifying the kinase as JNK2 and not MKK7 (Supplementary Figure S1a). In addition, transfected DCX is phosphorylated in vivo in 293 cells by endogenous kinases, since there was a mobility shift in the size of FLAG–DCX in the extract treated with alkaline phosphatase (CIP) (Figure 1C). Cotransfection of FLAG–DCX with JNK2–MKK7 resulted in a pronounced band shift that was reduced after CIP treatment (Figure 1C). Following CIP treatment the mobility of DCX was apparently increased and more than one DCX band was observed, suggesting the possibility of more than one phosphorylation site. Preliminary mass-spectrometry data confirmed that DCX is phosphorylated with some phosphorylation site(s) residing on a peptide 317–338 (data not shown). In agreement with this, mutant DCX (T331, S334A) was not phosphorylated in vitro by JNK (Supplementary Figure S1b). Single and double mutations may also change local protein conformation, which is crucial for phosphorylation. Therefore, we prepared p-specific antibodies by immunizing rabbits with two phosphorylated peptides: one including p-T321, S327, and the other including p-T331, S334. The antibodies' specificity was validated by their ability to recognize in vitro phosphorylated GST–DCX but not unphosphorylated recombinant protein (Supplementary Figure S1c,d). Using these antibodies in combination with mutated recombinant DCX allowed determining that in vitro JNK phosphorylated T321 and not S327 (Supplementary Figure S1c), but both T331 and S334 are phosphorylated. Furthermore, the phospho-antibodies recognized DCX well in lysates from transfected cells with the constitutively active JNK but much less when the kinase dead version was used (Supplementary Figure S1e,f). Both phospho-specific DCX antibodies immunostained only transfected cells, in a pattern similar to the FLAG tag used (T331, T331, S334, Supplementary Figure S1g–e). Moreover, combined with the specific JNK inhibitor (SP600125, Bennett et al, 2001), it was possible to demonstrate that DCX is phosphorylated by JNK on these sites in primary hippocampal neurons (Figure 1D).
Figure 1.
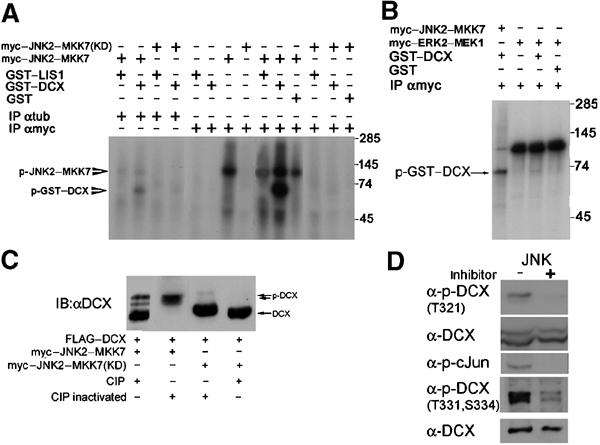
DCX is a JNK substrate. (A) DCX is a substrate of JNK2–MKK7. Myc-tagged JNK2–MKK7 kinase active or dead (KD) were immunoprecipitated from transfected cells and used for in vitro phosphorylation assays using γ-ATP32, with the autoradiogram shown here. The kinase source was immunoprecipitated by anti-myc antibodies or anti-tubulin antibodies as a control. The recombinant proteins used in the assay were GST–DCX, GST–LIS1, and GST. (B) Erk2 does not phosphorylate DCX. ERK2–MKK1 was used in addition to JNK2–MKK7. Note that although the autophosphorylation of JNK2–MKK7 is less than that of ERK2–MKK1, GST–DCX is phosphorylated mainly by the first kinase and insignificantly by the second. (C) DCX is phosphorylated in vivo. Transfected DCX is phosphorylated in cells, since alkaline phosphatase (CIP) treatment reduced the mobility of DCX. In the presence of activated JNK, a significant mobility shift is noted that is reduced with CIP treatment. (D) DCX is phosphorylated in rat primary hippocampal neurons. Phosphorylation was detected in vivo by Western blot analysis using two sets of anti-p-DCX antibodies (designated T321, or T331, S334). Addition of the JNK inhibitor SP600125 resulted in abolishment of the signal of p-DCX as well as of p-cJun, although the amount of total proteins loaded was similar (see total DCX).
DCX interacts with JNK
The possibility of physical interactions between JNK and DCX was tested. JNK2–MKK7 and FLAG–DCX co-immunoprecipitated from cells transfected with the myc-tagged kinase dead version by anti-myc antibodies (Figure 2A) or anti-FLAG antibodies (Figure 2B), and similar results were obtained with a constitutively active kinase (Figure 2C). Recombinant DCX and recombinant JNK interacted directly without the presence of other mediators (Figure 3B). The interaction domains mapped to either repeat of the DC motif (pep1 and pep2) (Sapir et al, 2000) using GST pulldowns from transfected cell extract or in brain extracts (Figure 2D and E). An in vivo interaction was suggested by co-immunoprecipitation from brain extracts (Figure 3C). A preferential interaction of DCX with JNK2 rather than JNK1was noticed in pulldown assays (Figure 2E). Binding of DCX to JNK (in the DC domain(s), but not in the C-terminus domain) (Figure 2D and E) is essential for its phosphorylation (data not shown). This fits the usual spatial distinction between the binding domain of the kinase (the JNK-docking domain) residing in the DC motif and the phosphorylated sites residing in the C-terminal region of DCX.
Figure 2.
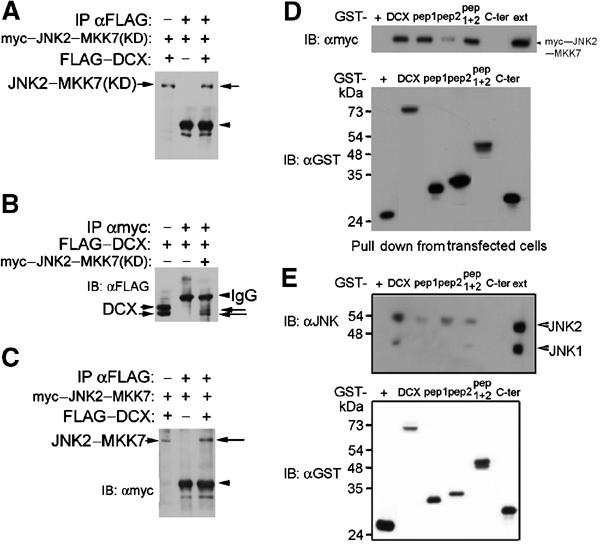
DCX and JNK interact. (A–C) DCX and JNK co-immunoprecipitated from transfected cells. Cells were transfected with FLAG-tagged DCX, myc-tagged JNK2–MKK7 kinase dead or active as indicated. In each of the blots, the left lane is the extract, the middle lane is immunoprecipitations from cells transfected with one plasmid, and the right lane is the test immunoprecipitations. (A, C) Immunoprecipitations with anti-FLAG antibodies, immunoblot with anti-myc antibodies. (B) Immunoprecipitations with anti-myc antibodies, immunoblot with anti-FLAG antibodies. (D) Pull down experiments from transfected cells. Cells transfected with myc–JNK2–MKK7 kinase active were subject to GST pulldown using the following GST fusion proteins (order left to right): GST, DCX, pep1 (amino acids 51–135), pep2 (amino acids 178–259), pep1+2 (amino acids 51–259), Cter (from amino acid 273 to end). Note that both pep1 and pep2 interacted independently with myc–JNK2–MKK7. (E) Pull down experiments from P7 mouse brain extracts. The following were the recombinant proteins (order left to right): GST, GST–DCX, GST–pep1, GST–pep2, GST–pep1+2, GST–Cter. The GST proteins were checked by immunoblot (lower blots in D, E).
Figure 3.
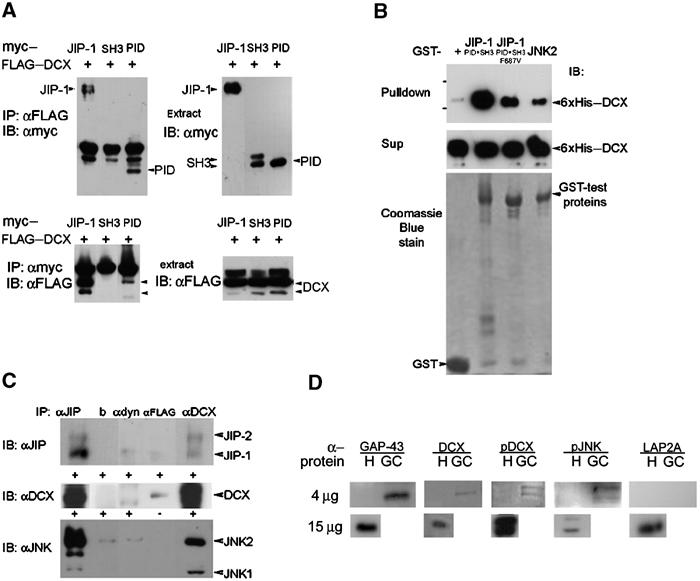
DCX interacts with JIP-1 and the interaction domain is in the DC motif and the PID domain, respectively. (A) Cells transfected with FLAG–DCX, and with myc-tagged JIP-1, the SH3 domain of JIP-1, or the PID domain of JIP-1 were subjected to immunoprecipitations using anti-FLAG antibodies (top left panel) or anti-myc antibodies (bottom left panel). The expression of each of the proteins was verified by Western blot analysis (both right panels). The interaction domain mapped within the PID domain. (B) Recombinant proteins 6xHis-tagged DCX were pulled down using the corresponding GST-tagged proteins (GST control, JIP-1 (SH3+PID domain), JIP-1 (SH3+PID domain mutated F687V), JNK2). The levels of the input proteins were similarly judged by Coomassie blue stain (lower panel) or anti-His Western blot (middle panel). (C) DCX, JIP and JNK co-immunoprecipitated from mouse brain extracts. P7 mouse brain was used for immunoprecipitations using monoclonal anti-DCX antibodies (228), anti-FLAG antibodies (Sigma), anti-dynein antibodies (Sigma), beads, or anti-JIP goat polyclonal antibodies (E19, Santa Cruz), followed by Western blot analysis using anti-JIP1 mouse monoclonal antibodies (BD Transduction Laboratories), anti-JNK2 mouse monoclonal antibodies (D-2, Santa Cruz), and anti-DCX rabbit polyclonal antibodies (Shmueli et al, 2001). The negative controls were anti-FLAG, anti-dynein, and beads only. DCX antibodies immunoprecipitated JIP-1 and JIP-2 (top panel), JIP-1 antibodies immunoprecipitated DCX (middle panel), and DCX antibodies immunoprecipitated JNK1 and JNK2 (lower panel). (D) DCX, p-DCX, and p-JNK are enriched in growth cones. Homogenates (H) (4 or 15 μg) and growth-cone preparations (GC) (4 μg) were separated on gels and Western blotted with the following antibodies: GAP-43 (positive control, enriched in growth cones), DCX, p-DCX, p-JNK (these proteins are also enriched in growth cones), and LAP2A (negative control, nuclear protein).
JNK is known to interact with several scaffold proteins that are capable of assembling a JNK signaling module. Therefore, the possible interaction of DCX with one of these scaffold proteins, JIP-1, was examined.
DCX interacts with JIP-1
DCX and JIP-1 co-immunoprecipitated from transfected cells (Figure 3A). DCX co-precipitated with JIP-2, but not with JLP (Lee et al, 2002) (data not shown). The interaction of DCX mapped within the protein-interaction domain (PID) of JIP-1 (Figure 3A). In DCX, the interaction domain mapped within the DC motif and either pep1 or pep2 was capable of precipitating JIP-1 (Supplementary Figure S2a-b). Two mutations found in lissencephaly patients did not affect this interaction (Supplementary Figure S2a-b); either a point mutation (S47R) or a truncation mutation (246X) removing part of the second DC repeat and the C-terminus were tested. The interaction between DCX and JIP-1 is direct, evident from pulldown of recombinant proteins (Figure 3B). We tested whether a point mutation (F687V) in the PID previously reported to affect the interaction of JIP-1 with rhoGEF (Meyer et al, 1999) affected the interaction with DCX. It was noticeable that the mutation reduced the interaction by either pulldown assays (Figure 3B) or using the yeast two-hybrid system (Supplementary Figure S2c). The interactions between DCX, JIP-1, and JNK exist in vivo demonstrated by co-immunoprecipitation of the proteins from embryonic brain extracts (Figure 3C). In addition, enrichment of DCX, pDCX, and pJNK was noticed in growth-cone preparations (Figure 3D). These results suggest that DCX may be part of the large JNK signaling module in vivo.
Module components coexist and are activated in the developing brain
To elucidate the possible in vivo interactions, the temporal and spatial expressions of p-DCX, p-JNK, JIP, ApoER2, reelin, MUK, and p-cJun were examined in the developing mouse brain. p-DCX and p-JNK were coexpressed in the cortical intermediate zone (IZ) and the ventricular zone (VZ) of the cortical plate, and both were expressed in the marginal zone (Figure 4A–C), with a higher degree of colocalization when p-specific antibodies were used in comparison to regular antibodies (data not shown). JIP (−1/2) expression was observed in the IZ where some cells coexpressed DCX (Figure 4D–F). No expression of JIP was observed in the marginal zone. MUK, a MAPKKK of JNK (Hirai et al, 1996) that interacts with JIPs (Whitmarsh et al, 1998; Ito et al, 1999), was concentrated in the subventricular zone, IZ, and subplate, as reported (Hirai et al, 2002) (Figure 4G–I). p-c-Jun (a substrate of JNK) and MUK were coexpressed only in a subset of cells (Figure 4I), suggesting the existence of additional JNK activators enabling c-Jun phosphorylation. ApoER2, which also interacts with JIP-1, was expressed at low levels in the VZ as reported (Luque et al, 2003). The expression markedly increased in the IZ and the VZ of the cortical plate and was very high in the marginal zone where it colocalized with reelin (Figure 4J–L).
Figure 4.
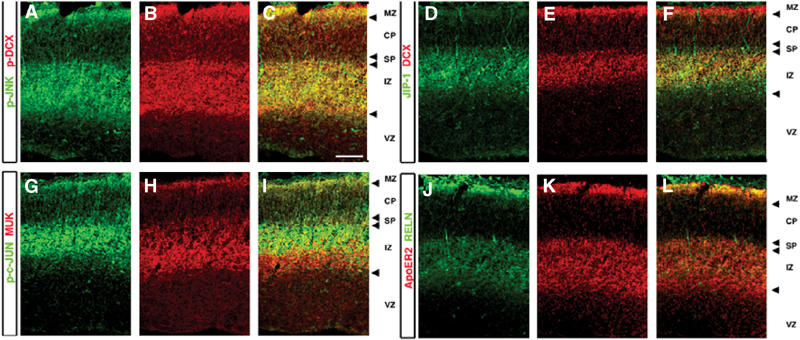
Localization of the JNK module in the developing cerebral cortex. (A–L) Coronal sections of E15.5 cortex stained with antibodies against components of the JNK module: pJNK (Thr 183, Tyr 185) p-DCX (A–C) DCX, JIP1 (D–F), MUK the MAPKKK and a substrate of JNK, phosphorylated c-JUN (G–I), ApoER2, reelin (Reln) (J–L). Images represent one optical slice (2.5 μm).
Interactions with signaling module components affect the localization of DCX
In primary neurons, DCX localization includes the tips of the neurites (Francis et al, 1999) (Figure 5A, white arrows). In these primary hippocampal neurons, JIP-1 appeared in a typical punctate manner as reported (Kim et al, 2002), and partial colocalization of DCX with JIP-1 was observed (Figure 5C).
Figure 5.
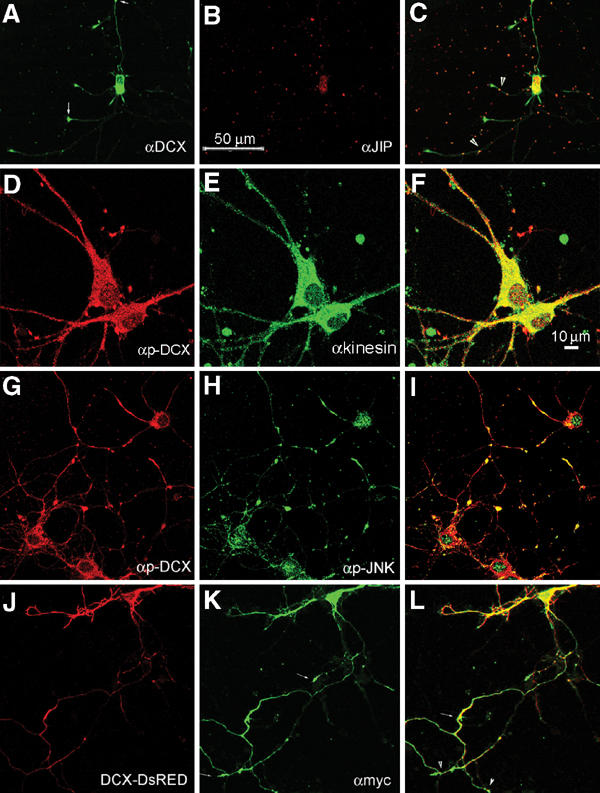
Localization of DCX and the JNK signaling module molecules in rat primary hippocampal neurons. (A–C) Colocalization of DCX and JIP-1 in some primary hippocampal neurons. DCX stained in green; some of the tips are marked with arrowheads (A), JIP in red (B) and some of the overlapping dots are indicated (arrowheads) (C). (D–F) Colocalization of p-DCX (D) and conventional kinesin (E), overlap in (F). (G–I) Colocalization of p-DCX (G) and p-JNK (H), merge in (I), in primary hippocampal neurons pretreated with kainic acid. (J–L) DCX–DsRED and JIP-1 colocalized in transfected neurons: DCX–DsRED (J) myc-tagged-JIP-1 stained with anti-myc antibodies (in green, K), merge (L), note the labeled tips of the neurites (small arrows, L).
Since kinesin interacts with JIP-1, its colocalization with p-DCX was examined (Figure 5D–F). In addition, there is noticeable colocalization between p-DCX and p-JNK (Figure 5G–I). The degree of colocalization between DCX and JIP increased when both genes were cotransfected (Figure 5J–L). Interestingly, JIP-1 also localized to the tips of the neurites (Figure 5K, small arrows), as has been reported in rat cortical neurons (Pellet et al, 2000) and in NE-115, and PC12 cells (Meyer et al, 1999) with overexpressed JIP-1. DCX–DsRED is well distributed in the transfected cells (Figures 5J and 6A–C); however, it did not localize to the tips of the neurites as the endogenous protein (compare Figure 5J with Figure 5C). Since JIP-1 interacts with kinesin (Verhey et al, 2001), we anticipated that DCX is mobilized along the neurites as part of this complex. Indeed, overexpression of the JIP-1 PID (lacking the last 11 amino acids essential for interaction with kinesin) (Verhey et al, 2001) resulted in accumulation of DCX–DsRED closer to the cell soma (Figure 6D–F). This is in striking contrast with the wide distribution of DCX–DsRED (Figure 6A–C).
Figure 6.
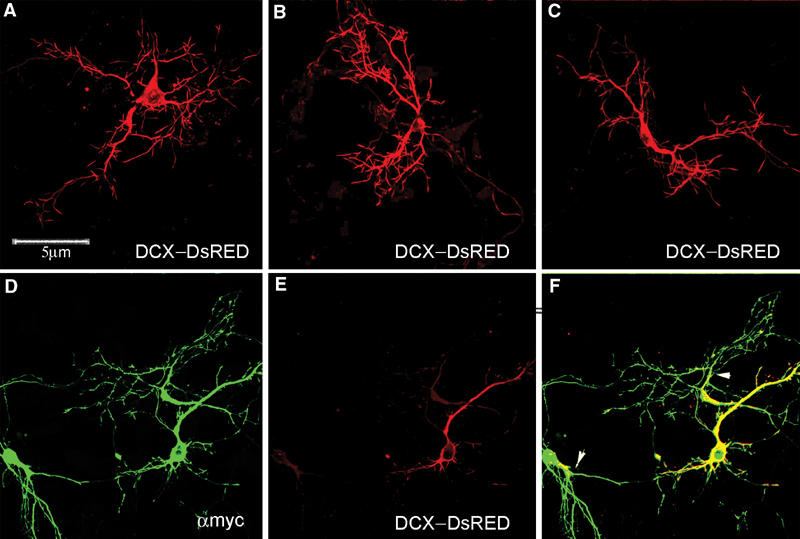
Expression of dominant-negative JIP-1 results in mislocalization of DCX–DsRED. (A–C) DCX–DsRED is well distributed in transfected primary hippocampal neurons. (D–F) Cotransfection of DCX–DsRED with myc–JIP-1 dominant negative. myc–JIP-1 dominant negative is well distributed in the neurons (in green, D), and DCX–DsRED remains less distributed (in red, E) as indicated in the overlap (F, arrowheads).
Next, we tested whether DCX's phosphorylation affects its intracellular localization. To this end, p-DCX antibodies were used to stain rat primary hippocampal neurons where JNK was basally activated (Figure 7A) (high levels of p-JNK were detected, data not shown). DCX (Figure 7A) invaded the actin-rich region of the growth cone (Figure 7C, note the overlap in yellow). When JNK activity was inhibited, the growth-cone localization of DCX was significantly reduced in the actin-rich domain (Figure 7D–I) using both sets of pDCX antibodies. These results suggested that DCX's phosphorylation by JNK might affect its interaction with partner proteins in the growth cone.
Figure 7.
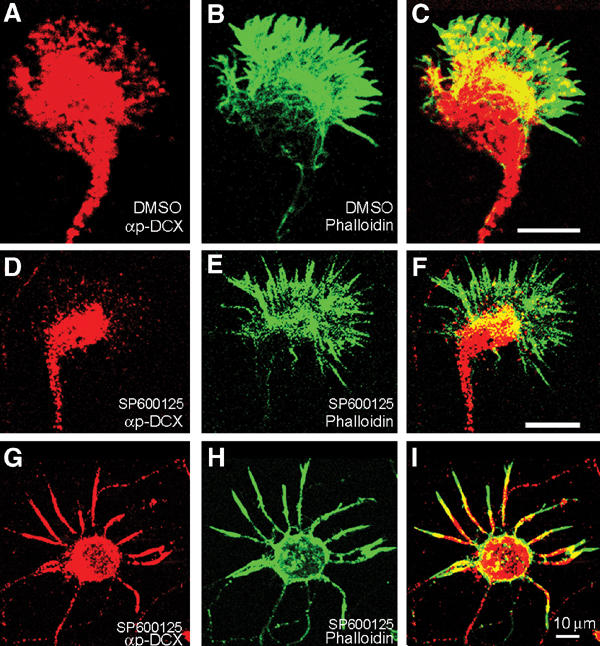
p-DCX intracellular localization. (A) Rat primary hippocampal growth cone stained with anti-p-DCX, T331, S334 (red) and (B) phalloidin-FITC (green); note the high degree of overlap (C) (yellow). (D–I) Growth cones treated with a specific JNK inhibitor and then stained with anti-p-DCX, T331, S334 (D), or T321 (G) (red) and phalloidin-FITC (green, E, H); note the reduced overlap (F, I, in comparison to C).
DCX's phosphorylation by JNK affects neurite outgrowth
The first event occurring in migrating neurons involves neurite extension, followed by translocation of nucleus and cytoplasmic components (Rakic, 1971). Therefore, we tested whether DCX's phosphorylation by JNK affects neurite outgrowth. In response to NGF, PC12 cells differentiate into neuron-like cells with the formation and elongation of neurites. This differentiation is accompanied by the activation of JNK as well as by an increased expression and phosphorylation of c-Jun (Waetzig and Herdegen, 2003). Indeed, inhibition of JNK by SP600125 resulted in minimal neurite outgrowth in PC12 cells (Supplementary Table S1). To test the specific effects of DCX's phosphorylation by JNK during neurite formation and extension, we analyzed the effect of transfecting wild-type DCX, DCX T331,S334A, or T321A (mimicking unphospho-form), and DCX T331,S334E (mimicking phospho-form), in PC12 cells, N2A cells, and primary cerebellar neurons (T331,S334 mutations). In all these different cells, neurite length was affected in comparison to wild-type DCX; the unphospho-forms reduced neurite length, while the phospho-form increased neurite length (Figure 8A, Supplementary Figures S3, S4, S5 and S6). The number of neurites per cell generally decreased with overexpression of the unphospho-forms, and increased with overexpression of the phospho-form (Figure 8B, Supplementary Figures S3, S4 and S5).
Figure 8.
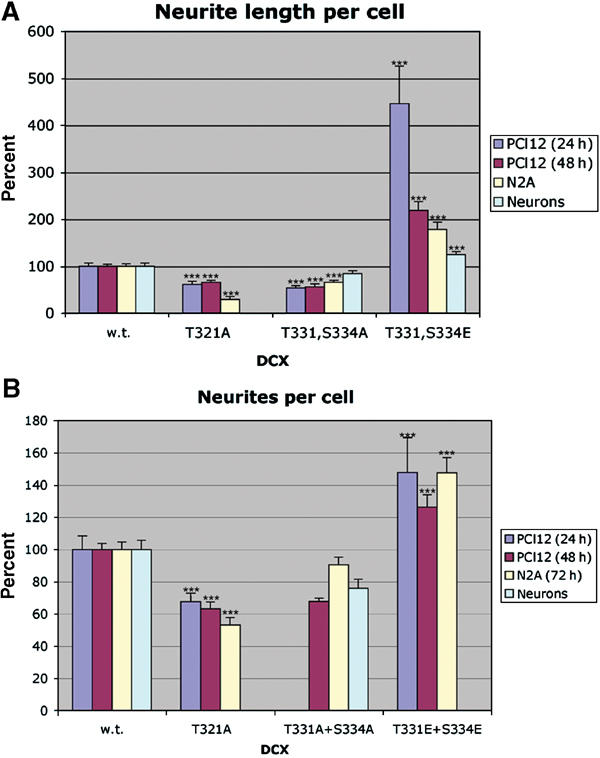
Effect of unphospho- and phospho-mimicking DCX mutants on neuronal cells. PC12, N2A, and primary cerebellar cells were transfected with the indicated DCX mutant constructs, and cells were analyzed for neurite length using image J (A) and number of neurites per cell (B). The s.e. is indicated in the bars. ***P<0.001.
DCX's phosphorylation by JNK affects neuronal motility
To test the possible effect of DCX's phosphorylation by JNK on neuronal migration, primary cerebellar neurons were transfected with T331, S334A or T331,S334E mutated DCX. Transfected neurons were followed by time-lapse microscopy. The deficient migration of T331, S334A cells in comparison to T331, S334E cells was characterized by both a reduced maximal velocity and prolonged periods at rest (Table I), suggesting that DCX's phosphorylation by JNK is instrumental in neuronal migration.
Table 1.
The effect of overexpression of DCX phospho-mutants on sites phosphorylated by JNK on the migration of cerebellar neurons
| Average velocity (μm/min) | Average velocity without pause (μm/min) | % of time at pause | n | |
|---|---|---|---|---|
| T331,S334A | 2.8±0.2 | 3.51±0.2 | 23.2±1.3 | 37 |
| T331,S334E | 3.81±0.2 | 4.19±0.2 | 12.1±1.3 | 64 |
|
P |
0.0041 |
0.038 |
<0.0001 |
|
| Primary cerebellar neurons were transfected with either unphospho-mimicry form T331,S334A or phospho-mimicry form T331,S334E. Neurons were followed by time-lapse microscopy and analyzed using the Delta-Vision system software. Note that the phospho-mimicry T331,S334E transfected neurons moved faster, and spent less time in pause than those transfected with the unphospho-mimicry form. | ||||
Discussion
DCX is a downstream target of JNK
The current study indicates that DCX is a downstream target of JNK. A physical interaction between JNK and DCX was demonstrated, albeit the usual transient enzyme–substrate interactions. Furthermore, a classical distinction between JNK docking sites (pep1 or pep2 of the DC motif) and the phosphorylation sites (in the C-terminal region of DCX) has been defined. The docking site(s) identified here bears no sequence similarity with previously recognized JNK docking sites (Kallunki et al, 1996; May et al, 1998; Tanoue et al, 2000). Furthermore, it was evident that ERK was not capable of phosphorylating DCX, suggesting that indeed there are distinct sites for each kinase (Jacobs et al, 1999).
DCX motif as a platform for multiple interactions
In addition, DCX interacts with JIP, and its interaction with this scaffold protein probably facilitates and specifies its phosphorylation. DCX utilizes the same domains for interacting with JIP-1, JNK, MTs (Sapir et al, 2000), and LIS1 (Caspi et al, 2000). The same domain is also necessary but not sufficient for its interaction with neurofascin (Kizhatil et al, 2002). As the domain is a tandem repeat, it may engage JIP-1 using one repeat element, and JNK via the other. Likewise, the PID in JIP-1 is bound to multiple proteins: DCX, rhoGEF (Meyer et al, 1999), APP (Alzheimer's amyloid precursor) (Matsuda et al, 2001), and ApoER2 (Stockinger et al, 2000) (reviewed by Herz and Bock, 2002) which is one of the receptors for reelin. Therefore, connecting DCX to JIP-1 provides a possible crosstalk with the reelin pathway. This crosstalk is unlikely to be linear due to the difference in mutant phenotypes.
Importance of the signaling module
JIP-1 serves as a scaffold protein for the different signaling components, and is a cargo for kinesin (reviewed by Weston and Davis, 2002). The interaction of DCX with JIP-1 and the latter with kinesin determines its mobilization to the tips of the neurites. Therefore, it is likely that JIP-1 plays a pivotal role in bringing together the kinase (JNK) with its substrate (DCX) to the growth cone.
The staining patterns observed in the developing brain included two domains of activation, one that is composed mainly of migrating neurons with a high expression of JIP, p-JNK, p-DCX, p-c-Jun, ApoER2, and MUK, and the other in the marginal zone with a high expression of reelin, p-JNK, p-DCX, and ApoER2. We suggest that the kinase activity of JNK has an important role in cortical lamination through the activity of phosphorylated substrates. One of these key molecules may be DCX, and indeed the colocalization between p-DCX and p-JNK is quite remarkable.
JNK and migration
The active migration of neurons involves neurite extension toward the target destination, followed by the translocation of nuclei and cytoplasmic components (Rakic, 1971). Environmental cues are captured and interpreted by the growth cone, a specialized structure that developed at the edge of the leading neurite. The notion that MTs participate in growth-cone function has been suggested (Gordon-Weeks, 2004). The localized concentration of p-DCX in the growth cone led to the possibility that it may play a role in neurite extension. This fits well with the proposed role of MAPs in neurite extension (Dehmelt and Halpain, 2004). Mimicking JNK phosphorylation of DCX increased the number of neurites extended and the length of the neurites. Phosphorylation of MAPs in the growth cone may be a general mode of controlling MT dynamics and hence neurite outgrowth. Supporting this notion is the finding that S/T-P phosphorylation of Tau affects neurite outgrowth (Biernat and Mandelkow, 1999). Inhibiting JNK's activity abolished neurite outgrowth, suggesting that additional JNK substrates participate in this signaling pathway.
Abnormal JNK activity impaired radial migration (Hirai et al, 2002; Kawauchi et al, 2003) in the developing cortex. JNK regulates migration via a wide range of different substrates. JNK phosphorylates paxillin, and expression of a mimicry unphosphorylated mutant inhibited cellular migration (Huang et al, 2003). Our results demonstrate faster movement and decreased pause time for the DCX mutant mimicking phosphorylated residues, in comparison to the DCX mutant mimicking the unphosphorylated form. Therefore, we suggest that in migrating neurons, DCX is one of the important JNK substrates.
DCX and molecular motor functions
Our results imply that DCX's interaction with JIP-1 facilitates its phosphorylation and regulates its intracellular localization in neurons through interacting with kinesin. We propose that the different intracellular localizations of DCX may affect the tight balance between the MT associated plus- and minus-end motor proteins (Figure 9 model), and that these coordinated activities control the movement of migrating neurons. Neurons undergoing migration along radial glia have a distinct morphology, with a growth-cone-like structure frequently present at the end of the leading process (Rakic, 1972; Edmondson and Hatten, 1987; O'Rourke et al, 1992; Nadarajah et al, 2001). Phosphorylated DCX is localized to growth cones in a complex with JIP and JNK via the activity of kinesin. p-DCX may be part of a signaling complex starting from reelin binding to ApoER2 that associates with JIP resulting in JNK phosphorylation and consequently phosphorylates DCX. Once the signal is terminated, DCX that is dephosphorylated is mobilized from neurite tips and associates with MT bundles, where it will recruit more LIS1 (Caspi et al, 2000) that will in turn regulate the dynein motor complex driving retrograde transport. In addition, dynein activity assists in nuclear movement in an MT cage surrounding the nucleus (Hatten, 2002). In this structure, there are high concentrations of LIS1, dynein, NudE, and NudEL (reviewed by Gupta et al, 2002). During neuronal migration, the cell undergoes major morphological changes, and membranes are moved into areas of the leading edge. Therefore, membrane trafficking and cell motility should be highly integrated with cytoskeleton dynamics. Thus, DCX's interaction with the μ subunits of AP-1, AP-2 (Friocourt et al, 2001) may also be relevant to the proper fusion of vesicles needed for normal neuronal migration.
Figure 9.
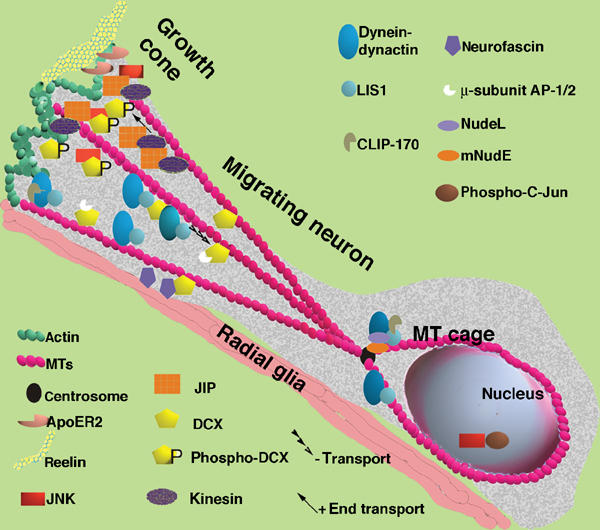
Model of a neuron migrating along radial glia. This model is based on earlier models (Morris et al, 1998; Feng and Walsh, 2001; Gupta et al, 2002; Hatten, 2002) and incorporates the finding and hypotheses derived from this paper. The migrating neuron has an elongated structure with a growth cone. There is more p-DCX located in the growth cone where it interacts with JNK and JIP. JIP is mobilized there by kinesin. JIP also interacts with ApoER2 that binds to the extracellular matrix protein reelin, and to kinesin that is a plus-end directed motor. DCX also interacts with the membranal protein neurofascin, and with the μ-subunits of the AP-1/2 complexes. At the MT plus-end tips we can also find CLIP-170, which recruits LIS1, and the dynein–dynactin complex. The dynein–dynactin retrograde motor is recruited to MTs with LIS1 and DCX followed by enhanced activity of this motor. Nucleokinesis is assisted by the activity of the dynein motor that is associated with the MT cage and the centrosome (there also mNudE and NudeL can be found). Within the nucleus, the transcription factor c-Jun is phosphorylated by JNK. The activity of JNK may thereby indirectly regulate the differential activities of kinesin and dynein.
The movement of neurons along radial glia is discontinuous, with periods of rapid forward movements and pauses (Edmondson and Hatten, 1987; O'Rourke et al, 1992; Nadarajah et al, 2001). Forward extension is coupled to mobility of vesicles into growing growth cones using the kinesin molecular motor. It is an open question as to whether DCX may be instructive in loading kinesin's cargo complex necessary for its mobilization. Radial migration is the most recently evolved neuronal migration (Hatten, 2002). Therefore, it may well be that while in fruitflies and in nematodes abnormal activity in this pathway results primarily in mislocalization of synaptic markers (Sunday driver (Bowman et al, 2000) and UNC-16 (Byrd et al, 2001), respectively), in mammals this pathway is coupled with the basic paradigm of neuronal migration.
Materials and methods
Plasmids, antibodies, and reagents
Myc–JNK2–MKK7 (kinase active and kinase dead) was received from Dr Kerkhoff, University of Würzburg (Otto et al, 2000). ERK2–MEK1 was received from Dr Cobb, University of Texas Southwestern (Robinson et al, 1998). JIP-1 and dominant-negative kinesin constructs were received from Dr Verhey, Harvard Medical Center (Verhey et al, 2001). JIP-2 constructs were received from Dr Nimpf, University of Vienna (Stockinger et al, 2000). Ha–JNK2 was received from Professor Zeger from the Weizmann Institute of Science. GST–c-Jun was received from Professor Wallach from the Weizmann Institute of Science. GST–DCX (Horesh et al, 1999) was mutated using PCR-based site-directed mutagenesis with the following primers (and their reverse complement primers):
T321A: 5′-CCTCCAGCAGCCAGCTCTCTGCCCCCAAGTCTAAG CAGTCT-3′.
S327A: 5′-CCCCCAAGTCTAAGCAGGCTCCCATCTCTACGCCCACC-3′.
T331A: 5′-CTAAGCAGTCTCCCATCTCTGCGCCCACCAGTCCTGG CAGC-3′.
S334A: 5′-CTCCCATCTCTACGCCCACCGCTCCTGGCAGCCTCCG GAAGC-3′.
GST–JIP1 (SH3+PID) was cloned and the PID domain was mutated using the following primer: 5′-cagagtccgtggggagagcaGtccagcagttctacaagca g-3′. GST–JNK2 kinase active and kinase dead was subcloned from myc–JNK2–MKK7. Both JIP1 (SH3+PID), wild and mutated forms, and GST–JNK2 (kinase dead) were subcloned into pEG202 and pJG4-5 vectors.
Rabbit anti-GST–DCX polyclonal antibodies were produced by injecting recombinant GST–DCX and purified as described (Harlow and Lane, 1988). Mouse anti-DCX monoclonal antibodies (228, IgM) were produced against GST–DCX using conventional methods (Harlow and Lane, 1988). Rabbit polyclonal antibodies anti-JIP-1b (Matsuda et al, 2001) were a gift from Dr Nishimoto (KEIO, Tokyo, Japan). Monoclonal-anti-conventional kinesin (SUK4) were a gift from Dr Gelfand, University of Urbana. Monoclonal anti-GAP43 were a gift from Professor Segal, The Weizmann Institute. Monoclonal antireelin antibodies (G10) were a gift from Dr Goffinet, University of Louvain Medical School. Affinity-purified rabbit anti-MUK antibodies were a gift from Dr Hirai, Yokohama City University School of Medicine, Japan. Rabbit anti-ApoER2 were a gift from Dr Nimpf, University of Vienna. Mouse monoclonal antibodies anti-JIP-1 were purchased from BD Transduction Laboratories; goat polyclonal antibodies JIP (E19), rabbit polyclonal antibodies anti-JNK (FL), mouse monoclonal JNK (D2), mouse monoclonal p-JNK (G7), mouse monoclonal p-c-Jun (KM-1), mouse anti-myc tag monoclonal antibodies (clone 9E10), rabbit affinity-purified polyclonal anti-GST antibodies (clones 2–5) were from Santa Cruz, CA; mouse monoclonal antibodies anti-β tubulin, mouse monoclonal antibodies anti-poly histidine (clone HIS-1), mouse anti-FLAG M2 monoclonal antibodies were from Sigma, Rehovot, Israel; mouse monoclonal antibody HA.11 (BabCO) for immunostaining (F7) or for immunoprecipitations were from Santa Cruz, CA. Rhodamine-conjugated affinipure goat anti-mouse, Cy3-conjugated affinipure goat anti-rabbit IgG (H+L), fluorescein (FITC)-conjugated affinipure goat anti-rabbit IgG (H+L) were from Jackson Immunoresearch (West Grove, PA); and Alexa Fluor® 488 goat anti-mouse IgG (H+L) was from Molecular Probes.
Hippocampal cultures: These were described previously (Brann et al, 2002). Neurons were transfected as described previously (Craig, 1991) with removal of the DNA mixture after 70 min, or using the Amexa® electroporation protocol. Inhibition of JNK activity was carried out by applying 50 μM SP600125 (Bennett et al, 2001) for 40 min before cell harvesting.
Growth-cone preparation: This was done according to previously published protocols (Gordon-Weeks and Lockerbie, 1984; Meiri and Gordon-Weeks, 1990; Mansfield et al, 1991).
Immunostaining: Neurons were stained as described (Schwarz and Futerman, 1996); coverslips were mounted with Vectashield or HardSet (Vector, CA) and examined using a confocal microscope (Radiance 2000, Bio Rad). The slides from the different subgroups were analyzed in a blind fashion, and the pictures are typical of the group indicated. E15.5 ICR embryos for immunostaining were perfused using 4% PFA and postfixed in 4% PFA for 30 min Embryos were cryoprotected in 20% sucrose-PBS and coronal sections (20 μm) were collected and stained. Data were analyzed using a Zeiss LSM 510 confocal microscope.
Kinase assays: 293 cells were transfected with plasmid encoding Myc–JNK2–MKK7 fusion protein, kinase active or kinase dead. The kinase assays were performed essentially as described (Zhang et al, 2000).
Protein interactions: GST pulldowns were prepared as described (Sapir et al, 2000). Transfections and immunoprecipitations of 293 cells were done as described (Caspi et al, 2000). Yeast transformation and detection of interactions were performed according to manual (OriGene Technologies, Inc.).
Neurite outgrowth: Transfected PC12, N2A, or primary cerebellar neurons were pictured at time points indicated with fluorescently tagged DCX wild-type or mutant constructs. The lengths of neurites per cell were analyzed using ImageJ software. The measurements were subject to ANOVA statistical analysis using JMP sofware.
Time-lapse microscopy: Migration was visualized by fluorescent and phase-contrast video microscopy of live cultures using the Delta-Vision system. The temperature on the microscope stage was maintained at 37°C using gradient control. Each recording session lasted 7.5 h, and one frame was taken every 9 min. The analysis was carried out using the Delta-Vision system package. Statistical analysis (t-test) was carried out using JMP software.
Supplementary Material
Supplementary Figure S1 a) The phosphorylation of GST-DCX is by JNK and not by MKK7. Cells were transfected with HA-JNK and prior to the immunoprecipitations JNK was activated by arsenate. GST-DCX was phosphorylated by the activated kinase (indicated). In this autoradiogram, the presence of phosphorylated JNK is marked as well. b) In vitro on phosphorylation of GST-DCX and GST-DCX mutated in single or double putative phosphorylation sites. GST-DCX wild type or mutated in the designated amino acids, and c-Jun (positive control) were phosphorylated in vitro by JNK. Note that the double mutant T331, S334A protein was not phosphorylated. c) DCX is phosphorylated in vitro on threonine 321 and on threonine 331 and serine 334 (right panel). GST-DCX wild type or mutated in the designated amino acids were phosphorylated in vitro by JNK, and the proteins were blotted and reacted with anti-p-DCX antibodies. The unphosphorylated protein was not recognized by the p-specific antibodies. Reduced reactivity was observed with DCX mutated in threonine 321, or mutated in threonine 321 and serine 327. Enhanced recognition was observed in case of DCX mutated in serine 327. Therefore, we conclude that antibodies recognize DCX phosphorylated on threonine 321. d) DCX is phosphorylated in vitro on threonine 331 and serine 334. GST-DCX wild type or mutated in the designated amino acids were phosphorylated in vitro by JNK, and the proteins were blotted and reacted with the indicated anti-p-DCX antibodies. The unphosphorylated protein was barely recognized by the p-specific antibodies. Reduced reactivity was observed with DCX mutated in threonine 321, or mutated in threonine 331 and serine 334. Our conclusion is that the antibodies recognize both p-threonine 321and p-serine 334. e) Phospho-specific DCX threonine 321 antibodies recognized the phospho-protein. HEK 293 cells were co-transfected with FLAG-DCX and constitutively active JNK kinase or the kinase-dead version (KD). Cell extract was analyzed by Western blot using p-DCX antibodies recognizing p-threonine 321. Note the reduced recognition in case of co-expression with the kinase-dead expression construct. f) Phospho-specific DCX threonine 331 and serine 334 antibodies recognized the phospho-protein. HEK 293 cells were co-transfected with FLAG-DCX and constitutively active JNK kinase or the kinase-dead version (KD). Cell extract was analyzed by Western blot using p-DCX antibodies recognizing p-threonine 331 and serine 334. Note the reduced recognition in case of co-expression with the kinase-dead expression construct. g) HEK 293 cells were transfected with FLAG-DCX and constitutively active JNK kinase. The pattern of immunostaining using phospho-specific DCX threonine 321 antibodies was similar to that observed by immunostaining with anti-FLAG antibodies. Similar results were obtained with phospho-specific DCX threonine 331 and serine 334 antibodies (data not shown).
Supplementary Figure S2 DCX interacts with JIP. a,b) Cells were transfected with myc-JIP-1 and with either FLAG-tagged constructs of DCX S47R, 247X, pep1, pep2. Myc-tagged JIP-1 was immunoprecipitated using anti-FLAG antibodies (a), or the FLAG-tagged constructs were immunoprecipitated with anti-myc antibodies (b). The expression of each of the plasmids was verified by Western blot analysis (lower panels, extract). c) Yeast expression constructs with DCX, JIP-1 (SH3+PID), JIP-1 (SH3+PID, 687 mutation), and JNK2 were tested for interaction in the two-hybrid system. DCX and JIP-1 strongly interacted and the PID mutation reduced the interaction in both directions tested. No interaction was observed with JNK2.
Supplementary Figure S3 Typical transfected DCX PC12 cells (24 hrs after transfection). The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted. The number of analyzed cells for the 24 hr time point were for T321A; 87 cells, T331, S334A; 85 cells, wild-type DCX; 130 cells, and T331, S334E; 34 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S4 Typical transfected DCX PC12 cells (48 hrs after transfection). The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted, The number of analyzed cells for the 48 hr time point were for T321A; 87 cells, T331, S334A; 116 cells, wild-type DCX; 170 cells, and T331, S334E; 40 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S5 Typical transfected DCX N2A cells. The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted, the number of analyzed cells for each transfection were for T321A; 125 cells, T331, S334A; 92 cells, wild-type DCX; 149 cells, and T331, S334E; 138 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S6 Typical transfected DCX-DsRed primary cerebellar cells. Primary cerebellar neurons were transfected with either unphospho-mimicry form T331, S334A, (top panel), wild-type DCX (middle panel) and the phospho-mimicry form T331, S334E (bottom panel). In each transfection the length of neurons was measured, the number of analyzed cells for each transfection were for T331, S334A; 68 cells, wild-type DCX; 56 cells, and for T331, S334E; 61 cells. The results of the averages are shown in Figure 8.
Supplementary Table S1
Acknowledgments
We thank Professor Benny Geiger and Ronen Zaidel-bar for help in time-lapse microscopy. We thank several colleagues for sharing reagents: Dr Kerkhoff, University of Würzburg; Dr Cobb, University of Texas Southwestern; Dr Verhey, Harvard Medical Center; Dr Nimpf, University of Vienna; Dr Nishimoto, KEIO, Tokyo; Dr Hirai, Yokohama City University School of Medicine; Dr Goffinet, University of Louvain Medical School; Dr Gelfand, University of Urbana; Dr Eyal Ben-Gal, Techion; Dr Eyal Scheter and Professors Zeger, Wallach, Segal, and Kimchi, The Weizmann Institute of Science. This research was supported in part by the Israeli Science Foundation (grant no. 19/00), the Benozyio Institute for Molecular Medicine, the Forcheimer center, and the Kekst center. OR is an incumbent of the Aser Rothstein Career Development Chair in Genetic Diseases.
References
- Aicardi J (1989) The lissencephaly syndromes. Int Pediat 4: 118–126 [Google Scholar]
- Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D'Arcangelo G, Clark GD (2003) Interaction of reelin signaling and Lis1 in brain development. Nat Genet 35: 270–276 [DOI] [PubMed] [Google Scholar]
- Barth PG (1987) Disorders of neuronal migration. Can J Neurol Sci 14: 1–16 [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA 98: 13681–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM (1999) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell 10: 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS (2000) Kinesin-dependent axonal transport is mediated by the Sunday driver (SYD) protein. Cell 103: 583–594 [DOI] [PubMed] [Google Scholar]
- Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M (2002) Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J Biol Chem 277: 9812–9818 [DOI] [PubMed] [Google Scholar]
- Brummendorf T, Kenwrick S, Rathjen FG (1998) Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol 8: 87–97 [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y (2001) UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 32: 787–800 [DOI] [PubMed] [Google Scholar]
- Caspi M, Atlas R, Kantor A, Sapir T, Reiner O (2000) Interaction between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet 9: 2205–2213 [DOI] [PubMed] [Google Scholar]
- Corbo JC, Deuel TA, Long JM, LaPorte P, Tsai E, Wynshaw-Boris A, Walsh CA (2002) Doublecortin is required in mice for lamination of the hippocampus but not the neocortex. J Neurosci 22: 7548–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM (1991) Transfecting cultured neurons. In Culturing Nerve Cells, Banker G, Goslin K (eds), pp 79–111. Cambridge, MA: MIT Press [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999) Reelin is a ligand for lipoprotein receptors. Neuron 24: 471–479 [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374: 719–723 [DOI] [PubMed] [Google Scholar]
- Dehmelt L, Halpain S (2004) Actin and microtubules in neurite initiation: are MAPs the missing link? J Neurobiol 58: 18–33 [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J (1998) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar hetrotropia and lissencephaly syndrome. Cell 92: 51–61 [DOI] [PubMed] [Google Scholar]
- Edmondson JC, Hatten ME (1987) Glial-guided granule neuron migration in vitro: a high-resolution time-lapse video microscopic study. J Neurosci 7: 1928–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Walsh CA (2001) Protein–protein interactions, cytoskeletal regulation and neuronal migration. Nat Rev Neurosci 2: 408–416 [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23: 247–256 [DOI] [PubMed] [Google Scholar]
- Friocourt G, Chafey P, Billuart P, Koulakoff A, Vinet MC, Schaar BT, McConnell SK, Francis F, Chelly J (2001) Doublecortin interacts with mu subunits of clathrin adaptor complexes in the developing nervous system. Mol Cell Neurosci 18: 307–319 [DOI] [PubMed] [Google Scholar]
- Friocourt G, Koulakoff A, Chafey P, Boucher D, Fauchereau F, Chelly J, Francis F (2003) Doublecortin functions at the extremities of growing neuronal processes. Cereb Cortex 13: 620–626 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA (1998) Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92: 63–72 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23: 257–271 [DOI] [PubMed] [Google Scholar]
- Gordon-Weeks PR (2004) Microtubules and growth cone function. J Neurobiol 58: 70–83 [DOI] [PubMed] [Google Scholar]
- Gordon-Weeks PR, Lockerbie RO (1984) Isolation and partial characterisation of neuronal growth cones from neonatal rat forebrain. Neuroscience 13: 119–136 [DOI] [PubMed] [Google Scholar]
- Gupta A, Tsai LH, Wynshaw-Boris A (2002) Life is a journey: a genetic look at neocortical development. Nat Rev Genet 3: 342–355 [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D (1988) Antibodies, A Laboratory Manual. Cold Spring Harbor: Cold Sring Harbor Laboratory [Google Scholar]
- Hatten ME (2002) New directions in neuronal migration. Science 297: 1660–1663 [DOI] [PubMed] [Google Scholar]
- Herz J, Bock HH (2002) Lipoprotein receptors in the nervous system. Annu Rev Biochem 71: 405–434 [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J (1999) Direct binding of reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24: 481–489 [DOI] [PubMed] [Google Scholar]
- Hirai S, Izawa M, Osada S, Spyrou G, Ohno S (1996) Activation of the JNK pathway by distantly related protein kinases, MEKK and MUK. Oncogene 12: 641–650 [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S (2002) MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development 129: 4483–4495 [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A (1998) Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet 19: 333–339 [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S, Nakao K, Katsuki M, Hayashizaki Y (1995) The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet 10: 77–84 [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (2000) Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26: 93–96 [DOI] [PubMed] [Google Scholar]
- Horesh D, Sapir T, Francis F, Caspi M, Grayer Wolf S, Elbaum M, Chelly J, Reiner O (1999) Doublecortin, a stabilizer of microtubules. Hum Mol Genet 8: 1599–1610 [DOI] [PubMed] [Google Scholar]
- Hortsch M (2000) Structural and functional evolution of the L1 family: are four adhesion molecules better than one? Mol Cell Neurosci 15: 1–10 [DOI] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K (2003) JNK phosphorylates paxillin and regulates cell migration. Nature 424: 219–223 [DOI] [PubMed] [Google Scholar]
- Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI (1999) JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol 19: 7539–7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13: 163–175 [PMC free article] [PubMed] [Google Scholar]
- Kallunki T, Deng T, Hibi M, Karin M (1996) c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87: 929–939 [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Yi, Hoshino M (2003) The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J 22: 4190–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV (2002) Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 35: 697–709 [DOI] [PubMed] [Google Scholar]
- Kim MH, Cierpicki T, Derewenda U, Krowarsch D, Feng Y, Devedjiev Y, Dauter Z, Walsh CA, Otlewski J, Bushweller JH, Derewenda ZS (2003) The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat Struct Biol 10: 324–333 [DOI] [PubMed] [Google Scholar]
- Kizhatil K, Wu YX, Sen A, Bennett V (2002) A new activity of doublecortin in recognition of the phospho-FIGQY tyrosine in the cytoplasmic domain of neurofascin. J Neurosci 22: 7948–7958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22: 667–676 [DOI] [PubMed] [Google Scholar]
- Lee CM, Onesime D, Reddy CD, Dhanasekaran N, Reddy EP (2002) JLP: a scaffolding protein that tethers JNK/p38MAPK signaling modules and transcription factors. Proc Natl Acad Sci USA 21: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque JM, Morante-Oria J, Fairen A (2003) Localization of ApoER2, VLDLR and Dab1 in radial glia: groundwork for a new model of reelin action during cortical development. Brain Res Dev Brain Res 140: 195–203 [DOI] [PubMed] [Google Scholar]
- Mansfield SG, Diaz-Nido J, Gordon-Weeks PR, Avila J (1991) The distribution and phosphorylation of the microtubule-associated protein MAP 1B in growth cones. J Neurocytol 20: 1007–1022 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yasukawa T, Homma Y, Ito Y, Niikura T, Hiraki T, Hirai S, Ohno S, Kita Y, Kawasumi M, Kouyama K, Yamamoto T, Kyriakis JM, Nishimoto I (2001) c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer's amyloid precursor protein with JNK. J Neurosci 21: 6597–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May GH, Allen KE, Clark W, Funk M, Gillespie DA (1998) Analysis of the interaction between c-Jun and c-Jun N-terminal kinase in vivo. J Biol Chem 273: 33429–33435 [DOI] [PubMed] [Google Scholar]
- Meiri KF, Gordon-Weeks PR (1990) GAP-43 in growth cones is associated with areas of membrane that are tightly bound to substrate and is a component of a membrane skeleton subcellular fraction. J Neurosci 10: 256–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Liu A, Margolis B (1999) Interaction of c-Jun amino-terminal kinase interacting protein-1 with p190 rhoGEF and its localization in differentiated neurons. J Biol Chem 274: 35113–35118 [DOI] [PubMed] [Google Scholar]
- Morris NR, Efimov VP, Xiang X (1998) Nuclear migration, nucleokinesis and lissencephaly. Trends Cell Biol 8: 467–470 [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL (2001) Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci 4: 143–150 [DOI] [PubMed] [Google Scholar]
- O'Rourke NA, Dailey ME, Smith SJ, McConnell SK (1992) Diverse migratory pathways in the developing cerebral cortex. Science 258: 299–302 [DOI] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajiman K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K (1995) The reeler gene-associated antigen on Cajal–Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 14: 899–912 [DOI] [PubMed] [Google Scholar]
- Otto IM, Raabe T, Rennefahrt UE, Bork P, Rapp UR, Kerkhoff E (2000) The p150-Spir protein provides a link between c-Jun N-terminal kinase function and actin reorganization. Curr Biol 10: 345–348 [DOI] [PubMed] [Google Scholar]
- Pellet JB, Haefliger JA, Staple JK, Widmann C, Welker E, Hirling H, Bonny C, Nicod P, Catsicas S, Waeber G, Riederer BM (2000) Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur J Neurosci 12: 621–632 [DOI] [PubMed] [Google Scholar]
- Rakic P (1971) Neuron–glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electron microscope study in Macacus rhesus. J Comp Neurol 141: 283–312 [DOI] [PubMed] [Google Scholar]
- Rakic P (1972) Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol 145: 61–84 [DOI] [PubMed] [Google Scholar]
- Reiner O (2000) LIS1: Let's Interact Sometimes…(part 1). Neuron 28: 633–636 [DOI] [PubMed] [Google Scholar]
- Reiner O, Carrozzo R, Shen Y, Whenert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH (1993) Isolation of a Miller–Dieker lissencephaly gene containing G protein ß-subunit-like repeats. Nature 364: 717–721 [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Stippec SA, Goldsmith E, White MA, Cobb MH (1998) A constitutively active and nuclear form of the MAP kinase ERK2 is sufficient for neurite outgrowth and cell transformation. Curr Biol 8: 1141–1150 [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF (1999) Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev 89: 115–124 [DOI] [PubMed] [Google Scholar]
- Sapir T, Horesh D, Caspi M, Atlas R, Burgess HA, Grayer Wolf S, Francis F, Chelly J, Elbaum M, Pietrokovski S, Reiner O (2000) Doublecortin mutations cluster in evolutionary conserved functional domains. Hum Mol Genet 5: 703–712 [DOI] [PubMed] [Google Scholar]
- Schwarz A, Futerman AH (1996) The localization of gangliosides in neurons of the central nervous system: the use of anti-ganglioside antibodies. Biochim Biophys Acta 1286: 247–267 [DOI] [PubMed] [Google Scholar]
- Shmueli O, Gdalyahu A, Sorokina K, Nevo E, Avivi A, Reiner O (2001) DCX in PC12 cells: CREB-mediated transcription and neurite outgrowth. Hum Mol Genet 10: 1061–1070 [DOI] [PubMed] [Google Scholar]
- Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, Schneider WJ, Nimpf J (2000) The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J Biol Chem 275: 25625–25632 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Adachi M, Moriguchi T, Nishida E (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol 2: 110–116 [DOI] [PubMed] [Google Scholar]
- Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG (2000) Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem 275: 34442–34450 [DOI] [PubMed] [Google Scholar]
- Tsukada M, Prokscha A, Oldekamp J, Eichele G (2003) Identification of neurabin II as a novel doublecortin interacting protein. Mech Dev 120: 1033–1043 [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol 152: 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T (2003) A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J Biol Chem 278: 567–572 [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ (2002) The JNK signal transduction pathway. Curr Opin Genet Dev 12: 14–21 [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281: 1671–1674 [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Kovalenko A, Cantarella G, Wallach D (2000) Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity 12: 301–311 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 a) The phosphorylation of GST-DCX is by JNK and not by MKK7. Cells were transfected with HA-JNK and prior to the immunoprecipitations JNK was activated by arsenate. GST-DCX was phosphorylated by the activated kinase (indicated). In this autoradiogram, the presence of phosphorylated JNK is marked as well. b) In vitro on phosphorylation of GST-DCX and GST-DCX mutated in single or double putative phosphorylation sites. GST-DCX wild type or mutated in the designated amino acids, and c-Jun (positive control) were phosphorylated in vitro by JNK. Note that the double mutant T331, S334A protein was not phosphorylated. c) DCX is phosphorylated in vitro on threonine 321 and on threonine 331 and serine 334 (right panel). GST-DCX wild type or mutated in the designated amino acids were phosphorylated in vitro by JNK, and the proteins were blotted and reacted with anti-p-DCX antibodies. The unphosphorylated protein was not recognized by the p-specific antibodies. Reduced reactivity was observed with DCX mutated in threonine 321, or mutated in threonine 321 and serine 327. Enhanced recognition was observed in case of DCX mutated in serine 327. Therefore, we conclude that antibodies recognize DCX phosphorylated on threonine 321. d) DCX is phosphorylated in vitro on threonine 331 and serine 334. GST-DCX wild type or mutated in the designated amino acids were phosphorylated in vitro by JNK, and the proteins were blotted and reacted with the indicated anti-p-DCX antibodies. The unphosphorylated protein was barely recognized by the p-specific antibodies. Reduced reactivity was observed with DCX mutated in threonine 321, or mutated in threonine 331 and serine 334. Our conclusion is that the antibodies recognize both p-threonine 321and p-serine 334. e) Phospho-specific DCX threonine 321 antibodies recognized the phospho-protein. HEK 293 cells were co-transfected with FLAG-DCX and constitutively active JNK kinase or the kinase-dead version (KD). Cell extract was analyzed by Western blot using p-DCX antibodies recognizing p-threonine 321. Note the reduced recognition in case of co-expression with the kinase-dead expression construct. f) Phospho-specific DCX threonine 331 and serine 334 antibodies recognized the phospho-protein. HEK 293 cells were co-transfected with FLAG-DCX and constitutively active JNK kinase or the kinase-dead version (KD). Cell extract was analyzed by Western blot using p-DCX antibodies recognizing p-threonine 331 and serine 334. Note the reduced recognition in case of co-expression with the kinase-dead expression construct. g) HEK 293 cells were transfected with FLAG-DCX and constitutively active JNK kinase. The pattern of immunostaining using phospho-specific DCX threonine 321 antibodies was similar to that observed by immunostaining with anti-FLAG antibodies. Similar results were obtained with phospho-specific DCX threonine 331 and serine 334 antibodies (data not shown).
Supplementary Figure S2 DCX interacts with JIP. a,b) Cells were transfected with myc-JIP-1 and with either FLAG-tagged constructs of DCX S47R, 247X, pep1, pep2. Myc-tagged JIP-1 was immunoprecipitated using anti-FLAG antibodies (a), or the FLAG-tagged constructs were immunoprecipitated with anti-myc antibodies (b). The expression of each of the plasmids was verified by Western blot analysis (lower panels, extract). c) Yeast expression constructs with DCX, JIP-1 (SH3+PID), JIP-1 (SH3+PID, 687 mutation), and JNK2 were tested for interaction in the two-hybrid system. DCX and JIP-1 strongly interacted and the PID mutation reduced the interaction in both directions tested. No interaction was observed with JNK2.
Supplementary Figure S3 Typical transfected DCX PC12 cells (24 hrs after transfection). The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted. The number of analyzed cells for the 24 hr time point were for T321A; 87 cells, T331, S334A; 85 cells, wild-type DCX; 130 cells, and T331, S334E; 34 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S4 Typical transfected DCX PC12 cells (48 hrs after transfection). The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted, The number of analyzed cells for the 48 hr time point were for T321A; 87 cells, T331, S334A; 116 cells, wild-type DCX; 170 cells, and T331, S334E; 40 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S5 Typical transfected DCX N2A cells. The mutations are indicated on the panels. Three of the four mutations were on the backbone of DCX-DsRed contruct. T321A was on the backbone of FLAG-DCX, to visulalize transfected cells, cells were co-transfected in a 1:8 ratio with EGFP. In each transfection the length of neurons per cell was measured, and the number of neurites per cell was counted, the number of analyzed cells for each transfection were for T321A; 125 cells, T331, S334A; 92 cells, wild-type DCX; 149 cells, and T331, S334E; 138 cells. The results of the averages are shown in Figure 8.
Supplementary Figure S6 Typical transfected DCX-DsRed primary cerebellar cells. Primary cerebellar neurons were transfected with either unphospho-mimicry form T331, S334A, (top panel), wild-type DCX (middle panel) and the phospho-mimicry form T331, S334E (bottom panel). In each transfection the length of neurons was measured, the number of analyzed cells for each transfection were for T331, S334A; 68 cells, wild-type DCX; 56 cells, and for T331, S334E; 61 cells. The results of the averages are shown in Figure 8.
Supplementary Table S1