

Summary
Background
Von Willebrand factor (VWF) binds to subendothelial collagen at sites of vascular injury. Laboratory testing for von Willebrand disease (VWD), however, does not always include collagen binding assays (VWF:CB) and standard VWF:CB assays use type I and/or type III collagen rather than type VI collagen.
Objectives
We report here on several mutations that exclusively alter binding to type VI collagen.
Patients/methods
Healthy controls and index cases from the Zimmerman Program for the Molecular and Clinical Biology of VWD were analyzed for VWF antigen (VWF:Ag), VWF ristocetin cofactor activity, and VWF:CB with types I, III, and VI collagen. VWF gene sequencing was performed for all subjects.
Results
Two healthy controls and one type 1 VWD subject were heterozygous for an A1 domain sequence variation, R1399H, and displayed a selective decreased binding to type VI collagen but not types I and III. Expression of recombinant 1399H VWF resulted in absent binding to type VI collagen. Two other VWF A1 domain mutations, S1387I and Q1402P, displayed diminished binding to type VI collagen. An 11 amino acid deletion in the A1 domain also abrogated binding to type VI collagen.
Conclusions
VWF:CB may be useful in diagnosis of VWD, as a decreased VWF:CB/VWF:Ag ratio may reflect specific loss of collagen binding ability. Mutations that exclusively affect type VI collagen binding may be associated with bleeding, yet missed by current VWF testing.
Keywords: von Willebrand factor, von Willebrand disease, type VI collagen, gene mutation
Introduction
Von Willebrand factor (VWF) plays a crucial role in maintaining vascular integrity by binding to collagen in exposed subendothelium and helping to recruit platelets to sites of injury. Defects in the quantity or quality of von Willebrand factor lead to von Willebrand disease (VWD), a condition associated in most cases with mild to moderate bleeding. Quantitative defects are classified as type 1 (mild-moderate deficiency) and type 3 (severe deficiency). Type 2 VWD includes qualitative defects, a category that encompasses both gain and loss of function in terms of platelet binding as well as defects in multimerization, collagen binding, or factor VIII binding [1]. Type 2M VWD includes those mutations that do not affect multimer distribution but alter VWF-platelet interactions; this category also currently includes collagen binding defects.
Although collagen binding is a critical part of VWF function, clinical testing for VWD does not usually include assays to quantitate the interaction between VWF and collagen. When collagen binding assays are performed, they typically utilize type I, type III, or a combination of types I and III collagen [2]. In clinical practice, diagnosis of von Willebrand disease is often excluded if the routine VWF testing is normal, potentially leading to costly workups for rare bleeding disorders, or no diagnosis for patients with a history of bleeding symptoms.
Type VI collagen is present in the vascular subendothelium and co-localizes with VWF [3,4]. This relationship appears to occur in the superficial subendothelium, provoking early VWF-dependent adhesion of platelets under low shear conditions [5]. VWF binds to type VI collagen via the A1 domain, whereas it binds to types I and III collagen via the A3 and A1 domains [6,7]. The clinical importance of type VI collagen binding to VWF in promoting hemostasis is unknown, and no mutations have been reported previously to affect VWF-collagen type VI interactions.
In the current study, investigation of type VI collagen binding was performed for subjects enrolled in a large study of VWD. Defects in type VI collagen binding were observed for subjects with type 1 and type 2M VWD as well as in several healthy controls. To the best of our knowledge, this is the first report of specific defects in VWF binding to type VI collagen.
Methods
Subject recruitment
Subjects were recruited into the Zimmerman Program for the Molecular and Clinical Biology of von Willebrand Disease as previously described [8]. VWD subjects were eligible if they had a pre-existing diagnosis of VWD of any type. Healthy controls were required to have no history of bleeding or diagnosed VWD and were recruited from the local population surrounding 7 of the 8 Primary Clinical Centers. Each institution participating in the Zimmerman Program had the study approved by their Institutional Review Board. Informed consent was obtained from participants [8].
Data on bleeding symptoms was obtained through a bleeding questionnaire, which included questions from the European Molecular and Clinical Markers for the Diagnosis and Management of Type 1 VWD (MCMDM-1 VWD) study and enabled a bleeding score to be calculated [9]. Blood was collected in 3.2% sodium citrate for coagulation testing. An additional sample was obtained for DNA extraction.
Clinical laboratory VWF assays
Von Willebrand factor antigen (VWF:Ag), VWF ristocetin cofactor activity (VWF:RCo), VWF collagen binding with type III collagen (VWF:CB), and VWF multimers were performed by the BloodCenter of Wisconsin Hemostasis Reference Laboratory on all samples as previously described [8]. Platelet aggregation studies were performed on one subject reported below in the clinical laboratory of the University of Iowa using citrated platelet-rich plasma adjusted to a platelet count of 300,000. Platelet aggregation was measured using the Chronolog platelet aggregometer (Havertown, PA) with collagen type I (Chronolog) at a concentration of 1, 2 and 5 μg/mL as the agonist.
Collagen binding assays
Binding to collagen type III was tested by the BloodCenter of Wisconsin clinical hemostasis reference laboratory using type III human placental collagen (Southern Biotech, Birmingham, AL) plated at a concentration of 1 μg/mL. Collagen type I binding was tested using type I human placental collagen (Sigma, St. Louis, MO) plated at 5 μg/mL. A polyclonal anti-VWF antibody (Dako) was used for detection in both ELISA assays. Type VI collagen binding was tested using a kit obtained from Technoclone (Technozyme VWF:CBA ELISA for type VI collagen, Technoclone, Austria, distributed in the United States by Diapharma, West Chester, OH). Results were normalized to an international plasma standard (SSC/ISTH standard lot #3). As no standard value was available for type VI VWF:CB, the plasma standard was arbitrarily assigned a value of 100 U/dL. Measurement of the coefficient of variation (CV) for the type VI VWF:CB assay showed intra- and inter-assay CVs of 5% (at low and low-normal VWF levels) up to 10% at higher VWF levels.
DNA and RNA preparation and sequencing
DNA was purified from EDTA-anticoagulated whole blood. All healthy controls and type 1 VWD index cases had PCR amplification and semi-automated bidirectional sequencing performed for the entire coding sequence of VWF, including intron-exon boundaries, as previously described [8]. Primer sequences are available on request. For family linkage studies, several regions of the VWF gene were analyzed for variable number of tandem repeats (VNTR), including the promoter and intron 40 [10,11].
Platelet RNA was prepared from a frozen platelet pellet. First, whole blood was collected in EDTA and spun at 650 × g for 5 minutes at room temperature (RT). Prostaglandin E1 (Sigma) at a final concentration of 50 ng/mL was added to the platelet-rich plasma. The resulting solution was spun again at 16,000 × g for 10 minutes at RT, the pellet resuspended in Phillips buffer (96.5 mM NaCl, 85.7 mM glucose, 1.1 mM EDTA, 8.5 mM Tris pH 7.4) with prostaglandin E1 at a final concentration of 50 ng/mL, and spun again at 16,000 × g for 10 minutes at RT. The platelet pellet was frozen for later RNA extraction. RNA was extracted from the platelet pellet using the Qiagen RNeasy Mini kit (Qiagen, Valencia, CA) and cDNA transcribed using the Superscript III kit (Invitrogen, Carlsbad, CA) and primers specific to exons 18 and 28 of the VWF gene. DNA sequencing was then performed to evaluate for expression of one or both alleles.
Recombinant VWF constructs
Recombinant VWF containing the sequence variations of interest was synthesized using the Stratagene QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Full length wild-type or mutant VWF in the pCIneo expression vector was expressed in HEK293T cells as previously described [12] and supernatants collected at 72 hours. An expression vector lacking the VWF construct was also used as a negative control. To mimic the heterozygous state, a mixture of 50% wild-type and 50% mutant DNA was also transfected. Expression studies were also done using all wild-type or all mutant DNA as noted in the text.
Results
232 healthy control subjects enrolled in the Zimmerman Program were analyzed for binding to type VI collagen. The mean VWF:CB with type VI collagen was 135 U/dL with a calculated normal range (± 2 SD) of 55-280. The mean VWF:CB/VWF:Ag ratio was 1.21, with a calculated normal range of 0.76-1.65. 263 subjects were enrolled in the Zimmerman Program with a diagnosis of type 1 VWD. For the 154 subjects who had a VWF:Ag <60 on their enrollment sample, the mean VWF:CB with type VI collagen was 41 U/dL. The mean VWF:CB/VWF:Ag ratio was 1.09 with an observed range of 0-3.25. While most subjects had comparable ratios of VWF:CB/VWF:Ag for all the collagen subtypes analyzed, several subjects were remarkable for a selective decrease in binding to type VI collagen. A total of 4 subjects, 2 healthy controls and 2 type 1 VWD subjects, had the same VWF A1 domain sequence variation, R1399H.
R1399H VWF leads to selective decreased VWF:CB with type VI collagen
Two unrelated families enrolled in the Zimmerman Program with type 1 VWD had the same R1399H sequence variation (table 1). In family 1 (figure 1A), the proband and the proband’s mother had mild decrease in VWF levels and elevated bleeding scores of 6 and 15 as calculated according to the European Union scoring system, where a score of >3 is abnormal [13]. The mother displayed decreased binding to type VI collagen compared to the normal range seen in both healthy controls and other type 1 VWD subjects, with a VWF:CB/VWF:Ag ratio for type VI collagen of 0.61. No other sequence variation was found in the VWF coding sequence. The father had normal VWF sequence and elevated type VI collagen binding, with a ratio of 2.13. The significance of the elevated ratio is currently unclear; no mutation has been found in this subject’s VWF coding region to date, and no other family members are available to determine linkage of that phenotype.
Table 1.
Zimmerman Program Subjects with Decreased Type VI Collagen Binding
| VWF:Ag | VWF.RCo | VWF. CB (VI) | CB/Ag Ratio | BS | Mutation | |
|---|---|---|---|---|---|---|
| Family #1 | ||||||
| Proband | 60 | 56 | 51 | 0.85 | 6 | R1399H |
| Mother | 46 | 47 | 28 | 0.61 | 15 | R1399H |
| Father | 92 | 91 | 196 | 2.13 | 0 | WT |
| Family #2 | ||||||
| Proband | 54 | 53 | 0 | 0 | 8 | R1399H, c.2435delC |
| Father | 33 | 33 | 35 | 1.06 | −1 | c.2435delC |
| Mother | 114 | 106 | 96 | 0.84 | 0 | WT |
| PGF | 139 | 132 | 162 | 1.17 | −1 | WT |
| PGM | 38 | 32 | 43 | 1.13 | 8 | c.2435delC |
| Family #3 | ||||||
| Proband | 64 | 26 | 0 | 0 | 10 | Del 1392-1402 |
| Brother | 47 | 11 | 3.9 | 0.08 | 2 | Del 1392-1402 |
| Brother | 42 | <10 | 0 | 0 | −1 | Del 1392-1402 |
| Family #4 | ||||||
| Proband | 25 | 11 | 14 | 0.56 | 5 | S1387I |
| Sibling | 24 | 10 | 19 | 0.79 | 2 | S1387I |
| Mother | 25 | 11 | 23 | 0.92 | −1 | S1387I |
| Family #5 | ||||||
| Proband | 31 | 14 | 28 | 0.90 | 3 | Q1402P |
| Daughter | 30 | 18 | 22 | 0.73 | 2 | Q1402P |
| Son | 86 | 76 | 91 | 1.06 | 0 | WT |
| Mother | 37 | 15 | 21 | 0.57 | 6 | Q1402P |
VWF:Ag = VWF antigen, VWF:RCo = VWF ristocetin cofactor activity, VWF:CB (VI) = VWF collagen binding with type VI collagen, CB/Ag ratio = ratio of VWF:CB (VI) to VWF:Ag, BS = bleeding score, PGF = paternal grandfather, PGM = paternal grandmother, WT = wild-type.
Figure 1. Family pedigrees for type VI collagen binding mutations.
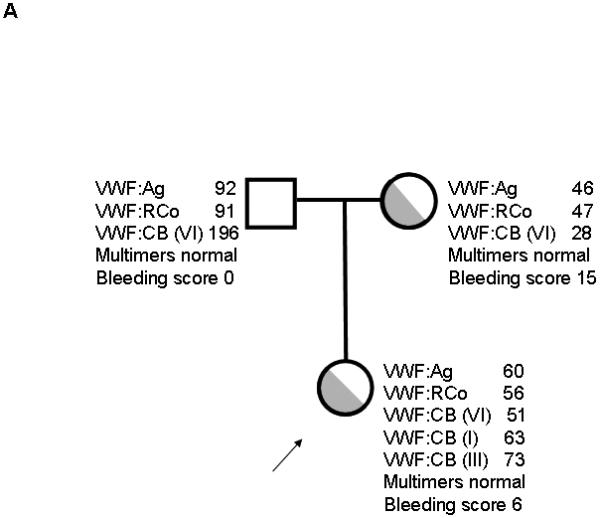
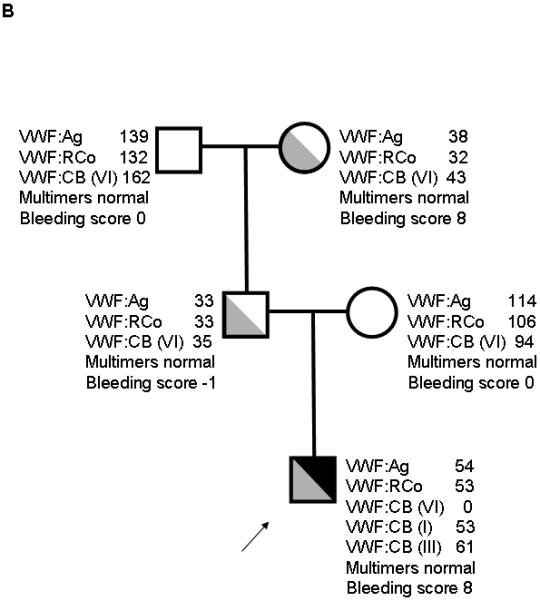
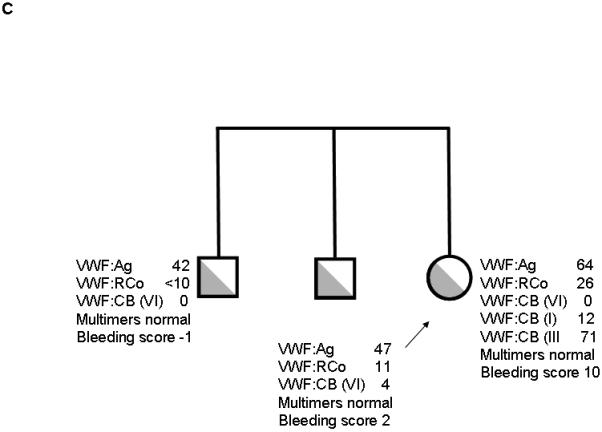

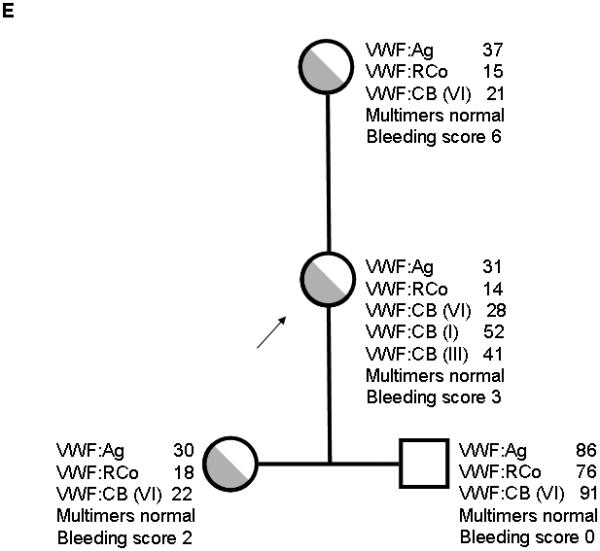
Figure 1A shows family 1, with heterozygous R1399H in the proband and mother. Figure 1B shows family 2, with heterozygous R1399H in the index case and heterozygous exon 18 deletion c.2435delC in the proband, father, and paternal grandmother. Figure 1C shows family 3 with deletion of amino acids 1392-1402 in the proband and siblings. Figure 1D shows family 4 with S1387I in the proband, sibling, and mother. Figure 1E shows family 5 with Q1402P in the proband, daughter, and mother. Arrows denote the proband in each family. VWF:Ag = VWF antigen, VWF:RCo = VWF ristocetin cofactor activity, VWF:CB (VI) = VWF collagen binding with type VI collagen, VWF:CB (I) = VWF collagen binding with type I collagen, VWF:CB (III) = VWF collagen binding with type III collagen.
In family 2 (figure 1B), the proband had VWF:Ag of 36 and VWF:RCo of 34 at the time of original diagnosis at age 2. Bleeding score was 8, as calculated according to the European Union scoring system [9]. VWF:CB with type VI collagen was undetectable despite normal ratios of VWF:CB to VWF:Ag when types I and III collagen were used. Platelet aggregation was normal in response to 3 different doses of collagen (1, 2, and 5 μg/mL). This subject was also heterozygous for R1399H. All other family members had normal VWF:CB with type VI collagen, and none shared the R1399H sequence variation. VNTR analysis confirmed presence of both a maternal and paternal allele. These results suggest that the R1399H is either a novel mutation in the proband or that the mother is a potential germline mosaic.
The proband is a compound heterozygote, for 1399H and a second mutation. Full-length sequencing revealed an exon 18 deletion, c.2435delC that results in premature truncation of VWF synthesis. The subject’s father and grandmother also had laboratory findings suggestive of mild deficiency of VWF, as shown in the family pedigree, and the c.2435delC deletion (figure 1B). This has been previously reported in type 1 VWD, and also in type 3 VWD either as homozygous or heterozygous in conjunction with another known or presumed defect [14-17]. This region contains a string of 6 cytosines in a row, creating a hotspot for deletion mutants [14,15]. Three subjects with type 3 VWD enrolled in the Zimmerman Program had the c.2435delC mutation and an allele with either another deletion or a premature stop codon. Four other subjects with type 1 VWD had the c.2435delC mutation with low VWF:Ag and VWF:RCo, but VWF:CB with types I, III, and VI collagen in those subjects was comparable to the VWF:Ag.
Given the presence of the c.2435delC mutation on one allele, and the fact that this mutation has been seen in type 3 VWD, we suspected that the absent type VI collagen binding was due to lack of expression of one allele (the paternal allele with c.2435delC) and expression only of the other allele containing 1399H. To validate this hypothesis, platelet RNA was purified and sequenced from the type 1 VWD proband described above. The platelet RNA results showed amplification of only one allele with the 1399H sequence (supplemental figure 1). In addition, expression of recombinant c.2435delC VWF resulted in no detectable VWF or VWF propeptide in cell culture supernatant (figure 2). No protein is expressed, due to the fact that no RNA is expressed from the c.2435delC allele.
Figure 2. Comparison of VWF antigen and propeptide for c.2435delC recombinant VWF.
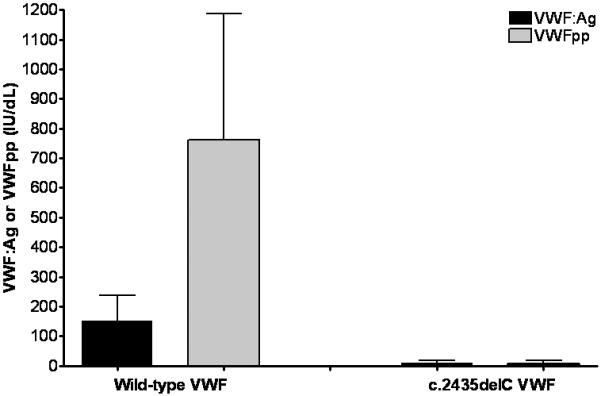
VWF:Ag and VWF propeptide (VWFpp) were measured for recombinant full length VWF and a construct containing the c.2435delC mutation. VWF:Ag is shown in black and VWFpp is shown in gray, both in IU/dL. Data represent a minimum of 3 separate transfections, and are given as the mean + 1 SD. VWFpp levels are higher than VWF:Ag due to the use of a plasma standard to determine concentration. Although VWFpp and VWF:Ag are secreted in an equimolar ratio, VWFpp half-life in plasma is much shorter [26]. This graph demonstrates lack of synthesis of either the propeptide or full-length VWF for the c.2435delC construct.
Two healthy control subjects were noted to have VWF:CB/VWF:Ag ratios with type VI collagen below the normal range, with ratios of 0.49 and 0.61. Both of these subjects had bleeding scores of −1 using the European Union scoring system [9], and normal binding to types I and III collagen. Both were heterozygous for the R1399H sequence variation. However, no further information is available with regard to minor bleeding symptoms other than the bleeding score obtained at study enrollment.
Recombinant 1399H VWF does not bind type VI collagen
To further explore the significance of R1399H VWF, we expressed 1399H using a full length recombinant VWF construct in HEK293T cells. The 1399H construct expressed normally compared to wild-type VWD, but demonstrated undetectable binding to type VI collagen when only homozygous 1399H VWF was present (figure 3). This suggests that the 1399H sequence variation affects the ability of VWF to bind type VI collagen. The location of this amino acid is on one face of the known A1 domain crystal structure (figure 4, supplemental video 1). Co-transfection of 1399R and 1399H DNA resulted in normal expression (as expected). Binding to type VI collagen was approximately 50% of that seen with only the wild-type 1399R construct (figure 3). These results suggests that in the heterozygous state, the R1399H sequence variation should result in about 50% type VI collagen binding, similar to that seen with the heterozygous normal controls, who had type VI VWF:CB/VWF:Ag results of 0.49 and 0.61 respectively. There was no evidence of a dominant negative phenotype in these studies.
Figure 3. Comparison of 1399R and 1399H rVWF collagen binding.
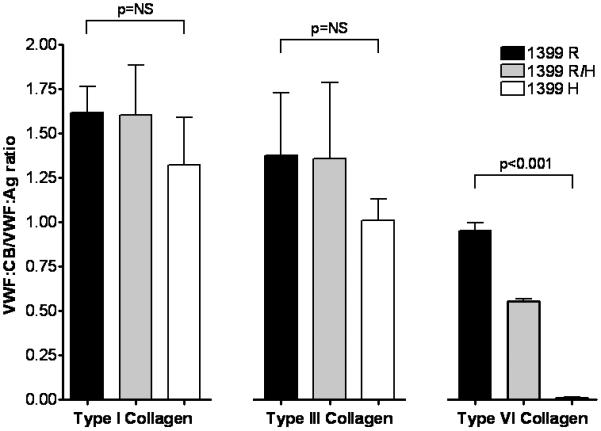
Recombinant full-length VWF was expressed in HEK293T cells using 100% wild-type DNA (1399R, shown in black), 100% variant DNA (1399H, shown in white), or a mixture of 50% of each DNA (1399R/H, shown in gray). Supernatants were analyzed for VWF:Ag and VWF:CB with types I, III, and VI collagen. Graphed here is the ratio of VWF:CB/VWF:Ag for each construct with each type of collagen. Data represent a minimum of 3 separate transfections, and are given as the mean + 1 SD.
Figure 4. Location of type VI collagen binding mutations in the crystal structure of the A1 domain.

Shown here is the VWF A1 domain crystal structure (1AUQ [27]) graphed using Pymol. The orange sphere represents the location of S1387, the blue sphere represents the location of R1399, and the yellow sphere represents the location of Q1402. The red spheres represent the 11 amino acid deletion from 1392 to 1402. The pink spheres represent the area between 1387 and 1392, which may also be a part of the type VI collagen binding region. The small grey spheres represent the cysteines forming the disulfide bond (1272-1458).
Abnormal type VI VWF:CB in Zimmerman Program type 2M VWD subjects
Three additional families with type 2M VWD and documented decreased VWF:RCo/VWF:Ag ratios were also noted to have decreased binding to type VI collagen. All subjects had mutations in the VWF A1 domain (table 1). The first subject (family 3, figure 1C) had absent binding to type VI collagen and an 11 amino acid deletion that has been previously described [12]. The other subjects were heterozygous for novel point mutations, S1387I (family 4, figure 1D) and Q1402P (family 5, figure 1E). The latter is one of the amino acids encompassed in the 11 amino acid deletion present in family 3 [12]. Collagen binding results with type VI collagen were decreased in most family members with these mutations, but not all (table 1). Bleeding scores, however, were higher in those subjects with decreased VWF:CB/VWF:Ag ratios.
Recombinant VWF was synthesized containing the 1387I and 1402P sequence variations. Both point mutations had a significant decrease in binding to type VI collagen (figure 5). Multimer distribution was normal (data not shown), supporting a specific defect in collagen type VI interactions as the cause of the reduced collagen binding. These mutations all occur on a single face of the A1 domain (figure 4, supplemental video 1) that likely represents the site of interaction with type VI collagen.
Figure 5. Type VI collagen binding for recombinant VWF A1 domain mutations.
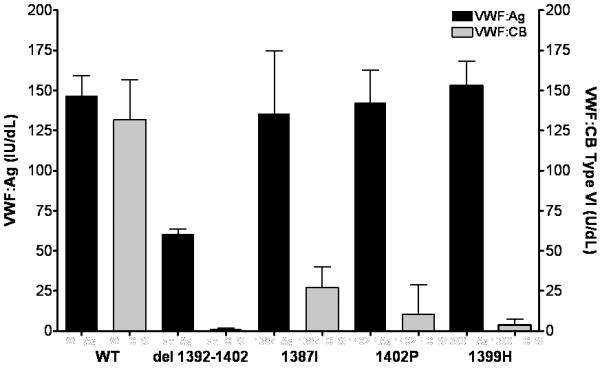
VWF:Ag and VWF:CB with type VI collagen were measured for recombinant full length VWF and constructs containing the del 1392-1402, 1387I ,1402P, and 1399H mutations. VWF:Ag (in IU/dL) is shown in black, graphed on the left y axis, and VWF:CB (VI) (in U/dL) is shown in gray, graphed on the right y axis. Data represent a minimum of 3 separate transfections, and are given as the mean + 1 SD.
Discussion
We report here on sequence variations in the VWF A1 domain that alter VWF binding to type VI collagen, which were associated with varying degrees of bleeding phenotypes in 5 families. These families were identified as part of the Zimmerman Program, which provides an opportunity to correlate genotype and phenotype in VWD. Traditional VWD screening would miss these defects in type VI collagen binding, which may be linked to pathologic bleeding.
In 3 of the 5 families, bleeding is clearly linked to the sequence variation, including both families with R1399H and the family with Q1402P. In families 3 and 4, the proband had significant bleeding while other affected family members did not. This may be due to the presence of other factors ameliorating the VWD phenotype, such as a concurrent procoagulant disorder. Most of the subjects with normal bleeding scores were male, removing the possibility of points acquired due to menorrhagia. In addition, the bleeding score is not necessarily representative of bleeding, particularly in young children. There was a trend toward higher bleeding scores in those subjects with a lower VWF:CB/VWF:Ag ratio for type VI collagen.
The R1399H sequence variation has previously been considered as a polymorphism, present in 0.015% to 2% of the North American population studied [18,19]. However, as shown by plasma and recombinant VWF studies here, 1399H VWF does result in markedly decreased binding to type VI collagen. Binding to types I and III collagen is preserved, emphasizing the importance of the A1 domain in type VI collagen interactions. Although the proband in family 2 had a second mutation leading to lack of expression of one allele and functional hemizygosity for 1399H, the proband in family 1 had no other sequence variations noted in the VWF coding sequence. The elevated bleeding scores in family 1 suggest a possible association with decreased VWF function. The two healthy controls with R1399H had normal bleeding scores. Further work is needed to better characterize the in vivo effect of R1399H on hemostasis.
The laboratory data supports the patient data, in that expression of the mutations in recombinant VWF also demonstrates a significant decrease in binding to type VI collagen for S1387I, R1399H, Q1402P, and the 11 amino acid deletion 1392-1402. Interpretation of these results in VWD patients is limited, however, because most of these subjects had low VWF antigens. None had levels below 30 IU/dL, the level cited in the National Heart, Lung, and Blood Institute guidelines as indicative of true VWD [20]. Although it is difficult to attribute all the symptoms to the collagen binding mutation, it may be that collagen mutations explain some of the bleeding seen in the cohort of patients with low VWF.
Mutations in the A3 domain that result in defective collagen binding to types I and III collagen are well described [21-23]. One of these, the H1786D mutation, abrogates binding to types I and III collagen but has no effect on binding to type VI collagen [23]. Two additional mutations, W1745C and S1783A, have been recently described by Riddell and colleagues, also in the A3 domain, with a profound effect on VWF:CB (with types I and III collagen) as compared to VWF:RCo [22]. None of these variants affected type VI collagen binding (data not shown). Type VI collagen is located in the subendothelium and demonstrates VWF-dependent platelet binding under conditions of low shear [5]. This interaction of VWF and type VI collagen then enables platelets to be recruited to sites of injury without stimulating vessel occlusion in the case of superficial injuries [24]. No defects in VWF have previously been reported to affect this interaction and contribute to VWD. Specific screening for type VI collagen binding defects may ultimately be required to identify affected patients.
The prevalence of mutations affecting VWF:CB is not known, given that collagen binding assays are not routinely performed in workup for the diagnosis VWD, or in workup of patients with bleeding but relatively preserved VWF:Ag and VWF:RCo. In the Zimmerman Program, two out of 154 type 1 VWD subjects were found to have collagen binding mutations, for a prevalence of 1.9%, including a previously described H1786D variant [23]. The R1399H sequence variation was present in 0.9% of the healthy control subjects and 1.3% of the type 1 VWD subjects. Collagen binding mutations were increased in those subjects enrolled as type 2M, with a prevalence of 30% in Zimmerman Program subjects. In the current diagnostic scheme, collagen binding mutations are classified as type 2M VWD, but may merit their own subgroup to better demonstrate their different pathology.
Accurate diagnosis of VWD and the causative defect in VWF has implications for treatment. Desmopressin is frequently administered for treatment of bleeding in patients with type 1 VWD, but may not be adequate for achieving hemostasis in patients with type 2 VWD [25]. In the case of mutations in collagen binding, increased release of VWF may occur, but may not be useful in achieving hemostasis if this merely results in increased release of defective VWF.
Some subjects with decreased VWF:CB have normal VWF:Ag and VWF:RCo and therefore would not be detected with initial screening tests. Without testing VWF:CB, isolated collagen binding defects that do not affect VWF expression will go undiagnosed. The prevalence of such mutations is not clear due to the fact that most patients with bleeding symptoms who have normal VWF:Ag and VWF:RCo will either receive no diagnosis or proceed to costly workups of platelet function and rare bleeding disorders depending on the extent of their symptoms. If the R1399H variant is present in 2% of Caucasians, then 1 in 10,000 (1/50 * 1/50 * 1/4) individuals could be homozygous for 1399H. Additional cases could be present in those heterozygous for R1399H with a null allele similar to the patient presented here. Defects in type VI collagen binding would not be identified by current VWD screening but could conceivably explain bleeding symptoms in patients undergoing workup for hemorrhagic disorders. Further studies are required, however, to determine the importance of type VI collagen in routine clinical laboratory testing.
Supplementary Material
Supplemental Figure 1: VWF exon 28 sequencing for R1399H proband. Genomic DNA and platelet RNA were purified from the proband with both c.2435delC and R1399H as described in the text. Exon 28 sequencing results are shown for the genomic DNA preparation in the top panel and the platelet RNA preparation in the bottom panel. The star denotes the nucleotide change for R1399H.
Supplemental Video 1. Video of the VWF A1 domain demonstrating location of type VI collagen binding mutations. This movie shows the VWF A1 domain crystal structure (1AUQ [27]) graphed using Pymol. The orange sphere represents the location of S1387, the turquoise sphere represents the location of R1399, and the pink sphere represents the location of Q1402. The blue strand represents the 11 amino acid deletion from 1392 to 1402. The small grey spheres represent the cysteines forming the disulfide bond (1272-1458).
Acknowledgements
The authors would like to acknowledge the Zimmerman Program investigators, staff, and subjects who participated in this study.
This work was supported by the National Institutes of Health K08 grant HL102260 (VF) and program project grant HL081588 (RRM). Support for VHF was also provided by a Mentored Research Award from the Hemophilia and Thrombosis Research Society and a Career Development Award from the National Hemophilia Foundation. Support for RRM was also provided through National Institutes of Health grants HL33721 and HL044612. This work was also supported in part by the Midwest Athletes Against Childhood Cancer and the Clinical & Translational Science Institute of Southeastern Wisconsin: NIH UL1RR031973.
Footnotes
Conflict of interest disclosures
JCG is a consultant for Archemix, Baxter, Bayer, and CSL Behring. SRL is a consultant for Novo Nordisk. RRM is a consultant for GTI Diagnostics, Baxter, Bayer, CSL Behring, and AstraZeneca. None of the other authors have conflicts of interest to disclose.
References
- [1].Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, Federici AB. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- [2].Favaloro EJ. An update on the von Willebrand factor collagen binding assay: 21 years of age and beyond adolescence but not yet a mature adult. Semin Thromb Hemost. 2007;33:727–44. doi: 10.1055/s-2007-1000364. [DOI] [PubMed] [Google Scholar]
- [3].Rand JH, Patel ND, Schwartz E, Zhou SL, Potter BJ. 150-kD von Willebrand factor binding protein extracted from human vascular subendothelium is type VI collagen. J Clin Invest. 1991;88:253–9. doi: 10.1172/JCI115285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rand JH, Wu XX, Potter BJ, Uson RR, Gordon RE. Co-localization of von Willebrand factor and type VI collagen in human vascular subendothelium. Am J Pathol. 1993;142:843–50. [PMC free article] [PubMed] [Google Scholar]
- [5].Ross JM, McIntire LV, Moake JL, Rand JH. Platelet adhesion and aggregation on human type VI collagen surfaces under physiological flow conditions. Blood. 1995;85:1826–35. [PubMed] [Google Scholar]
- [6].Hoylaerts MF, Yamamoto H, Nuyts K, Vreys I, Deckmyn H, Vermylen J. von Willebrand factor binds to native collagen VI primarily via its A1 domain. Biochem J. 1997;324:185–91. doi: 10.1042/bj3240185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pareti FI, Niiya K, McPherson JM, Ruggeri ZM. Isolation and characterization of two domains of human von Willebrand factor that interact with fibrillar collagen types I and III. J Biol Chem. 1987;262:13835–41. [PubMed] [Google Scholar]
- [8].Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, Branchford BR, Hoffmann RG, Abshire TC, Di Paola JA, Hoots WK, Leissinger C, Lusher JM, Ragni MV, Shapiro AD, Montgomery RR. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood. 2010;116:280–6. doi: 10.1182/blood-2009-10-249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tosetto A, Rodeghiero F, Castaman G, Goodeve A, Federici AB, Batlle J, Meyer D, Fressinaud E, Mazurier C, Goudemand J, Eikenboom J, Schneppenheim R, Budde U, Ingerslev J, Vorlova Z, Habart D, Holmberg L, Lethagen S, Pasi J, Hill F, Peake I. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD) J Thromb Haemost. 2006;4:766–73. doi: 10.1111/j.1538-7836.2006.01847.x. [DOI] [PubMed] [Google Scholar]
- [10].Peake IR, Bowen D, Bignell P, Liddell MB, Sadler JE, Standen G, Bloom AL. Family studies and prenatal diagnosis in severe von Willebrand disease by polymerase chain reaction amplification of a variable number tandem repeat region of the von Willebrand factor gene. Blood. 1990;76:555–61. [PubMed] [Google Scholar]
- [11].Zhang ZP, Deng LP, Blomback M, Anvret M. Dinucleotide repeat polymorphism in the promoter region of the human von Willebrand factor gene (vWF gene) Hum Mol Genet. 1992;1:780. doi: 10.1093/hmg/1.9.780. [DOI] [PubMed] [Google Scholar]
- [12].Mancuso DJ, Kroner PA, Christopherson PA, Vokac EA, Gill JC, Montgomery RR. Type 2M:Milwaukee-1 von Willebrand disease: an in-frame deletion in the Cys509-Cys695 loop of the von Willebrand factor A1 domain causes deficient binding of von Willebrand factor to platelets. Blood. 1996;88:2559–68. [PubMed] [Google Scholar]
- [13].Tosetto A, Castaman G, Rodeghiero F. Assessing bleeding in von Willebrand disease with bleeding score. Blood Rev. 2007;21:89–97. doi: 10.1016/j.blre.2006.04.002. [DOI] [PubMed] [Google Scholar]
- [14].Zhang ZP, Falk G, Blomback M, Egberg N, Anvret M. A single cytosine deletion in exon 18 of the von Willebrand factor gene is the most common mutation in Swedish vWD type III patients. Hum Mol Genet. 1992;1:767–8. doi: 10.1093/hmg/1.9.767. [DOI] [PubMed] [Google Scholar]
- [15].Schneppenheim R, Krey S, Bergmann F, Bock D, Budde U, Lange M, Linde R, Mittler U, Meili E, Mertes G. Genetic heterogeneity of severe von Willebrand disease type III in the German population. Hum Genet. 1994;94:640–52. doi: 10.1007/BF00206958. [DOI] [PubMed] [Google Scholar]
- [16].Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Hashemi Soteh M, Baronciani L, Hallden C, Guilliatt A, Lester W, Peake I. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD) Blood. 2007;109:112–21. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- [17).James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, Brown C, Andrews C, Labelle A, Chirinian Y, O’Brien L, Othman M, Rivard G, Rapson D, Hough C, Lillicrap D. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007;109:145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- [18].Cooney KA, Nichols WC, Bruck ME, Bahou WF, Shapiro AD, Bowie EJ, Gralnick HR, Ginsburg D. The molecular defect in type IIB von Willebrand disease. Identification of four potential missense mutations within the putative GpIb binding domain. J Clin Invest. 1991;87:1227–33. doi: 10.1172/JCI115123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sadler JE, Ginsburg D. A database of polymorphisms in the von Willebrand factor gene and pseudogene. For the Consortium on von Willebrand Factor Mutations and Polymorphisms and the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1993;69:185–91. [PubMed] [Google Scholar]
- [20].Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, Rick ME, Sadler JE, Weinstein M, Yawn BP. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA) Haemophilia. 2008;14:171–232. doi: 10.1111/j.1365-2516.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- [21].Ribba AS, Loisel I, Lavergne JM, Juhan-Vague I, Obert B, Cherel G, Meyer D, Girma JP. Ser968Thr mutation within the A3 domain of von Willebrand factor (VWF) in two related patients leads to a defective binding of VWF to collagen. Thromb Haemost. 2001;86:848–54. [PubMed] [Google Scholar]
- [22].Riddell AF, Gomez K, Millar CM, Mellars G, Gill S, Brown SA, Sutherland M, Laffan MA, McKinnon TA. Characterization of W1745C and S1783A: 2 novel mutations causing defective collagen binding in the A3 domain of von Willebrand factor. Blood. 2009;114:3489–96. doi: 10.1182/blood-2008-10-184317. [DOI] [PubMed] [Google Scholar]
- [23].Flood VH, Lederman CA, Wren JS, Christopherson PA, Friedman KD, Hoffmann RG, Montgomery RR. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost. 2010;8:1431–3. doi: 10.1111/j.1538-7836.2010.03869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu XX, Gordon RE, Glanville RW, Kuo HJ, Uson RR, Rand JH. Morphological relationships of von Willebrand factor, type VI collagen, and fibrillin in human vascular subendothelium. Am J Pathol. 1996;149:283–91. [PMC free article] [PubMed] [Google Scholar]
- [25].Mannucci PM. Treatment of von Willebrand’s Disease. N Engl J Med. 2004;351:683–94. doi: 10.1056/NEJMra040403. [DOI] [PubMed] [Google Scholar]
- [26].Haberichter SL, Balistreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, Manco-Johnson MJ, Gill JC, Montgomery RR. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108:3344–51. doi: 10.1182/blood-2006-04-015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Emsley J, Cruz M, Handin R, Liddington R. Crystal structure of the von Willebrand Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J Biol Chem. 1998;273:10396–401. doi: 10.1074/jbc.273.17.10396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: VWF exon 28 sequencing for R1399H proband. Genomic DNA and platelet RNA were purified from the proband with both c.2435delC and R1399H as described in the text. Exon 28 sequencing results are shown for the genomic DNA preparation in the top panel and the platelet RNA preparation in the bottom panel. The star denotes the nucleotide change for R1399H.
Supplemental Video 1. Video of the VWF A1 domain demonstrating location of type VI collagen binding mutations. This movie shows the VWF A1 domain crystal structure (1AUQ [27]) graphed using Pymol. The orange sphere represents the location of S1387, the turquoise sphere represents the location of R1399, and the pink sphere represents the location of Q1402. The blue strand represents the 11 amino acid deletion from 1392 to 1402. The small grey spheres represent the cysteines forming the disulfide bond (1272-1458).