

Abstract
Purpose.
To identify the causative mutations in two early-onset canine retinal degenerations, crd1 and crd2, segregating in the American Staffordshire terrier and the Pit Bull Terrier breeds, respectively.
Methods.
Retinal morphology of crd1- and crd2-affected dogs was evaluated by light microscopy. DNA was extracted from affected and related unaffected controls. Association analysis was undertaken using the Illumina Canine SNP array and PLINK (crd1 study), or the Affymetrix Version 2 Canine array, the “MAGIC” genotype algorithm, and Fisher's Exact test for association (crd2 study). Positional candidate genes were evaluated for each disease.
Results.
Structural photoreceptor abnormalities were observed in crd1-affected dogs as young as 11-weeks old. Rod and cone inner segment (IS) and outer segments (OS) were abnormal in size, shape, and number. In crd2-affected dogs, rod and cone IS and OS were abnormal as early as 3 weeks of age, progressing with age to severe loss of the OS, and thinning of the outer nuclear layer (ONL) by 12 weeks of age. Genome-wide association study (GWAS) identified association at the telomeric end of CFA3 in crd1-affected dogs and on CFA33 in crd2-affected dogs. Candidate gene evaluation identified a three bases deletion in exon 21 of PDE6B in crd1-affected dogs, and a cytosine insertion in exon 10 of IQCB1 in crd2-affected dogs.
Conclusions.
Identification of the mutations responsible for these two early-onset retinal degenerations provides new large animal models for comparative disease studies and evaluation of potential therapeutic approaches for the homologous human diseases.
Keywords: retina, mutation, GWAS
We describe two genome-wide association studies in two closely related dog breeds affected with retinal degeneration, the pathology of the diseases and the discovery of a novel deletion mutation in PDE6B and an insertion mutation in IQCB1 as the causality for these diseases.
Introduction
Hereditary retinal degenerations (HRD) are blinding disorders characterized by dysfunction and death of rod and cone photoreceptor cells. They are genetically and phenotypically heterogeneous, with causative mutations identified in 202 genes to date (RetNet; available in the public domain at https://sph.uth.tmc.edu/retnet, 2013). This heterogeneity has significant implications for gene discovery, not only because it is critical to identify patients with the same mutation if causality is to be resolved by large-scale association or case-control studies, but also to enable gene therapy to be implemented in gene-specific and even allele-specific manner.
We previously identified two early-onset autosomal recessive retinal degenerations in American Staffordshire Terrier dogs (AmStaff), and American Pit Bull Terrier dogs.1 In both diseases very young dogs (less than 1-year old) were affected by severe photopic and scotopic visual impairment, which progressed to more severe blindness in early adulthood. Because of the similarity of these two diseases, they were termed crd1 and crd2 (for cone–rod dystrophy 1, and 2, respectively). For the same reason, and because these two breeds of dog are physically similar and share common ancestry, a cross-breeding complementation test was undertaken to prove that the two diseases were nonallelic. Candidate gene analysis excluded the genes ABCA41 and AIPL1, GUCY2D, CRX, RDH5, CRB1, RDH12, TULP1, RPGRIP1, RPE65, RDS, and HGR4 (Goldstein O, et al., IOVS 2005;46:ARVO E-Abstract 3191) for both diseases. We now report results of genome-wide association and subsequent studies that identify the causative mutations for both diseases: a PDE6B deletion mutation causing crd1, and an IQCB1 insertional mutation causing crd2.
Methods
Animal Use
All procedures involving animal care were conducted in accordance with the guidelines of the Institute for Laboratory Animal Research (Guide for the Care and Use of Laboratory Animals); the United States Public Health Service (Public Health Service Policy on Humane Care and Use of Laboratory Animals); and the ARVO Resolution on the Use of Animals in Ophthalmic and Vision Research.
Sample Collection
Blood was collected for DNA extraction from (1) 61 colony-derived crd1-affected and unaffected dogs,1 including the proband, a purebred crd1-affected AmStaff, (Fig. 1A, dogs 1–61), (2) eight privately owned crd2-affected and nonaffected-related purebred American Pit Bull Terrier dogs (Fig. 1B, dogs 1–5 and 29–31), and 50 colony-derived crd2-affected and unaffected dogs1 (Fig. 1B, dogs 6–28 and 32–59), and (3) 110 privately owned pedigreed dogs from 19 breeds not known to segregate crd1 or crd2 (Supplementary Table S1).
Figure 1.

Canine pedigrees segregating crd1 and crd2. Solid symbols = affected; half-filled = unaffected obligate heterozygotes; empty = unaffected, either carriers or homozygous normal. Blue = animals genotyped on a SNPchip array for genome wide association study, as well as for the identified crd1 or crd2 mutation. Black numbered = dogs genotyped for informative SNPs and the mutation for linkage analysis. Arrows indicate the propositi for the mixed breed crd1 and crd2 colonies, and the respective informative pedigrees. (A) A crd1-affected AmStaff (dog 2, arrow) was bred to a homozygous normal dog (dog 1). All F1 pups were unaffected. Heterozygous offspring were intercrossed to generate informative litters. (B) A crd2-affected American Pit Bull Terrier (dog 3, arrow) was bred to 2 homozygous healthy dogs (32, 33). All F1 pups were unaffected. Heterozygous offspring were intercrossed or backcrossed to generate informative litters. Dogs 1, 2, 4, 5, 29, 30, and 31 are purebred Pit bull terrier dogs, relatives of the affected founder of the mixed-breed crd2 colony.
Clinical diagnoses were based on ophthalmoscopic examinations, and in selected individuals by electroretinography, as described previously.1
Morphologic Evaluation
From dogs selected for retinal morphologic examination, eyes were enucleated and processed using a triple-fixative protocol (3% glutaraldehyde-2% formaldehyde; 2% glutaraldehyde-1% osmium tetroxide; and 2% osmium tetroxide) as previously described.2,3 Evaluated dogs included: (1) crd1-and crd2-affecteds at selected ages, doubly heterozygous dogs (i.e., dogs heterozygous for both crd1 and crd2), (3) rcd1-affected dogs (rcd1-affected dogs carry a PDE6B mutation, see further discussion below), and (4) two progeny of a crd1xrcd1 crossbreeding.
After fixation, the posterior segment was trimmed into four quadrants extending from the optic disc to the ora serrata. Following dehydration, tissues were embedded in an epoxy resin (polyBed 812; Polysciences, Warrington, PA), sectioned at 1 μm (Supercut 2065 microtome; Leica, Deerfield, IL), and stained with azure II-methylene blue and a paraphenylenediamine counterstain. For each dog, 1-μm sections extending continuously from the optic disc to the ora serrata of superior, inferior, and temporal meridians were evaluated by light microscopy.
Genome-Wide Association Study (GWAS)
crd1.
Group Design.
Seventeen colony-derived-crd1-affected dogs were selected that included one purebred AmStaff, the colony proband (Fig. 1A, dog number 2). For the control group, 18 unaffected-related dogs were selected. Dogs were chosen based on DNA availability and quality, relatedness, and parental and offspring trios for future analysis, if needed (Fig. 1A, dogs colored in blue).
SNP Genotyping.
Samples were assayed using the Illumina HD Canine array, which genotypes 173662 SNP loci, following the manufacturer's standard protocol (Illumina, Inc., San Diego, CA). Genotypes were called using the GenomeStudio algorithm (Illumina, Inc.).
Association Analysis.
Genotype calls were converted into a Plink-format file and association was tested using the PLINK4 association command without pedigree or sex information (available in the public domain at http://pngu.mgh.harvard.edu/purcell/plink/). Genotype calls for canine chromosome 3 (CFA3) were retrieved from the files and assembled into a haplotype to identify the minimal Linkage Disequilibrium (LD) interval and the homozygosity block.
Homozygosity Blocks Analysis.
This analysis was undertaken on affected dogs only (n = 17) using Plink, with the following criteria: sliding window criteria: 1000 Kb, 50 SNPs, 5 missing calls, 1 heterozygous call, 0.05 threshold; homozygous segment criteria: 1000 Kb length, 100 SNPs, 50 density (Kb/SNP). The output was then filtered for chromosomes where at least 16 animals showed a minimum of one homozygous segment anywhere in the chromosome. The segments were then aligned for each chromosome, to identify those where at least 16 dogs shared a homozygous block. For such regions, genotype calls were retrieved to evaluate if all homozygous blocks were homozygous for the same haplotype. If so, then the haplotypes were compared with those observed in the control group.
crd2.
Group Design.
A total of 15 crd2-affected and 13 unaffected dogs were selected, including: (1) Three purebred crd2-affected dogs and two purebred obligate crd2-heterozygotes (Fig. 1B, dogs 1–5), and (2) 12 crd2-affected and 11 obligate crd2-heterozygous colony dogs (Fig. 1B, dogs 6–28). Affected dogs were chosen to be the least related to each other (no siblings) within the expanded colony-derived pedigree.
SNP Genotyping.
Samples were assayed using the Affymetrix canine SNP genotyping array version 2, which genotypes approximately 127000 SNP loci. The standard Affymetrix GeneChip Mapping 250K Sty Assay protocol was followed, using 250 ng genomic DNA. Genotypes were called using the MAGIC algorithm5 based on the intensity values contained in the Affymetrix CEL files.
Association Analysis.
Genotype calls for SNPs that passed the MAGIC quality control filters were then tested for association with the disease phenotype, using Fisher's exact test as follows. First, under or overrepresentation of disease phenotype was evaluated for all samples called homozygous for one of the SNP's two alleles (i.e., “allele 1”), in comparison to all other samples (i.e., a pooled class combining homozygotes for “allele 2” with all those called heterozygous). Then the test was repeated, comparing homozygotes for “allele 2” with all other samples, in this case, the pooled class combined homozygotes for “allele 1” with the heterozygotes. A Bonferroni correction for multiple tests was used to set the significance threshold at −Log10(P) ≥ 6.39 [0.05/(60245*2)], a highly conservative approach given the correlated tests and the conservative nature of this correction. Genotype calls for CFA33 were retrieved from the files and assembled into a haplotype to identify the minimal LD interval and the homozygosity block.
Homozygosity Blocks Analysis.
This analysis was undertaken on affected dogs only (n = 14) using Plink, with the following criteria: sliding window criteria: 50 Kb, 5 SNPs, 5 missing calls, 0 heterozygous calls, 0.05 threshold; homozygous segment criteria: 1000 Kb length, 20 SNPs, 50 density (Kb/SNP). The output was then filtered for those chromosomes where at least 13 animals showed a minimum of one homozygous segment. The segments were then aligned to identify chromosomes where at least 13 dogs shared a homozygous block. For such regions, genotype calls were retrieved to evaluate if all homozygous blocks were homozygous for the same haplotype.
Candidate Gene Analysis.
For both crd1 and crd2 studies, RNA was extracted from the retinas of both a 12-week-old affected dog and a healthy dog as previously described,6 affected and normal sequences were compared using Sequencher 4.2.2 Software (Gene Codes Corporation, Ann Arbor, MI).
crd1.
Within the identified homozygous block, the gene Phosphodiesterase 6B (PDE6B) was the outstanding candidate because of its known importance to phototransduction,7,8 and its previous causal association with hereditary retinal degenerations.9–11 This gene was screened by retinal-cDNA PCR amplification (Supplementary Table S2Ai). The mutation was confirmed by genomic-DNA-PCR of an affected, a carrier, and a healthy dog, followed by sequencing validation (Supplementary Table S2Aii). An allele-specific extension test to screen 105 dogs from 18 breeds (Supplementary Table S1) was then designed as: (1) a primer pair including a forward primer specific to the wild-type allele, to amplify a 186 bp fragment (the normal allele), and (2) a separate primer pair including a mutant-allele-specific reverse primer with a deliberate mismatch in the 3′ penultimate base, to amplify a 362 bp fragment (the mutant allele) (Supplementary Table S2Aiii).
crd2.
Within the identified homozygous block, the gene “IQ calmodulin-binding motif-containing protein 1” (IQCB1, also known as NPHP5) was the outstanding candidate because of its previously identified causal association with human hereditary retinal degeneration syndromes.12–14 This gene was evaluated by exon scanning on genomic DNA (Supplementary Table S2Bi) and by amplification of the coding sequence from a normal and an affected retinal cDNA (Supplementary Table S2Bii). Mutation screening was done by either direct sequencing using the Applied Biosystems Automated 3730 DNA Analyzer (Applied Biosystems, Foster City, CA; Supplementary Table S2Biii, primer pair 1) or by performing an allele-specific extension test using a specific reverse primer for each allele (Supplementary Table S2Biii, primer pairs 2 and 3). Primer pair 2 identifies the wild-type allele and amplifies a 225 bp fragment; primer pair 3 identifies the mutated allele and amplifies a 226 bp fragment. Polymerase chain reaction products were visualized on a 1.8% agarose gel stained with Ethidium Bromide using standard protocols.
Linkage Analysis
Linkage analysis was run using informative pedigrees of colony-derived dogs for each disease, using MultiMap15 as previously described.6,16
crd1.
One informative SNP (rs23644636) and the identified PDE6B mutation, were both genotyped on an extended set of dogs (23 affected, 26 obligate heterozygotes, 9 unaffected, and 3 healthy, Fig. 1A dogs 1–61).
crd2.
Seven dogs from the purebred study pedigree and 52 dogs from the colony segregating the disease were genotyped for a SNP on chromosome 12 (rs22195986) and for the identified IQCB1 mutation (Fig. 1B, dogs 1–59).
RNA Expression
RNA was extracted from the retinas of: (1) a 12-week-old crd1-affected dog, (2) 2.9-, 5.0- and 12.3-week-old crd2-affected dogs, and (3) 3.3-, 4.3-, 8.6-, 10.4-, 12-week-old healthy dogs, and from the spleen of a 22.1-week-old healthy dog. Total RNA membranes were generated as previously described17 from crd2-unaffected and affected retinas. An IQCB1 probe was produced by amplification of normal retinal cDNA using primer pair 5 in Supplementary Table S2Bii (exon 13 to 3′ UTR). The product was then cloned (TOPO TA cloning kit; Invitrogen, Carlsbad, CA), and used for blot hybridization. Hybridization was carried out using Ultrahyb solution (Ambion, Austin, TX) following the manufacturer's protocol. The Blot was exposed to X-ray film at −70°C for 11 days with two intensifying screens. Loading control was achieved by hybridizing a canine-specific beta-actin probe to the membranes under the same conditions and exposed to X-ray film for 4 to 6 hours.
PDE6B Compound-Heterozygosity Analysis
Because rod–cone dysplasia type 1 (rcd1) in Irish Setter dogs is caused by a previously described nonsense mutation in PDE6B,18 a crd1-affected dog was crossbred to an rcd1-affected dog to produce compound-heterozygous progeny. Retinal morphologic evaluation was undertaken, as described above, on eyes from two such pups aged 10 weeks.
Results
Morphology
In the healthy retina at 3-weeks postnatal age the photoreceptor nuclei were individually mostly spindle shaped and formed an outer nuclear layer (ONL) that was approximately 12- to 15-nuclei thick. The nascent inner segment layer (ISL) had formed, with cone inner segments larger but less numerous than those of rods. Few if any outer segments had yet developed (Fig. 2A).
Figure 2.

Light microscopic retinal morphology in healthy (row 1); crd1 (row 2); crd1xrcd1, rcd1, and crd1 (row 3); and crd2 affected dogs (row 4). (A) Retina of a 3-week-old healthy dog. Photoreceptor nuclei are mostly spindle shaped, forming an ONL approximately 12- to 15-nuclei thick. A nascent ISL has formed, with cone inner segments larger, but less numerous than those of rods. Few, if any, photoreceptor OS are detectable. (B) Retina of a 14-week-old healthy dog. The ONL comprises approximately 10 to 11 rows of rod nuclei and a single outermost row of cone nuclei. Photoreceptor IS and are of consistent proportions, tightly aligned, and orientated in parallel. (C) Retina of an 11-week-old crd1-affected dog. The ONL is only 5- to 7-layers thick. Inner segments are severely distorted, disorganized, and loosely packed, with cone IS less damaged than those of rods. Few, if any, OS are present. (D) Retina of a 20-month-old crd1-affected dog. Severe degeneration is evidenced by loss of rod and cone IS, OS, and nuclei. (E) Retina of a 10-week-old rcd1xcrd1 compound heterozygous dog. Degeneration, as evidenced by reduction of the ONL to 4 to 5 layers and absence of IS and OS, is much more severe than in the age matched crd1-affected (see [B, G]), and similar to the age-matched rcd1-affected retina (F). (F) Retina of a 14-week-old rcd1-affected dog. Outer nuclear layer is thinned to 3 to 4 layers, with no evidence of IS and OS. Degeneration is much more severe than in the age-matched crd1-affected (see [B, G]). (G) Retina of an 11-week-old crd1-affected dog for comparison with (E) and (F). (H) Retina of a 3-week-old crd2-affected dog. Broadly similar to age-matched healthy (A). Photoreceptor nuclei in the ONL are broadly similar to age-matched healthy (A), but IS are less well developed and OS are not apparent. (I) Retina of a 12-week-old crd2-affected dog. The ONL comprises only 6 to 7 layers; cone and rod IS and OS are present but abnormal, disorganized, and much reduced in number compared with an age-matched healthy retina (B). (J) Retina of a 20-month-old crd2-affected dog. No IS and OS are present, and the ONL is thinned to less than 1- to 2-cell layers. Scale bars: 10 μM.
By 14-weeks postnatal age, the healthy canine retina ONL was 10- to 11-nuclei thick, and the photoreceptor inner (IS) and outer (OS) segments were well developed, uniformly elongate and arrayed in serried layers (Fig. 2B).
crd1.
At 11-weeks postnatal age, the earliest time-point examined, the ONL of the crd1-affected retina was reduced to between 6 to 8 nuclei in thickness (Fig. 2C). Photoreceptor IS and OS were distinctly distorted, with rod IS more severely affected than those of cones. Relatively few OS of either rods or cones were recognizable, and the profiles that comprised the putative ISL and OSL (i.e., the layer between the outer limiting membrane and the RPE) were sparse and disarrayed (Fig. 2C). By 20 months of age, the crd1-affected retina was in an advanced state of degeneration, with less than 2 to 3 ONL cells (Fig. 2D).
Retinas of compound heterozygous (rcd1/crd1) dogs, examined at 10-weeks postnatal age were affected by a degenerative disease very similar to that of an rcd1-homozygous affected dog at approximately the same age: the rcd1/crd1 ONL was 4- to 5-nuclei thick (Fig. 2E) compared with 3 to 4 in rcd1 (Fig. 2F), and in both cases the ISL and OSL had IS and OS that were similarly and severely reduced in both number and size (Figs. 2E, 2F). In comparison, the 11-week-old crd1-affected retina (Fig. 2G), although distinctly degenerate, had an ONL that was between 6 to 8 nuclei in thickness, representing approximately twice as many remaining photoreceptors as in either the rcd1/crd1 or rcd1 retina. Similarly, although the ISL and OSL of the crd1-affected retina were distinctly degenerate at this age, they were also distinctly better preserved than in either the rcd1/crd1 or rcd1 retina.
crd2.
At 3.3-weeks postnatal age, the earliest time-point examined, the crd2-affected retina (Fig. 2H) had developed to a stage similar to that of the age-matched healthy retina (Fig. 2A). In particular, the number of cells in the ONL of the crd2-affected retina did not appear to be reduced. However, the ISL appeared a little less well developed than in the healthy control, and few if any OS were apparent.
By 12 weeks of age, (Fig. 2I), the ONL of the crd2-affected retina comprised only 5 to 7 layers; cone and rod IS and OS were present but distinctly abnormal, disorganized, and reduced in size and number compared with age-matched healthy retinas (Fig. 2B). At 20 months of age, no IS and OS were present in the crd2-affected retina, and the ONL was thinned to nowhere more than 2 cell layers (Fig. 2J).
In comparing different retinal regions of the same crd2-affected dog, and retinas of different crd2-affected dogs at the same age, it emerged that the stage of development and or degeneration was not always consistent either between regions, or between dogs. In particular, there were sometimes local regions where the retina was either markedly better developed or preserved, or less degenerate, than in other regions of the same eye, or compared with another age-matched affected retina. For example, at 3.3-weeks postnatal age, although the crd2-affected retina in the superior (Fig. 3A) and inferior quadrants (Fig. 2H) shows little IS and essentially no OS development, there is markedly more advanced development of both IS and OS in the temporal quadrant (Fig. 3B). In the latter region a minority of rods had developed outer segments, compared with none in the superior and inferior quadrants (Figs. 3B, 2H). A similar difference was observed in the retina of a 12-week-old crd2-affected retina. In the superior quadrant of this eye (Fig. 3C), the retina is distinctly more degenerate and less well developed than in the temporal quadrant (Fig. 3D), where cones are more numerous, and rod IS and OS, although strangely disarrayed, are much better preserved, or developed. However, these differences do not persist and by 34 weeks of age both central and temporal sections are showing severe degeneration of rods and cones (Figs. 3E, 3F), and this more uniform degeneration continues at later ages (Fig. 2J).
Figure 3.

Light microscopic retinal morphology in crd2-affected dogs exhibiting relative preservation or enhanced development at different ages and between different retinal quadrants. (A, C, E) Superior quadrant (central region, ∼midway from optic nervehead to ora serrata). (B, D, F) Temporal quadrant (central region, ∼midway from optic nervehead to ora serrata). (A, B) Retinas from 3-week-old crd2-affected dogs. Compare also with Figure 2H. Although the ONL is significantly thicker in the superior quadrant (A), IS and OS development is distinctly better preserved in the temporal quadrant (B). (C, D) Retinas from 12-week-old crd2-affected dogs. Compare also to Figure 2I. Preservation of the OS is noticeably better in the temporal quadrant (D), where the IS are more tightly organized and more cones are present, than in the central region (C) with its relatively sparse OSL and disorganized ISL. (E, F) Retinas from 34-week-old crd2-affected dogs. At this age the differences between the central (E) and temporal (F) quadrants are less noticeable, with more severe degenerative changes across the retina. Scale bars: 10 μM.
Double Heterozygous Analysis.
Prior to identification of the crd1 and crd2 mutations, a crd1-affected dog (Fig. 1A, dog number 2) was bred to a crd2-heterozygous dog (Fig. 1B, dog number 11) as a test for allelism, and produced 8 phenotypically healthy progeny.1 We subsequently bred a crd1-heterozygous dog to a crd2-affected dog, obtaining five further progeny. Once the crd1 and crd2 mutations were identified, these 13 dogs were genotyped for both mutations, to identify six dogs that were heterozygous for both the crd1 and the crd2 mutations. Clinical and ophthalmoscopic evaluation of these dogs showed no evidence of vision impairment or retinal abnormality. Retinal morphologic evaluation in two of these dogs at 20-weeks postnatal age found no pathologic changes (data not shown).
Genome-Wide Association Study
crd1.
Thirty-five dogs were genotyped for the 173662 SNPs on the HDCanine-Illumina-array. Thirty of these dogs had both parents represented in the experiment, and four dogs had only one parent. The proband, dog number 3 in Figure 1A, had neither parent analyzed. Analysis of genotype calls determined the heritability frequency at 0.9998 for both one-parent- and two-parents-genotyped cases, and the average number of errors was 24.8 SNPs (Supplementary Table S3). Genotype calls were converted to Plink-formatted files and association analysis was run comparing the 17 affected dogs to the 18 controls. The highest −Log10(P) value was 5.764, observed in seven SNPs on CFA3, in the interval 93927492-94484373 (Table 1; Fig. 4A). All affected dogs were homozygous for the same allele in each of these seven SNPs, while all the unaffected dogs were heterozygous, supporting recessive inheritance as suggested by pedigree analysis. All CFA3 genotype calls were then aligned, to identify a homozygous block where all affected dogs were homozygous for the same haplotype and all controls were not. A 1.05 Mb homozygous block was observed from 93639445-to the telomeric end of CFA3 represented by a SNP located at 94693816 and included 88 SNPs (Supplementary Table S4A). Within this block, SNP rs23644636 (CFA3: 94607062) was genotyped on the extended crd1-derived colony pedigree (Fig. 1A, dogs 1–61). Complete linkage was observed between the SNP and the disease, and a lod score of 13.847 where theta equals 0.00 was obtained.
Table 1.
Top Hits (−Log10(P) > 4) of GWAS in the crd1 Study
|
Number |
i. Sort by −Log10(P) |
ii. Sort by Location |
||
|
Location* |
–Log10(P) |
Location |
–Log10(P) |
|
| crd1 significant SNPs and their −Log10(P) values | ||||
| 1 | 93927492 | 5.764 | 93618479 | 4.644 |
| 2 | 94075783 | 5.764 | 93639445 | 4.644 |
| 3 | 94287447 | 5.764 | 93684805 | 5.024 |
| 4 | 94296622 | 5.764 | 93697691 | 5.384 |
| 5 | 94355737 | 5.764 | 93700678 | 5.384 |
| 6 | 94451861 | 5.764 | 93731523 | 5.384 |
| 7 | 94484373 | 5.764 | 93737251 | 5.384 |
| 8 | 94578249 | 5.715 | 93799182 | 4.663 |
| 9 | 94607062 | 5.715 | 93815630 | 4.663 |
| 10 | 93697691 | 5.384 | 93914980 | 4.663 |
| 11 | 93700678 | 5.384 | 93927492 | 5.764 |
| 12 | 93731523 | 5.384 | 94075783 | 5.764 |
| 13 | 93737251 | 5.384 | 94221985 | 4.571 |
| 14 | 93684805 | 5.024 | 94287447 | 5.764 |
| 15 | 93799182 | 4.663 | 94296622 | 5.764 |
| 16 | 93815630 | 4.663 | 94306027 | 4.663 |
| 17 | 93914980 | 4.663 | 94355737 | 5.764 |
| 18 | 94306027 | 4.663 | 94410234 | 4.663 |
| 19 | 94410234 | 4.663 | 94415960 | 4.571 |
| 20 | 94659212 | 4.663 | 94451861 | 5.764 |
| 21 | 94668519 | 4.663 | 94476356 | 4.239 |
| 22 | 93618479 | 4.644 | 94484373 | 5.764 |
| 23 | 93639445 | 4.644 | 94578249 | 5.715 |
| 24 | 94221985 | 4.571 | 94607062 | 5.715 |
| 25 | 94415960 | 4.571 | 94659212 | 4.663 |
| 26 | 94476356 | 4.239 | 94668519 | 4.663 |
In bold are the SNPs with the highest −Log10(P) values.
All SNPs are located on chromosome 3 (CFA3) and location is based on CanFam 2.0.
Figure 4.

Manhattan plots summarizing results of GWAS in two canine hereditary retinal degenerations, crd1 and crd2. x-axis = Canine chromosomes 1-38, plus the X-chromosome presented as chromosomes 39 or 39 and 41. Y-axis = probability statistic (−log10[observed P]). (A) crd1. Results of association analysis using PLINK software. The highest −log10(P) values are observed at the telomeric end of chromosome 3 (CFA3). The X chromosome is represented by number 39 and 41. (B) crd2. Results of association analysis using Fisher-exact test. The highest peak is observed on chromosome 33. A second peak at lower significance is observed on CFA12. The X chromosome is represented by the numbers 39 and 40.
crd2.
Of the approximately 127000 loci on the Affymetrix SNP chip, genotype calls for 60245 passed quality control testing filters of the algorithm “MAGIC.” Genotype calling for one affected sample did not pass quality control and was not further used in the analysis (dog 16 in Fig. 1B). The Fisher exact test ran on 14 affected and 13 obligate-heterozygous samples yielding two hits: one to chromosome 33 and one to chromosome 12 (Table 2; Fig. 4B), the latter with lower P values. The peak −Log10(P) value of 6.126 was shared by five SNPs, all located within a 4.4 Mb interval on CFA33 (22690750-27122415). None of the SNPs passed genome-wide significance thresholds (−Log10[P] > 6.39). All 14 affected dogs were homozygous for these SNPs as was one obligate-heterozygote (dog number 2 in Fig. 1B, a purebred Pit Bull Terrier). The remaining 12 obligate-heterozygotes in the control group were heterozygous for all five SNPs. This supported recessive inheritance as suggested from pedigree analysis, except for dog 2. Accordingly, all genotype calls on CFA33 were aligned to identify a homozygosity block, where all affected were homozygous for the same haplotype and all controls were not (Supplementary Table S4B). Three such regions were identified. The proximal block, 0.273 Mb long (22519503-22792968) comprised genotypes for eight SNPs, and included one of the five most significant SNPs (CFA33: 22690750) and only one refseq gene (GAP34). Another homozygous block at CFA33: 24222713-24890251, a 0.667 Mb interval, included 31 SNPs, three of which had the highest P value, but no refseq genes. The most distal block was 2.67 Mb long (CFA33: 26786412-29463543), comprised 47 SNPs, one of which had the highest P value, included four refseq genes (COX17, CD86, CASR, and ADCY5) and more than 15 non-refseq genes that are expressed in the eye. Except for dog number 2, all dogs in the control group were heterozygous for all SNPs in all three “homozygosity blocks.” Dog number 2 was homozygous for the same haplotype as its affected offspring, dog number 3, for an 18 Mb interval on CFA 33 (data partially shown in Supplementary Table S4B for the interval 22512 990-29709462).
Table 2.
Top hits (−Log10(P) > 4) of GWAS in the crd2 Study
|
Number |
i. Sort by −Log10(P) |
ii. Sort by Location |
||
|
Location* |
–Log10(P) |
Location |
–Log10(P) |
|
| crd2 significant SNPs and their −Log10(P) values | ||||
| 1 | Chr33. 22690750 | 6.126 | Chr12. 3464390 | 4.172 |
| 2 | Chr33. 24438552 | 6.126 | Chr12. 3865659 | 4.162 |
| 3 | Chr33. 24438637 | 6.126 | Chr33. 21465360 | 4.470 |
| 4 | Chr33. 24885473 | 6.126 | Chr33. 22690750 | 6.126 |
| 5 | Chr33. 27122415 | 6.126 | Chr33. 23215781 | 4.152 |
| 6 | Chr33. 29049963 | 5.469 | Chr33. 23804255 | 4.470 |
| 7 | Chr33. 29860172 | 5.223 | Chr33. 23882615 | 4.470 |
| 8 | Chr33. 30531791 | 5.008 | Chr33. 24438552 | 6.126 |
| 9 | Chr33. 30378609 | 4.703 | Chr33. 24438637 | 6.126 |
| 10 | Chr33. 21465360 | 4.470 | Chr33. 24478407 | 4.470 |
| 11 | Chr33. 23804255 | 4.470 | Chr33. 24564448 | 4.470 |
| 12 | Chr33. 23882615 | 4.470 | Chr33. 24574669 | 4.470 |
| 13 | Chr33. 24478407 | 4.470 | Chr33. 24885473 | 6.126 |
| 14 | Chr33. 24564448 | 4.470 | Chr33. 27122415 | 6.126 |
| 15 | Chr33. 24574669 | 4.470 | Chr33. 28668552 | 4.172 |
| 16 | Chr12. 3464390 | 4.172 | Chr33. 28882337 | 4.152 |
| 17 | Chr33. 28668552 | 4.172 | Chr33. 29049963 | 5.469 |
| 18 | Chr12. 3865659 | 4.162 | Chr33. 29860172 | 5.223 |
| 19 | Chr33. 30517730 | 4.162 | Chr33. 30378609 | 4.703 |
| 20 | Chr33. 23215781 | 4.152 | Chr33. 30517730 | 4.162 |
| 21 | Chr33. 28882337 | 4.152 | Chr33. 30531791 | 5.008 |
In bold are the SNPs with the highest −Log10(P) values.
Location is based on CanFam 2.0.
The same analysis was applied to the second hit on CFA12. Haplotype analysis showed that within the significant region, two affected animals were heterozygous through the whole interval (approximately 8 Mb interval), and one control dog was homozygous to the affected haplotype (data not shown). To further assess this hit, a SNP within this interval (rs22195986, CFA12: 3865659, −log10[P] = 4.162; Table 2) was genotyped in a larger crd2-informative pedigree and linkage analysis undertaken. A lod score of 0.738, at theta equals 0.406 indicted this CFA12 hit as a false positive.
Homozygosity Analysis
To evaluate the power of homozygosity analysis in simple Mendelian recessive diseases, we analyzed the calls from affected dogs only (17 crd1-affected dogs and 14 crd2-affected dogs), and looked for homozygous blocks that are greater than 1.0 Mb in each disease separately. We did not analyze the sex chromosomes.
crd1.
Runs of homozygosity blocks analysis in affected dogs showed three loci where at least 16 out of the 17 affected dogs shared a homozygous block under the criteria applied: CFA1: 3014448-4989300 (1.97 Mb); CFA2: 69772560-71266032 (1.49 Mb); and CFA3: 93388160-94693816 (1.3Mb) (Fig. 5A, Supplementary Table S5). Analysis of the genotype calls within these blocks showed that for the CFA1 locus, all affected dogs shared the same homozygous haplotype, but that haplotype was also observed in control dogs. At the CFA2 locus, a few heterozygous calls were observed within the identified block. A smaller homozygous block was observed between 70054331 and 70579965 (525.6 Kb), and for that interval the haplotype was shared among the control dogs as well. At CFA3, 16 affected dogs shared 1.278 Mb homozygous haplotype (93415696-94693816), and the 17th affected dog shared a smaller interval of 1.05 Mb. This dog was not included in the result output, since it had 88 SNPs supporting its homozygous segment (less than 100 SNPs, one of the criteria for the run). All the control dogs presented a different haplotype within this segment (Supplementary Table S4A).
Figure 5.

Schematic illustration of results of homozygosity block analysis on crd1 and crd2 disease implicated chromosomes. (A) crd1. Seventeen affected dogs (y-axis) were analyzed to identify homozygous blocks larger than 1 Mb. Horizontal black bars represent homozygous blocks observed on chromosome 3 (x-axis). The grey-shaded vertical box (far right) shows where 16 dogs share a homozygous block. Dog number 17 shares this same region as well, but is not represented here with a horizontal black bar since its block was supported by less than 100 SNPs, one of the criteria used in the analysis. (B) crd2. Fourteen affected dogs (y-axis) were analyzed to identify homozygous blocks larger than 1 Mb. Horizontal black bars represent the homozygous blocks observed on chromosome 33 (x-axis). The grey shaded vertical box shows where all 14 dogs share a homozygous block.
crd2.
Runs of homozygosity blocks analysis in affected dogs identified only one locus where all 14 affected dogs were homozygous under the criteria of the run: CFA33: 27065615-29382529 (2.3 Mb). This is the homozygous block bearing the mutation, with 47 SNPs supporting that block and no heterozygous calls (Fig. 5B, Supplementary Tables S4B, S5).
Candidate Genes Evaluation
crd1.
PDE6B screening.
The canine PDE6B gene is located within the 1.05 Mb minimal homozygosity block identified by association analysis. Mutations in this gene have been previously identified as causing hereditary retinal degenerations in Irish Setter18 and Sloughi dogs,19 as well as in humans9–11 and mice.20 The gene was amplified from an affected dog and compared with a healthy, using cDNA prepared from retinal RNA extracts and five primer pairs producing overlapping amplicons, covering the complete open reading frame. Nine SNPs were observed in the coding sequence of the gene in a healthy dog, when compared with the National Center for Biotechnology Information (NCBI) database mRNA sequence NM001002934 (Supplementary Table S6). The affected dog was homozygous for the NCBI database Boxer canine genomic reference sequence, suggesting that these SNPs are not causally associated with the disease. However, a three-bases-deletion was identified in exon 21 of the gene in the affected dogs (c.2404-2406del, CFA3: 94574289-94574291), in-frame with the protein (Fig. 6A). This mutation would result in a deletion of the amino acid asparagine at position 802 of the protein (p.802del).
Figure 6.

Normal and affected chromatograms for crd1 and crd2 diseases. Each codon within the open reading frame is translated to its amino acid as denoted by a letter above or under the chromatogram. (A) The upper chromatogram is the normal canine sequence for part of PDE6B. In red and bold are the amino acids (NN) corresponding to codons 802 and 803 (N = asparagine). Boxed are the six bases corresponding to these two codons (AACAAC). The lower chromatogram is the corresponding sequence from a crd1-affected dog. Only one AAC codon is observed at position 802-803 (boxed), which translates to an in-frame protein with one amino acid deletion (only one asparagine, labeled in bold and red). (B) The upper chromatogram is the normal canine sequence for part of IQCB1. The lower chromatogram is the corresponding sequence from a crd2-affected dog. A cytosine insertion is observed (red arrow), resulting in a stretch of three cytosines compared with two in the healthy dog. This results in a frame-shift, changing 12 amino acids (colored in red and bold), followed by a premature stop codon TAA (p.S319IfsX12).
To evaluate whether this deletion was a result of an RNA editing mistake, or a deletion in the DNA, primers located in introns 20 and 21 flanking the deletion were used to amplify genomic DNA from affected, obligate-heterozygous and healthy dogs. Sequence confirmed the presence of this three bases deletion in the affected genomic DNA, a deletion not observed in the healthy dog, and the chromatogram from a heterozygous dog showed overlapping sequence downstream from the deletion point (data not shown).
Linkage analysis and population screening.
All dogs in an extended crd1-derived colony pedigree were genotyped for the mutation. Complete linkage was observed between the mutation and the disease, with a lod score of 13.847 at theta equaled 0.00. The deletion was not found in 105 dogs from 18 different breeds not known to segregate crd1 (Supplementary Table S1).
crd2.
IQCB1 screening.
Canine IQCB1 is located within the 2.67 Mb homozygous block identified by association study. Mutations in this gene have been identified in human patients affected with Senior-Loken syndrome and retinitis pigmentosa (RP).12,21 Human IQCB1 has 15 exons (NM_001023570, transcript variant 1), 13 of which code for a 598 amino acid long protein (NP_001018864, isoform a). Another splice variant is reported with 12 exons (NM_001023571, transcript variant 3) that codes for a 465 amino acid protein (NP_001018865, isoform c). Variant 3 does not include exons 8, 9, and 10 of transcript variant 1, and was observed in embryonic stem and melanocytes. Blasting NM_001023570 against CanFam2 identified all 15 predicted canine exons. To evaluate the role of the gene in crd2 disease, cDNA from an affected retina, and genomic DNA from blood of an affected dog, were amplified and compared with healthy canine sequence.
Reverse transcriptase PCR products from affected and healthy retinal cDNA were assembled into a 2027 bp long fragment and compared. The sequence comprised 85 bp partial 5′ UTR, 1797 bp of the complete coding sequence and 145 bp partial 3′ UTR (Accession number KF366421; Supplementary Fig. S1A). The predicted wild-type canine IQCB1 protein is 598 amino acids long with 88.7% identity to the human counterpart (Supplementary Fig. S1B).
A one-base insertion of cytosine in exon 10 was observed in the affected retinal cDNA, resulting in a stretch of three cytosines compared with two in the wild-type allele (CFA33: 28120686-28120687, Fig. 6B, red arrow; Supplementary Fig. S1C). This insertion, c.952-953insC, causes a frame-shift that results in a change of 12 amino acids (amino acids 319-330) and introduction of a premature stop codon (p.S319IfsX12). This would yield a predicted 330 amino acid protein, in which only 318 are identical to normal (Fig. 6B; Supplementary Fig. S1D). This deletion was also identified in genomic DNA from an affected dog by amplifying exon 10 using primers located in introns 9 and 10 (primer pair 10, Supplementary Table S2Bi).
Linkage analysis and population screening.
A subset of dogs from the crd2 colony-derived pedigree, plus seven purebred dogs related to the proband (Fig. 1B, dogs 1–59) were genotyped for the mutation. No recombination was observed between the mutation and the disease locus and linkage analysis gave a lod score of 11.74 at theta equals 0.0. Dog number 2, despite having the same SNPchip genotype in the LD interval on CFA33 as its affected offspring, genotyped heterozygous for the IQCB1 mutation, in concordance with its nonaffected phenotype.
A control group of 86 dogs from 17 different breeds was genotyped for the mutation (Supplementary Table S1) and were all homozygous for the wild-type allele.
IQCB1 mRNA expression analysis.
To evaluate mRNA expression of the IQCB1 gene in a retina affected by this nonsense mutation, an RNA blot was generated containing mRNA from healthy and affected retinas at selected ages, as well as from a healthy spleen (Fig. 7). Very low levels of IQCB1 expression were detected in control retinas and only after 11 days of film exposure (Fig. 7, lanes 1, 3, 5, 7, 8). In the crd2-affected retina there were almost undetectable levels of IQCB1 at the three ages evaluated: 2.9 weeks, 5.0 weeks, and 12.3 weeks (Fig. 7, lanes 2, 4, 6). IQCB1 was not detected in the spleen. Beta actin shows insignificant variation in loading quantity among the retinal samples. This suggests that mutant IQCB1 mRNA is degraded and presumably by nonsense-mediated decay.
Figure 7.
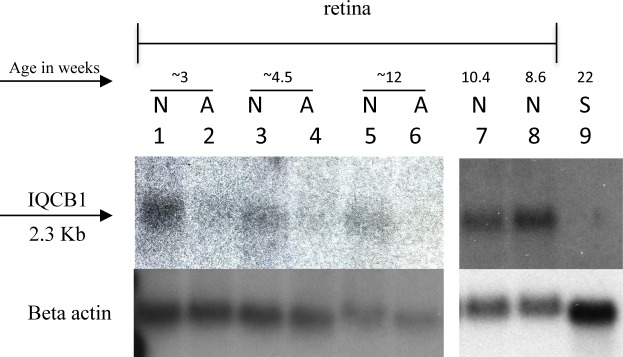
Northern blot demonstrating IQCB1 RNA expression in healthy and crd2-affected retina at specific ages. In samples from healthy retinas (N; lanes 1, 3, 5, 7, 8), low levels of IQCB1 mRNA are detected after 11 days of film exposure at all ages tested (3.3-weeks old, lane 1; 4.3-weeks old, lane 3; 12-weeks old, lane 5; 10.4-weeks old, lane 7; 8.6-weeks old, lane 8). In samples from crd2-affected retinas (A; lanes 2, 4, 6) IQCB1 expression is almost undetectable at the three different time-points evaluated (2.9-weeks old, lane 2; 5-weeks old, lane 4; and 12.3-weeks old, lane 6). No IQCB1 RNA was detected in a sample from 22-weeks-old healthy spleen (lane 9). Beta-actin control shows insignificant variation in loading quantity among the retinal samples.
Discussion
Genome-wide association studies have a wide range of application, from simple mendelian traits, to complex, within isolated populations and in more diverse cohorts, in humans and in animals.22–31 In the second quarter of 2012 only, 1350 studies were published with GWAS results at P values equal or lower than 5 × 10−8 (available in the public domain at www.genome.gov/gwastudies).
Canine populations mimic human isolates in their small, closed gene pools, resulting in a more homogenous genome and larger LD blocks within each breed, making them ideally suited for mapping disease-causative genes.17,32–35 Previously, we mapped by GWAS the gene responsible for crd3, a late onset cone–rod dystrophy segregating in the Irish Glen of Imaal Terrier breed of dog.36 In the current study, we mapped by GWAS the loci for two nonallelic but phenotypically similar diseases in two closely related breeds, in each study utilizing fewer than 20 dogs per case and control group, and exploiting mixed-breed colony-derived pedigrees.
The first disease, crd1, mapped to chromosome 3, enabling a mutation in PDE6B to be identified. The second, crd2, mapped to chromosome 33, and a mutation in IQCB1 was identified. Though the diseases show similar pathology, albeit with some differences in detail, the responsible genes are in two different pathways, and are expressed in different compartments of the photoreceptor cell: PDE6B in the rod photoreceptor OS, and IQCB1 in the connecting cilium.
PDE6B codes for one of the three subunits (alpha, beta, gamma in 1:1:2 ratio) required to assemble the heterotetrameric protein PDE6, a rod-specific essential component of the visual cascade. PDE6 mediates hydrolysis of cyclic guanine monophosphate (cGMP), closing ionic channels gated by cGMP and eventually generating a visual signal. Thus, cGMP-PDE is pivotal in phototransduction.37 This newly identified canine PDE6B mutation, a three-bases in-frame deletion, would produce a one amino acid deletion in the catalytic domain of the PDE6B protein (position 802, Fig. 8A, yellow arrow head). This amino acid is part of the enzyme activity domain PDEase_I-pfam00233, which starts at amino acid 556 and ends at amino acid 804 (Fig. 8A, green box). The deleted amino acid is highly conserved among species (Fig. 8B) and further domain conservation analysis suggests that the presence of an amino acid at this position is highly conserved as well (data not shown).
Figure 8.

PDE6B protein domain representation and partial protein alignment between seven species. The canine crd1-deletion is located in the PDEase-I domain, and it is highly conserved among species. (A) Schematic representation of PDE6B human protein (P35913) and its domain. Using the domain mapping of disease mutations bioinformatic tool,45 SNPs in PDE6B are shown by blue pins; reported mutations (n = 11) are shown in red pins; the inferred location of the crd1-mutation is shown by two yellow arrow-heads. Protein domains are shown in rectangles. The domain where the canine crd1- deletion is located (PDEase_I-pfam00233) is indicated by the green box. (B) Conservation of PDE6B protein. Partial PDE6B human protein sequence is aligned to homologous horse, bovine, mouse, xenopus, zebrafish, the dog wildtype, and affected protein sequence. Amino acid 802 (boxed N) is conserved in all seven species. The red W identifies the location of the nonsense mutation responsible for canine rcd1; the red K identifies the location of the insertional mutation responsible for PRA in the Sloughi dog breed, all located in exon 21. The deleted amino acid is denoted by (-) in the eighth sequence.
At least 25 different PDE6B mutations have been identified in humans and these account for approximately 4.5% of all autosomal recessive RPs (arRPs) in the United States.11 Most of those mutations are missense mutations, and although the downstream effect of these types of mutations has not been fully characterized, they may change the enzyme's function rather than eliminate it as nonsense mutations do. Since the type of mutation can influence the mode of inheritance, age of onset, rate of progression, and efficiency of gene therapy for the disease, it is important to identify animal models that harbor similar types of mutations to those found in human patients. Several mouse models for PDE6B deficiency are available. The rd1 mouse is homozygous for a PDE6B nonsense mutation and presents as a very early onset and rapid disease, with rod photoreceptor degeneration beginning by approximately 8-days postnatal, and complete by 4 weeks of age with no then remaining photoreceptors. On the other hand, rd10 mice, homozygous for a missense mutation in exon 13 (Arg560Cys) are affected with slower rod photoreceptor degeneration, beginning at 16 days of age in the central retina and at 20 days in peripheral retina, and proceeding to death of all photoreceptors by 60 days of age. Histologic analysis comparing rd1 and rd10 shows four ONL in rd10 mice compared with zero in rd1 at 24 days of age. In complementation testing, all F1 (rd1 x rd10) progeny were affected at an early age and identical in retinal appearance to rd1 at 66 days. The rd10 model is, thus, of great value for early-stage theraputic testing, balancing a significant retinal pathology with a slower rate of progression, and, thus, an enhanced opportunity to achieve rescue of cells.
The PDE6B mutation in crd1 dogs does not introduce a premature stop codon, as the three bases deletion is in-frame with the open reading frame, resulting in a one amino acid deletion (Fig. 8B). In contrast, the PDE6B mutation observed in rcd1 dogs is a nonsense mutation in codon 807 (Fig. 8B, circled W), just five amino acids downstream from the position of the crd1 mutation, and results in a protein truncated by 49 amino acids, and complete loss of enzyme function.18 We suggest that the two canine diseases, rcd1 and crd1, correspond to the rd1 and rd10 mouse models, respectively, in terms of the type of mutation, phenotype, age of onset, rate of progression, and the potential for gene therapy. Both rcd1 and rd1 have a nonsense mutation, while crd1 and rd10 do not, and no premature stop codon is introduced as a result of the latter two mutations. Functional assays showed complete loss-of-function in both rcd1 and rd1. In both, rod photoreceptor degeneration is early and fast. In contradistinction, crd1 affected dogs and rd10 mice show a relatively slower progression of cell death and offer important advantages for evaluating gene therapies particularly for human RP caused by missense rather than null PDE6B mutations.
Though phenotypically crd2 is very similar to crd1 disease, the genetic mutations underlying the two diseases were different. By genome-wide association we were able to identify the locus for canine crd2 on chromosome 33, to a 2.67 Mb interval. The use of obligate heterozygotes as a control group reduced the association signal, but did not change the ability to identify the causative locus. The usage of the broader colony-derived pedigree was essential to distinguish between the false positive association signal (hit on CFA12) and real positive hits (hit on CFA33). This emphasizes the importance of informative pedigrees to better evaluate statistics results, or to better prioritize loci if identified by a homozygosity approach. Interestingly, one purebred obligate carrier had genotype calls identical to its purebred affected offspring at the disease locus that extended over an 18 Mb interval (dog number 15, Supplementary Table S4). This suggests that when a mutation arises on a chromosome with a haplotype that is widespread in a population of limited diversity, the resolution of the SNPchip array can be limited, and GWAS challenging, particularly if only closely related individuals are available for the study.
The gene IQCB1, identified herein as mutated in canine crd2, codes for one member of a group of up to 13 different ciliary proteins collectively called Nephronophthisis (NPHP) proteins.38 Mutant NPHP proteins cause a broad spectrum of diseases collectively termed ciliopathies and characterized by pathologies including renal cysts, liver fibrosis, and retinal degeneration and others.38,39
Mutations in IQCB1 (NPHP5) consistently cause retinal disease, as is also the case for CEP290 (NPHP6).40 Eleven different mutations in IQCB1, all of which resulted in a predicted truncated protein, were identified in human patients with Senior-Loken syndrome (SLSN) a disorder including both nephronophthisis and RP, whereas IQCB1 mutations were not found in other nephronophthisis patients without RP12,21 (available in the public domain at http://www.hgmd.org). Plausible disease-causing genotypes in IQCB1 were identified in nine patients affected by Leber congenital amaurosis (LCA) without nephrononphthisis.14 These patients suffer from significant vision loss and nystagmus in the first few months of life. None exhibited evidence of renal disease in the first decade of life but two were diagnosed with NPHP at age 13 years. Similar results were reported in a different cohort of LCA patients from The Netherlands, with frame-shift and nonsense IQCB1 mutations identified in 11 patients, and a highly variable onset of renal failure.13 Nonophthalmologic problems consistent with a ciliopathy were not observed and have not been reported in crd2-affected dogs. Similarly, in the standard wire-haired dachshund breed of dog, where a mutation in NPHP4 has been causally associated with a cone–rod dystrophy, no kidney involvement is reported.41 However, in both these diseases, careful evaluation of older dogs is clearly warranted.
The IQCB1 protein interacts with calmodulin and the RP GTPase regulator (RPGR) protein. The encoded protein has a central coiled-coil region and four IQ domains: coil–coil domain (aa 336-362); IQ1 (aa 294-317), IQ2 (aa 318-338), IQ3 (aa 387-416), IQ4 (aa 417-437). It is localized to the primary cilia of renal epithelial cells and connecting cilia of photoreceptor cells. The canine insertional mutation in exon 10 of IQCB1 results in a frame-shift and a premature stop codon 39 bases after the insertion. RNA expression analysis suggests that the mutated RNA is degraded in the affected retina, and therefore the protein is not transcribed. If any RNA escapes the degradation by nonsense-mediated decay, the predicted protein would have only the first 318 amino acids, missing IQ2, IQ3, and IQ4 domains, and the coiled-coil domain. The reported human mutations are similar in that that all of them are nonsense mutations or small insertions or deletions resulting in a premature stop codon and a predicted truncated protein. Most of these mutations would result in the absent of one of the IQ domains or the coiled-coil domain. The 88.7% homology between the dog and human IQCB1 proteins (Supplementary Fig. S2) further supports canine crd2 disease as a valuable model for SLSN and LCA diseases.
Ciliopathies feature a broad spectrum of organ involvement. It has recently become evident that the spectrum can vary by at least two mechanisms: multiple allelism and modifiers. For example, a polymorphic coding variant of RPGRIP1L (NPHP8) was found to be associated with the development of retinal degeneration in patients with ciliopathies caused by mutations in other genes42; NPHP6 and AHl1 modify recessive NPHP1 mutations to express a more severe phenotype.43 Stone et al.14 showed that although all patients had seemingly similar disease alleles in that they were all nonsense mutations or frame-shifts, which might reasonably be expected to result in complete loss of function of the protein, variation in disease phenotype was still observed. One explanation might be the effect of genetic background, that is, variations in other genes that do not themselves adversely affect an individual's phenotype, but do alter the pathogenicity of a disease-causing allele. Future studies on the crd2 canine model might shed light on those genetic alleles and their effect on the renal involvement in this disease.
Stone and his group had also shown, by optical coherence tomography (OCT) imaging (Fourier-domain OCT system, RTVue-100; Optovue, Inc., Fremont, CA), partial preservation of foveal photoreceptors in two patients with IQCB1 mutations.14 This is especially encouraging for future therapeutic interventions. In young crd2-affected dogs retina also, the temporal quadrant tends to preferentially develop and preserve photoreceptors. The crd2-dog, thus, has significant appeal as a model for gene-therapy of SLSN and LCA diseases caused by IQCB1 mutations, and a valuable complement to the rd16;Nrl−/− double-mutant mouse model.44
Supplementary Material
Acknowledgements
Supported by National Institutes of Health Grants EY006855, EY17549, R24GM082910, 2PNEY018241-06; The Foundation Fighting Blindness; and Van Sloun Fund for Canine Genetic Research
Disclosure: O. Goldstein, None; J.G. Mezey, None; P.A. Schweitzer, None; A.R. Boyko, None; C. Gao, None; C.D. Bustamante, None; J.A. Jordan, None; G.D. Aguirre, Optigen LLC (I, C); G.M. Acland, Optigen LLC (I, C)
References
- 1. Kijas JW, Zangerl B, Miller B, et al. Cloning of the canine ABCA4 gene and evaluation in canine cone-rod dystrophies and progressive retinal atrophies. Mol Vis. 2004; 10: 223–232 [PubMed] [Google Scholar]
- 2. Zeiss CJ, Ray K, Acland GM, Aguirre GD. Mapping of X-linked progressive retinal atrophy (XLPRA), the canine homolog of retinitis pigmentosa 3 (RP3). Hum Mol Genet. 2000; 9: 531–537 [DOI] [PubMed] [Google Scholar]
- 3. Acland GM, Aguirre GD. Retinal degenerations in the dog. IV: early retinal degeneration in Norwegian elkhounds. Exp Eye Res. 1987; 44: 491–521 [DOI] [PubMed] [Google Scholar]
- 4. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J Hum Genet. 2007; 81: 559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyko AR, Quignon P, Li L, et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010; 8: e1000451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldstein O, Guyon R, Kukekova A, et al. COL9A2 and COL9A3 mutations in canine autosomal recessive oculoskeletal dysplasia. Mamm Genome. 2010; 21: 398–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Catty P, Pfister C, Bruckert F, Deterre P. The cGMP phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions. J Biol Chem. 1992; 267: 19489–19493 [PubMed] [Google Scholar]
- 8. Stryer L. Visual excitation and recovery. J Biol Chem. 1991; 266: 10711–10714 [PubMed] [Google Scholar]
- 9. McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993; 4: 130–134 [DOI] [PubMed] [Google Scholar]
- 10. Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet. 1994; 7: 64–68 [DOI] [PubMed] [Google Scholar]
- 11. McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A. 1995; 92: 3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Otto EA, Loeys B, Khanna H, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005; 37: 282–288 [DOI] [PubMed] [Google Scholar]
- 13. Estrada-Cuzcano A, Koenekoop RK, Coppieters F, et al. IQCB1 mutations in patients with Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2011; 52: 834–839 [DOI] [PubMed] [Google Scholar]
- 14. Stone EM, Cideciyan AV, Aleman TS, et al. Variations in NPHP5 in patients with nonsyndromic Leber congenital amaurosis (LCA) and Senior-Loken syndrome. Arch. Ophthalmol. 2011; 129: 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matise TC, Perlin M, Chakravarti A. Automated construction of genetic linkage maps using an expert system (MultiMap): a human genome linkage map. Nat Genet. 1994; 6: 384–390 [DOI] [PubMed] [Google Scholar]
- 16. Kukekova AV, Goldstein O, Johnson JL, et al. Canine RD3 mutation establishes rod-cone dysplasia type 2 (rcd2) as ortholog of human and murine rd3. Mamm Genome. 2009; 20: 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldstein O, Zangerl B, Pearce-Kelling S, et al. Linkage disequilibrium mapping in the domestic dog breeds narrows the progressive rod-cone degeneration interval and identifies ancestral disease-transmitting chromosome. Genomics. 2006; 88: 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suber ML, Pittler SJ, Qin N, et al. Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase β–subunit gene. Proc Natl Acad Sci. 1993; 90: 3968–3972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dekomien G, Runte M, Gödde R, Epplen JT. Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Cytogenet Cell Genet. 2000; 90: 261–267 [DOI] [PubMed] [Google Scholar]
- 20. Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A. 1991; 88: 8322–8326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otto EA, Helou J, Allen SJ, et al. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat. 2008; 29: 418–426 [DOI] [PubMed] [Google Scholar]
- 22. Hu X, Pickering E, Liu YC, et al. Meta-Analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer's disease. PLoS One. 2011; 6: e16616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Palmer ND, McDonough CW, Hicks PJ, et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS One. 2012; 7: e29202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenny EE, Pe'er I, Karban A, et al. A genome-wide scan of Ashkenazi Jewish crohn's disease suggests novel susceptibility loci. PLoS Genet. 2012; 8: e1002559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Begum F, Ghosh D, Tseng GC, Feingold E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 2012; 40: 3777–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bolormaa S, Pryce JE, Hayes BJ, Goddard ME. Multivariate analysis of a genome-wide association study in dairy cattle. J Dairy Sci. 2010; 93: 3818–33 [DOI] [PubMed] [Google Scholar]
- 27. Duijvesteijn N, Knol EF, Merks JW, et al. A genome-wide association study on androstenone levels in pigs reveals a cluster of candidate genes on chromosome 6. BMC Genet. 2010; 11: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill EW, McGivney BA, Gu J, Whiston R, Machugh DE. A genome-wide SNP-association study confirms a sequence variant (g.66493737C>T) in the equine myostatin (MSTN) gene as the most powerful predictor of optimum racing distance for Thoroughbred racehorses. BMC Genomics. 2010; 11: 552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnston SE, McEwan JC, Pickering NK, et al. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol Ecol. 2011; 20: 2555–2566 [DOI] [PubMed] [Google Scholar]
- 30. Awano T, Johnson GS, Wade CM, et al. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009; 106: 2794–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abasht B, Lamont SJ. Genome-wide association analysis reveals cryptic alleles as an important factor in heterosis for fatness in chicken F2 population. Anim Genet. 2007; 38: 491–498 [DOI] [PubMed] [Google Scholar]
- 32. Sutter NB, Bustamante CD, Chase K, et al. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007; 316: 1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parker HG, VonHoldt BM, Quignon P, et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science. 2009; 325: 995–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cadieu E, Neff MW, Quignon P, et al. Coat variation in the domestic dog is governed by variants in three genes. Science. 2009; 326: 150–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ostrander EA, Franklin H. Epstein Lecture. Both ends of the leash–the human links to good dogs with bad genes. N Engl J Med. 2012; 367: 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldstein O, Mezey JG, Boyko AR, et al. An ADAM9 mutation in canine cone-rod dystrophy 3 establishes homology with human cone-rod dystrophy 9. Mol Vis. 2010; 16: 1549–1569 [PMC free article] [PubMed] [Google Scholar]
- 37. Yau KW, Baylor DA. Cyclic GMP-activated conductance of retinal photoreceptor cells. Ann Rev Neurosci. 1989; 12: 289–327 [DOI] [PubMed] [Google Scholar]
- 38. Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol. 2011; 2011: 527137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D'Angelo A, Franco B. The dynamic cilium in human disease. Pathogenetics. 2009; 2: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009; 20: 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wiik AC, Wade C, Biagi T, et al. A deletion in nephronophthisis 4 (NPHP4) is associated with recessive cone-rod dystrophy in standard wire-haired dachshund. Genome Res. 2008; 18: 1415–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khanna H, Davis EE, Murga-Zamalloa CA, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009; 41: 739–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baala L, Romano S, Khaddour R, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007; 80: 186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cideciyan AV, Rachel RA, Aleman TS, et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet. 2011; 20: 1411–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peterson TA, Adadey A, Santana-Cruz I, Sun Y, Winder A, Kann MG. DMDM: domain mapping of disease mutations. Bioinformatics. 2010; 26: 2458–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.