

Abstract
There is accumulating evidence that excitotoxicity and oxidative stress resulting from excessive activation of glutamate (N-methyl-d-aspartate) NMDA receptors are major participants in striatal degeneration associated with 3-nitropropionic acid (3NP) administration. Although excitotoxic and oxidative mechanisms are implicated in 3NP toxicity, there are conflicting reports as to whether NMDA receptor antagonists attenuate or exacerbate the 3NP-induced neurodegeneration. In the present study, we investigated the involvement of NMDA receptors in striatal degeneration, protein oxidation and motor impairment following systemic 3NP administration. We examined whether NMDA receptor antagonists, memantine and ifenprodil, influence the neurotoxicity of 3NP. The development of striatal lesion and protein oxidation following 3NP administration is delayed by memantine but not affected by ifenprodil. However, in behavioral experiments, memantine failed to improve and ifenprodil exacerbated the motor deficits associated with 3NP toxicity. Together, these findings suggest caution in the application of NMDA receptor antagonists as a neuroprotective agent in neurodegenerative disorders associated with metabolic impairment.
Keywords: N-Methyl-d-aspartate, 3-Nitropropionic acid, Memantine, Ifenprodil, MK-801, Metabolic impairment, Excitotoxicity
Introduction
3NP, an irreversible inhibitor of succinate dehydrogenase (SDH) of the Krebs cycle and complex II of the electron transport chain, has been utilized to reproduce the cognitive and motor deficits associated with metabolic disorders such as Huntington’s disease (HD) [1-3]. Systemic administration of 3NP has been widely used to generate selective striatal degeneration in primate and rodent models, producing characteristics reminiscent of those seen in HD [4-6].
Although the mechanisms responsible for selective striatal damage are not known, it has been widely accepted that 3NP inhibition of SDH activity is the primary cause of 3NP-induced toxicity. However, the selective striatal degeneration seen in this model is not due to more severe metabolic impairment in the striatum since 3NP inhibits SDH activities uniformly throughout the brain [7-9]. Many reports have strongly implicated secondary excitotoxic mechanisms in 3NP-induced neurotoxicity [10-13]. Inhibition of SDH by 3NP results in the reduction of cellular ATP necessary for the maintenance of ion channels and impairs the ability of neurons to maintain a physiologically relevant resting membrane potential. The loss of membrane potential results in the release of Mg2+ block from the NMDA receptors and facilitates the NMDA receptor activation by the ambient levels of glutamate, leading to massive influx of extracellular Ca2+ into the cytosol and initiating a cascade of events with neuronal death as the final outcome [10-12, 14-17]. Consistent with this hypothesis, the removal of the glutamatergic corticostriatal pathway by decortication protects against striatal damage induced by 3NP [18], and systemic administration of 3NP exacerbates the damage caused by intrastriatal NMDA injection [19, 20]. Others have demonstrated that the secondary excitotoxicity may not be the cause of neurotoxicity in 3NP model [21]. According to this view, the primary effect of 3NP is to compromise Ca2+ sequestration by mitochondria, which ultimately results in calpain proteases activation and cell death. Therefore cell membrane depolarization is not necessarily the trigger for increasing intracellular Ca2+ concentration, but it is the inability of mitochondria to sequester cytosolic Ca2+ and maintain intracellular Ca2+ homeostasis that ultimately result in cellular demise.
Oxidative stress is also believed to be a likely co-factor that may play an essential role in glutamate toxicity since oxidative stress is intimately linked to NMDA receptor activation by coupling to neuronal nitric oxide synthase (nNOS) via the post synaptic density protein 95 (PSD-95) [22]. The calcium influx through the NMDA receptors enables nNOS to mediate the production of nitric oxide [23]. Nitric oxide can rapidly react with the superoxide radicals (O−) to form peroxynitrite, a toxic reactive nitrogen species capable of damaging proteins and DNA [24-26]. Tyrosine residues of proteins are highly susceptible to nitration by peroxynitrite, generating 3-nitrotyrosine which has been widely utilized as a marker for peroxynitrite production and protein oxidation in vivo [24, 25, 27-29]. The nitration of tyrosine residues of proteins is an irreversible process, resulting in alteration of structure and function of proteins [30, 31]. Peroxynitrite can also damage DNA, leading to the activation of poly(ADP-ribose) polymerase-1 (PARP-1), a nuclear enzyme involved in DNA repair [32].
In the present study, we investigated whether the reduction in NMDA receptor activity following treatment with NMDA receptor antagonists, MK-801, memantine and ifenprodil protects the striatal neurons against 3NP-induced neurotoxicity. The results indicate variable affects of NMDA receptor antagonists in the 3NP model, ranging from delayed neurodegeneration in the case of memantine to exacerbation of 3NP toxicity with ifenprodil and MK-801.
Experimental Procedure
3NP and memantine were purchased from Sigma-Aldrich (St. Louis, MO). Ifenprodil was from Tocris Cookson Inc. (Ellisville, MO). Osmotic mini pumps were obtained from Durect Corporation (Cupertino, CA). The anti-nitrotyrosine mouse monoclonal IgG (clone 1A6 Cat# 05-233) was purchased from Upstate biotechnology (Lake Placid, NY). The monoclonal anti-poly (ADP-ribose) antibody against ADP-ribosylated proteins (Cat# SA-250) was obtained from BioMol Research Laboratories (Plymouth, PA). All other reagents were from Sigma-Aldrich (St. Louis, MO).
Animals
Male Sprague-Dawley rats were obtained from Harlan Labs (Indianapolis, IN). Animals were housed individually and maintained on a 12 h dark/light with food and water available ad libitum. They were allowed to acclimate to the experimental environment for a minimum of one week prior to the experimental procedure. All experimental protocols involving animals were approved by the University of Kentucky Institutional Animal Use and Care Committee and are in accordance with the guidelines published in the NIH Guide for the Care and Use of Laboratory Animals and the Society for Neuroscience Guidelines for the Use of Animals in Neuroscience Research.
Drug Administration
To study the involvement of oxidative stress in 3NP toxicity, Sprague-Dawley rats were injected with 3-NP (20 mg/kg/day, i.p.) or saline for 1–5 days. At the end of each treatment day, a group of animals (n = 6 per day per group) were anesthetized with pentobarbital (60 mg/kg), decapitated, and the brain was removed immediately. One hemisphere was incubated in 4% paraformaldehyde for 24 h at 4°C followed by 24 h incubation in 30% sucrose in phosphate buffer solution (pH 7.4) for immunohistochemical analysis, and the other hemisphere was dissected and the striatum, was removed, frozen on dry ice and stored at −80°C for Western blot analysis.
To study the effects of NMDA receptor antagonists following 3NP administration, the osmotic mini pumps were loaded with physiological saline or ifenprodil (5 mg/kg/day) or memantine (5 mg/kg/day) or MK-801 (3 mg/kg/day) at room temperature and then incubated for 6 h in 0.9% saline at 37°C. The pumps were implanted subcutaneously in animals under sodium pentobarbital anesthesia 24 h prior to the first 3NP injection. 3NP (20 mg/kg/day, pH 7.4) was dissolved in physiological saline and injected directly into intraperitoneal cavity. In control animals, 3NP injection was replaced with saline injection.
Western Blotting
The striatum was homogenized in Tris-buffered saline (50 mM Tris–HCl, 150 mM NaCl, pH 7.5) containing protease inhibitor (l mM leupeptin, 25 mM EDTA, 1 μM pepstatin A, 200 μM AEBSF) then centrifuged at 14,000 × g for 5 min and supernatant was collected. For ADP-ribosylated proteins, 0.1% SDS, 1% Nonidet P-40 and 20% glycerol were added to the homogenizing buffer. Protein concentrations were determined using the BCA protein Assay, and equal samples were loaded on a gradient SDS-PAGE gel (20 μg/ lane). For 3-nitrotyrosine analysis, 10 μl of nitrotyrosine immunoblotting control (Cat # 05-233, Upstate Biotechnology) was utilized as a positive control, while as a negative control, the primary antibody was excluded in a sister blot. SDS-PAGE was performed according to the method of Laemmli [33] using a mini-gel apparatus (Bio-Rad, Hercules CA). Following SDS-PAGE, polypeptides were transferred electrophoretically onto 0.45 μm nitrocellulose membranes. The membranes were blocked for 60–120 min in 5% fat-free milk in TTBS and incubated with the primary antibody in TTBS overnight at room temperature. The membranes were then incubated with peroxidase-conjugated goat anti-mouse or horse anti-rabbit IgG (Jackson Immunoresearch Laboratories, West Grove, PA) for 60 min at room temperature. The blots were developed in SuperSignal West Pico chemiluminescent substrate (Pierce Chemical, Rockford, IL) for 1 minute and exposed to Kodak T-Max X-ray film.
Locomotor Activity
To monitor the effects of NMDA receptor antagonists on locomotor activities (ambulatory and stereotypical locomotion) following 3NP treatment, we utilized a cage activity-monitoring device. Three days prior to osmotic pump implants, the locomotor activities of animals were assessed to establish a standard base line. Based on the preliminary studies, the animals were divided into three groups of similar levels of locomotor activities. Group one received osmotic pumps containing physiological saline (n = 11), whereas groups two and three received osmotic pumps containing memantine (n = 8) or ifenprodil (n = 7), respectively. One hour after each 3NP injection, animals were placed individually in clear plastic cages with fresh bedding that were lowered into a self standing frame with 16 invisible infrared light beams located one inch apart along the length of the cage. Activities of animals were monitored by collecting the beam status using a Windows-based AccuScan system software (Columbus, OH). The AccuScan system automatically traced the movement of animals and calculated ambulatory (i.e. three consecutive beam breaks), stereotypical (i.e. repeated same-beam breaks) and total activity for 12 h (3 h light/9 h dark) for 5 days. The longer period of monitoring during the dark cycle was conducted since our preliminary data indicated that the rats were the most active in the beginning and the end of dark cycle.
Lesion Analysis
For the fixed tissue, each brain hemisphere was sectioned at 25 μm intervals using a cryostat instrument. Every 4th section was mounted on slides and stained with cresyl violet. The sections were imaged using a digital camera, and the total striatal volume and the lesion volume were measured and the percent spared tissue was calculated for each animal. The lesion area was identified by absent or pale cresyl violet staining. The cell loss in the lesion area was confirmed by microscopic examination.
Statistical Analysis
All values are expressed as the mean values ± the standard error of the mean of n observations. For Western blot quantitative analysis, comparisons among groups were made by two-way ANOVA followed by Fisher’s PLSD t-test. The percent spared striatal tissues in different groups were compared using an unpaired student t-test. The locomotor activity within each experimental group was analyzed by repeated measured ANOVA and among different experimental groups by two-way ANOVA followed by the Fisher PLSD post hoc comparisons. For all statistical tests performed, a probability level of P < 0.05 was considered significant.
Results
3NP Neurotoxicity and NMDA Receptor Antagonists
Preliminary studies demonstrated marked behavioral abnormalities and internal bleeding associated with subcutaneous administration of MK-801 via osmotic mini pumps when infused at the minimally effective dose of 3 mg/kg/day [34]. When combined with 3NP, MK-801 treatment was lethal within 24–48 h. The toxicity of MK-801 prevented proper evaluation of MK-801 in 3NP-induced toxicity and therefore was excluded from all subsequent analyses. In contrast to MK-801, no toxic side effects or behavioral abnormalities were associated with either memantine or ifenprodil administration in the absence of 3NP.
Animals treated with 3NP that were carrying the saline-filled osmotic pumps, first demonstrated striatal degeneration on the third day following 3NP treatment, as revealed by cresyl violet staining. The lesions first appeared on the lateral striatum, spreading to the dorsal/medial striatum by day four and five. Administration of ifenprodil did not attenuate the striatal lesion formation. However, memantine treatment delayed the lesion formation up to 4 days following 3NP treatment. By the end of day 5, the percent spared tissue was statistically similar in all treatment groups (Fig. 1a, b).
Fig. 1.
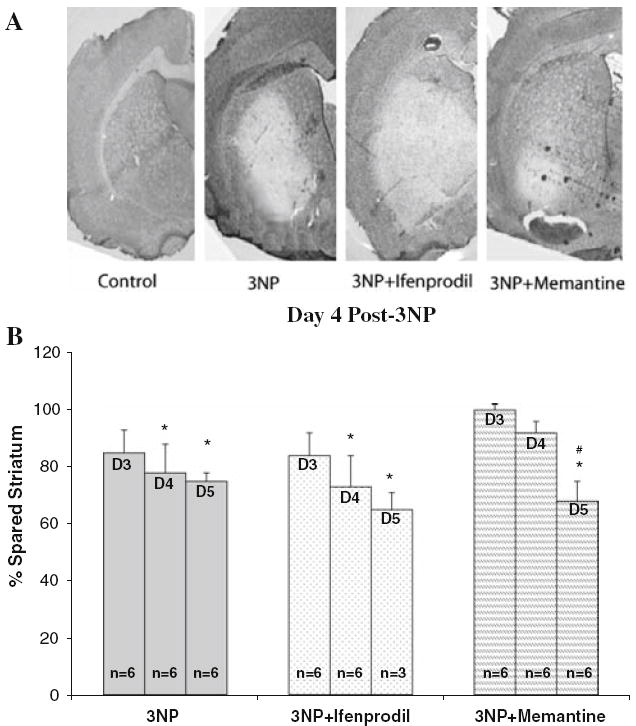
Effects of NMDA antagonists on the percent spared tissue in rats subjected to 3NP treatment (20 mg/kg/day, i.p.). (a) Four days of 3NP injection results in selective striatal degeneration. Ifenprodil does not protect against 3NP toxicity, while memantine delays the formation of striatal lesions; (b) quantitative analysis of the percent spared tissue following systemic 3NP administration for 5 days, and the effect of NMDA antagonists, memantine and ifenprodil, on striatal degeneration indicate a significant decline in percent spared striatal tissue in 3NP plus saline and 3NP plus ifenprodil following 4 days of treatment with 3NP, while the application of memantine delays the lesion formation until the day 5 post-3NP treatment. Percent spared striatum (y-axis) was determined in comparison to the saline-injected group. *P < 0.05 vs. percent saline treated control (no 3NP treatment); #P < 0.05 vs. days 3 and 4 within the same treatment group
Behavioral Activity Monitoring
The mean ambulatory and stereotypical activity scores prior to the start of 3NP administration were 10477 ± 100 and 8098 ± 194, respectively (mean ± SEM). One day following the first injection of 3NP, total locomotor activity declined by 40–55%, thus the data represented in Fig. 2 is based on the activity measurement post-3NP treatment (Fig. 2a, b). There were no significant statistical differences within each treatment group up to 2 days after 3NP treatment. However, 3 days following 3NP administration, the 3NP plus ifenprodil treatment resulted in a significant decline in both ambulatory and stereotypical activity compared to day one post-3NP within the same group. The declines in both ambulatory and stereotypical activities worsened by the end of fourth day, and no animals in the 3NP plus ifenprodil group survived the last day of locomotor analysis. Similar to the 3NP plus saline group, the ambulatory and stereotypical activity of animals in the 3NP plus memantine group gradually declined as compared to day one post-3NP but did not reach significance until the last day of the experiments. The locomotor activity of 3NP plus memantine group was consistently similar to that of 3NP plus saline with no significant difference on any day following 3NP treatment. The results of the total ambulatory and stereotypical activities during both the light (3 h) and the dark cycles (9 h) are illustrated in Fig. 2a and b.
Fig. 2.
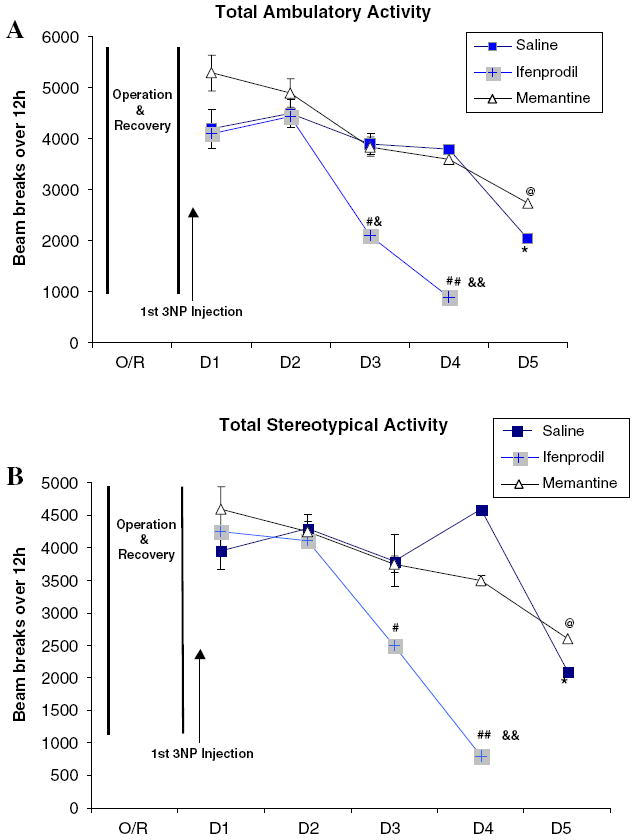
Locomotor activity was monitored using 16 invisible infrared light beams. Ambulatory activity (a) was measured by a minimum of three consecutive beam breaks, while stereotypical locomotion was measured by counting the repeated same-beam breaks (b). Total activity represents total count of beam breaks over 12 h of monitoring (3 h light and 9 h dark). In the behavioral study, the rate of mortality among animals in 3NP plus ifenprodil group was 100% on day 5 post-3NP administrations, and results were not included in the data analysis. Statistical analyses are only performed for the total stereotypical and ambulatory activities (*P < .05 vs. 3NP plus saline treated rats at day 1; @P < .05 vs. 3NP plus memantine treated rats at day one; &P < .05, &&P < .01 vs. 3NP plus ifenprodil treated rats at day one; #P < .05, ##P < .01 vs. rats in 3NP plus saline and 3NP plus memantine (D = Day, O/R = operation and recovery)
3-Nitrotyrosine Levels
Up to 48 h following 3NP administration, no significant elevations in 3-nitrotyrosine levels were detected in any of the treatment groups. Three days following 3NP treatment, there was a mild elevation in 3-nitrotyrosine immunoreactivity in the striatum of 3NP plus saline and 3NP plus ifenprodil groups. However, quantitative Western blot analysis did not indicate a statistical significance in either group compared to saline-treated control animals. By the fourth day of 3NP treatment, the levels of 3-nitrotyrosine in the striatum of 3NP plus saline and 3NP plus ifenprodil treatment groups reached maximum levels and were significantly higher than that of control animals (n = 6 per group, P < 0.05). Memantine treatment delayed the increase in 3-nitrotyrosine levels and did not significantly differ from that of the control animals until day five (n = 6, P < 0.01). On day 5, there was a sharp rise in 3-nitrotyrosine immunoreactivity in 3NP plus memantine group which was significantly higher than values obtained within the same group on day 3 and 4 (P < 0.01 and P < 0.05, respectively) (Fig. 3a, b).
Fig. 3.
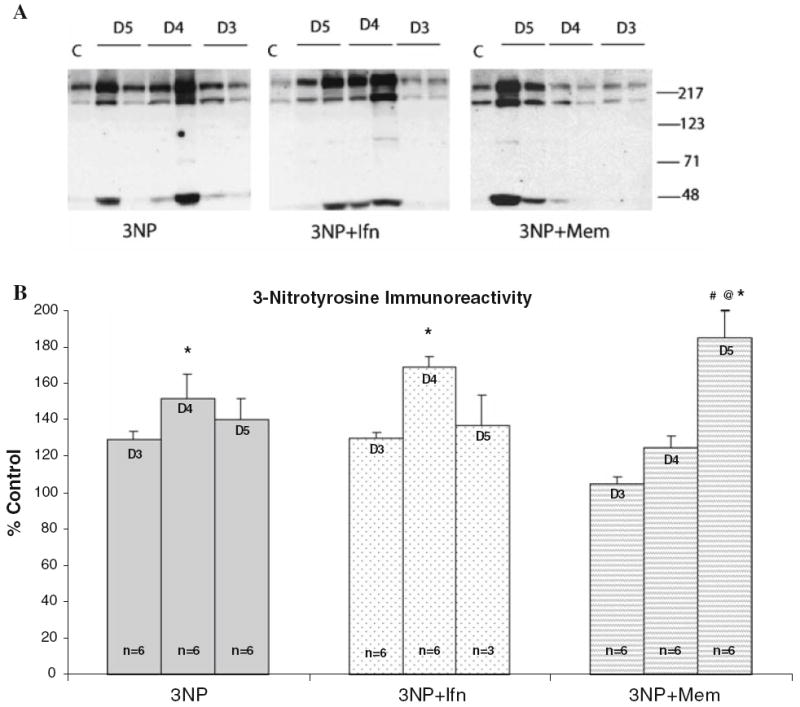
3-Nitrotyrosine levels following 3-NP treatment. (a) Representative Western blots indicate increased levels of 3-nitrotyrosine in the striatum in all treatment groups; (b) quantitative analysis of Western blot results for 3-nitrotyrosine indicates increase in 3-nitrotyrosinated protein levels in all treatment groups; however, rats treated with 3NP plus memantine (Mem) exhibited a delay in increased 3-nitrotyrosine levels compared to 3NP plus saline or 3NP plus ifenprodil (Ifn). *P < .05 vs. saline treated control (no 3NP treatment); @P < .05 vs. day 4 post-3NP within the same treatment group; #P < .01 vs. day 3 post-3NP within the same treatment group
ADP-Ribosylation Levels
To investigate the effect of 3NP in the presence or absence of NMDA receptor antagonists on poly(ADP-ribose) polymerase-1 (PARP-1) activity, we examined the levels of ADP- ribosylated proteins throughout striatum in all treatment groups. Our results indicated that there were no significant differences in the total levels of ADP-ribosylated proteins among the treatment groups (Fig. 4a, b). Furthermore, within each treatment group, there were no significant alterations in the total amount of ADP-ribosylated protein on any day following 3NP treatment.
Fig. 4.
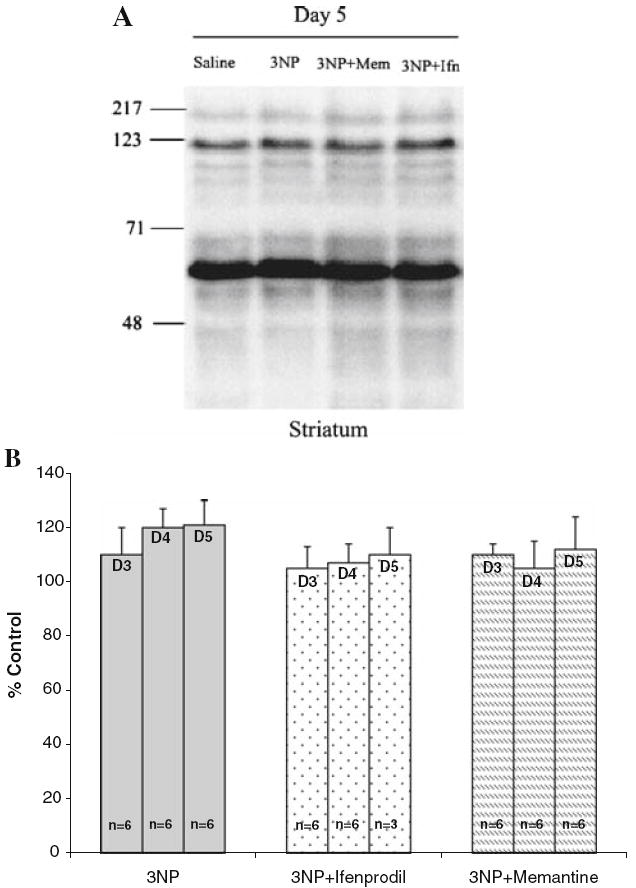
Poly ADP-ribosylation in the striatum following 3NP treatment. (a) Representative Western blot from rats treated with saline or 3NP or 3NP plus ifenprodil or 3NP plus memantine; (b) quantitative analysis of total poly ADP-ribosylated proteins in the striatum of rats treated with 3NP and NMDA antagonists indicate no significant change in the total amount of ADP ribosylated proteins among the treatment groups
Discussion
To elucidate the role of NMDA receptor activity in 3NP toxicity, we have utilized a noncompetitive NMDA receptor antagonist, MK-801, a low affinity NMDA antagonist, memantine, and a selective inhibitor of NMDA receptors containing NR2B subunits, ifenprodil. Since our pilot studies demonstrated severe toxicity associated with the subcutaneous administration of MK-801 (3 mg/kg/day), we excluded MK-801 from any subsequent experimental analysis. This observation agrees with earlier reports demonstrating severe toxic side-effects associated with MK801 in vivo [35].
Ifenprodil interacts with a voltage-dependent polyamine-regulated site on the NR2B subunit of NMDA receptors [36], which are predominantly expressed in the forebrain, including cortex, hippocampus and striatum [37-39]. Since it has been previously demonstrated that ifenprodil at higher concentration also inhibits monoamine oxidase [40], alpha-1-adrenergic receptors [41] and 5-hydroxytryptamine-3 receptors [42], we were compelled to choose a low concentration of ifenprodil in our studies compared to previous reports [43]. However, given that ifenprodil exacerbates the 3NP toxicity, it is unlikely that higher doses would have been beneficial in the 3NP model. Memantine is an uncompetitive low affinity NMDA antagonist with moderate efficacy and voltage-dependent blocking kinetics, which has been used clinically in treatment of Alzheimer’s disease and Parkinson’s disease [44-50]. It is believed that because of its relatively low affinity and fast on-off rate, memantine is released from many blocked channels rapidly, thereby allowing a baseline physiological activity of NMDA receptors while blocking the excessive activity of the receptors [51, 52]. The dose of 5 mg/kg/day was selected for the current study since it has been previously demonstrated that this dosage provides effective concentration of memantine in serum [53].
Of note is an apparent variability in the onset of biochemical, histological and behavioral signs of 3NP toxicity within each group. The variable toxic effects of systemic injection of 3NP on oxidative stress markers, motor impairment and striatal lesion formation have been well documented [4, 18, 54-56]. The cause for this variability remains to be elusive although some strains of rats demonstrate a greater variability to 3NP than others [57, 58].
We have previously demonstrated that the inhibition of NMDA receptors in vitro shifts the cell death mechanisms from necrosis to apoptosis, but does not attenuate the total neuron death in primary hippocampal neurons following 3NP treatment [59, 60]. However, in vivo, there are conflicting reports as to whether NMDA receptor antagonists are protective [56, 61-63] or are not protective [4, 64, 65] in the 3NP-induced toxicity. In the current study, analysis of the percent spared striatal tissue demonstrates that, in rats, the low affinity, uncompetitive NMDA receptor antagonist memantine delays the striatal degeneration and protein oxidation associated with 3NP-induced neurotoxicity. However, in the behavioral experiments, the neuroprotective effects of memantine were not evident. In contrast to memantine, ifenprodil exacerbated the neurotoxicity associated with 3NP treatment. Given the selectivity of ifenprodil for the NR2B subunit, the results suggest that this subtype of NMDA receptor is not primarily involved in the neurodegenerative processes associated with 3NP treatment, and it is likely that other NMDA receptor subunits (i.e. NR1or NR2A) or other glutamate receptors may be involved in 3NP-induced neurotoxicity. Supporting data for this hypothesis has been provided by Stefani and colleagues who have demonstrated a robust increase in intracellular calcium concentration in medium spiny neurons in the striatum through AMPA receptors following glutamate insult [66]. This finding supports the current hypothesis since medium spiny neurons are the most vulnerable neurons in the striatum in 3NP-induced neurotoxicity model [4].
Previous reports have demonstrated that the poly ADP-ribosyation of PARP-1 is essential for DNA repair following oxidative DNA damage [67]. Yet, 3NP, in the presence or absence of NMDA receptor antagonists, did not affect the levels of poly ADP-ribosylated proteins. This finding argues against the involvement of poly(ADP-ribose) polymerase-1 (PARP-1) in the 3NP model. However, PARP-1 is one of the first enzymes degraded during specific apoptotic [32, 68, 69] and necrotic models [70, 71]. Therefore, it is likely that the lack of increased poly ADP-ribosylated proteins in the striatum is a result of PARP-1 inactivation due to its cleavage via the activation of proteases following 3NP treatment [72, 73].
Evidence is accumulating that NMDA receptor activity as well as oxidative stress may significantly contribute to the striatal vulnerability in the metabolic impairment models. Supporting data includes attenuation of striatal lesion volume following removal of the corticostriatal pathway [18], by NMDA receptor antagonists [61], by the application of nNOS inhibitor 7-nitroindazole (7-NI) [74, 75]. However, there is also considerable evidence arguing against an exclusive involvement of NMDA receptors in 3NP-induced neurodegeneration. Contrasting evidence includes lack of neuroprotection by NMDA receptor antagonists in vitro [74] and in vivo [64] as well as alteration in the mode of cell death but not survival [59, 60]. Moreover, in clinical trials, NMDA receptor antagonists have failed to provide a neuroprotective treatment in situations of acute energy failure [76, 77]. In addition, several reports argue that NMDA receptor blockade does not affect the oxidative stress produced by malonate, a reversible inhibitor of SDH [78, 79]. Furthermore, Jacquard and colleagues have recently demonstrated that 3NP neurotoxic effect is primarily due to an improper sequestration of Ca2+ by mitochondria and consequent calpain proteases activation rather than indirect excitotoxicity where mitochondria defects results massive increase in calcium influx through NMDA receptors [21].
The disparity of previous reports and the lack of protection of motor impairment by NMDA receptor antagonists in the current study can be explained by several observations. First, the NMDA receptor activity is closely interconnected with AMPA/KA receptor activity since all three types of ionotropic glutamate receptors can localize on the same neuron [80]. It is possible that in vivo antagonists of NMDA receptors trigger a compensatory elevation and/or release of agonists from presynaptic endings, subsequently overactivating AMPA/KA or metabotropic glutamate receptors and promote neuronal death by allowing excessive Ca2+ entry into neurons [64, 81]. Second, both memantine and ifenprodil inhibition of NMDA receptors occurs, at least partially, in a voltage-dependent manner. Since 3NP treatment results in the failure of ATP dependent ion pumps, membrane depolarization is likely one of the primary outcomes of 3NP toxicity. It is plausible that the efficacy of NMDA receptor antagonists is compromised by changes in the membrane potential due to the lack of ability to produce ATP. However, this hypothesis does not account for exacerbation of motor deficits in 3NP plus ifenprodil treatment group. Finally, if the primary toxic effect of 3NP is due deregulation of Ca2+ sequestration by mitochondria rather than ATP production and cell membrane depolarization [21], NMDA receptor antagonists will only have minimal protective effect and will not be able to protect against 3NP-induced neurotoxicity.
In summary, the co-administration of ifenprodil with 3NP did not protect or delay the striatal degeneration or protein oxidation and exacerbated the motor deficits associated with 3NP toxicity. Conversely, co-administration of memantine with 3NP delayed the formation of striatal lesion and protein oxidation up to 4 days following 3NP treatment; however, there was no significant improvement in locomotors deficit associated with 3NP toxicity. Taken together, these findings suggest that in human conditions associated with acute energy failure, the application of NMDA receptor antagonists as a neuroprotective agent should be assessed carefully. Additional work will be necessary to elucidate the exact role of NMDA receptors in conditions such as those seen in energy impairment diseases.
Contributor Information
Payman Nasr, Department of Biological Sciences, Kent State University, Ashtabula, OH 44004, USA, pnasr@kent.edu.
Timothy Carbery, Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY 40536, USA.
James W. Geddes, Spinal Cord and Brain Injury Research Center, University of Kentucky, Lexington, KY 40536, USA
References
- 1.Beal MF. Neurochemistry and toxin models in Huntington’s disease. Curr Opin Neurol. 1994;7:542–547. doi: 10.1097/00019052-199412000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Palfi S, Ferrante RJ, Brouillet E, et al. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington’s disease. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/S0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins BG, et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brouillet E, Jenkins BG, Hyman BT, et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 6.Borlongan CV, Koutouzis TK, Freeman TB, et al. Hyper-activity and hypoactivity in a rat model of Huntington’s disease: the systemic 3-nitropropionic acid model. Brain Res Brain Res Protoc. 1997;1:253–257. doi: 10.1016/S1385-299X(96)00037-2. [DOI] [PubMed] [Google Scholar]
- 7.Gould DH, Wilson MP, Hamar DW. Brain enzyme and clinical alterations induced in rats and mice by nitroaliphatic toxicants. Toxicol Lett. 1985;27:83–89. doi: 10.1016/0378-4274(85)90123-7. [DOI] [PubMed] [Google Scholar]
- 8.Pang Z, Umberger GH, Geddes JW. Neuronal loss and cytoskeletal disruption following intrahippocampal administration of the metabolic inhibitor malonate: lack of protection by MK-801. J Neurochem. 1996;66:474–484. doi: 10.1046/j.1471-4159.1996.66020474.x. [DOI] [PubMed] [Google Scholar]
- 9.Brouillet E, Guyot MC, Mittoux V, et al. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- 10.Novelli A, Reilly JA, Lysko PG, et al. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 11.Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733–738. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- 12.Beal MF, Hyman BT, Koroshetz W. Do defects in mitochondrial energy metabolism underlie the pathology of neurodegenerative diseases? Trends Neurosci. 1993;16:125–131. doi: 10.1016/0166-2236(93)90117-5. [DOI] [PubMed] [Google Scholar]
- 13.Volbracht C, Van Beek JV, Zhu C, et al. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J NeuroSci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- 14.Nowak L, Bregestovski P, Ascher P, et al. Magnesium gates glutamate-activated channels in mouse central neurons. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 15.Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 16.Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- 17.Olney JW. Excitotoxicity: an overview. Can Dis Wkly Rep. 1990;16(Suppl 1E):47–57. Discussion 57–58. [PubMed] [Google Scholar]
- 18.Beal MF, Brouillet E, Jenkins B, et al. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 19.Simpson JR, Isacson O. Mitochondrial impairment reduces the threshold for in vivo NMDA-mediated neuronal death in the striatum. Exp Neurol. 1993;121:57–64. doi: 10.1006/exnr.1993.1071. [DOI] [PubMed] [Google Scholar]
- 20.Calabresi P, Gubellini P, Picconi B, et al. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001;21:5110–5120. doi: 10.1523/JNEUROSCI.21-14-05110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacquard C, Trioulier Y, Cosker F, et al. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB. 2006;20:245–259. doi: 10.1096/fj.05-5085fje. [DOI] [PubMed] [Google Scholar]
- 22.Christopherson KS, Hillier BJ, Lim WA, et al. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- 23.Sattler R, Xiong Z, Lu WY, et al. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 24.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 25.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 26.Halliwell B. What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett. 1997;411:157–160. doi: 10.1016/S0014-5793(97)00469-9. [DOI] [PubMed] [Google Scholar]
- 27.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 28.Ischiropoulos H, al-Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-U. [DOI] [PubMed] [Google Scholar]
- 29.Beal MF, Ferrante RJ, Browne SE, et al. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 30.Berlett BS, Friguet B, Yim MB, et al. Peroxynitrite-mediated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylylation: relevance to signal transduction. Proc Natl Acad Sci USA. 1996;93:1776–1780. doi: 10.1073/pnas.93.5.1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong SK, Yim MB, Stadtman ER, et al. Peroxynitrite disables the tyrosine phosphorylation regulatory mechanism: Lymphocyte-specific tyrosine kinase fails to phosphorylate nitrated cdc2 (6–20) NH2 peptide. Proc Natl Acad Sci USA. 1996;93:3377–3382. doi: 10.1073/pnas.93.8.3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ha HC, Snyder SH. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis. 2000;7:225–239. doi: 10.1006/nbdi.2000.0324. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Vezzani A, Serafini R, Stasi MA, et al. Kinetics of MK-801 and its effect on quinolinic acid-induced seizures and neurotoxicity in rats. J Pharmacol Exp Ther. 1989;249:278–283. [PubMed] [Google Scholar]
- 35.Lipton SA. Prospects for clinically tolerated NMDA antagonists: open-channel blockers and alternative redox states of nitric oxide. Trends Neurosci. 1993;16:527–532. doi: 10.1016/0166-2236(93)90198-U. [DOI] [PubMed] [Google Scholar]
- 36.Gallagher MJ, Huang H, Grant ER, et al. The NR2B-specific interactions of polyamines and protons with the N-methyl-D-aspartate receptor. J Biol Chem. 1997;272:24971–24979. doi: 10.1074/jbc.272.40.24971. [DOI] [PubMed] [Google Scholar]
- 37.Laurie DJ, Bartke I, Schoepfer R, et al. Regional, developmental and interspecies expression of the four NMDAR2 subunits, examined using monoclonal antibodies. Brain Res Mol Brain Res. 1997;51:23–32. doi: 10.1016/S0169-328X(97)00206-4. [DOI] [PubMed] [Google Scholar]
- 38.Butler TW, Blake JF, Bordner J, et al. (3R, 4S)-3-[4-(4-fluorophenyl)-4-hydroxypiperidin-1-yl]chroman-4, 7-diol: a conformationally restricted analogue of the NR2B subtype-selective NMDA antagonist (1S, 2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)- 1-propanol. J Med Chem. 1998;41:1172–1184. doi: 10.1021/jm9707986. [DOI] [PubMed] [Google Scholar]
- 39.Goebel DJ, Poosch MS. NMDA receptor subunit gene expression in the rat brain: a quantitative analysis of endogenous mRNA levels of NR1Com, NR2A, NR2B, NR2C, NR2D and NR3A. Brain Res Mol Brain Res. 1999;69:164–170. doi: 10.1016/S0169-328X(99)00100-X. [DOI] [PubMed] [Google Scholar]
- 40.Arai Y, Nakazato K, Kinemuchi H, et al. Inhibition of rat brain monoamine oxidase activity by cerebral anti-ischemic agent, ifenprodil. Neuropharmacology. 1991;30:809–812. doi: 10.1016/0028-3908(91)90190-M. [DOI] [PubMed] [Google Scholar]
- 41.Kurihara J, Tamaoki S, Kato H. Blockade of alpha 2-adrenoceptors protects the vagal baroreflex system from transient global cerebral ischemia in dogs. Eur J Pharmacol. 1993;240:73–76. doi: 10.1016/0014-2999(93)90547-U. [DOI] [PubMed] [Google Scholar]
- 42.McCool BA, Lovinger DM. Ifenprodil inhibition of the 5-hydroxytryptamine3 receptor. Neuropharmacology. 1995;34:621–629. doi: 10.1016/0028-3908(95)00030-A. [DOI] [PubMed] [Google Scholar]
- 43.Chenard BL, Menniti FS. Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharm Des. 1999;5:381–404. [PubMed] [Google Scholar]
- 44.Ambrozi L, Danielczyk W. Treatment of impaired cerebral function in psychogeriatric patients with memantine–results of a phase II double-blind study. Pharmacopsychiatry. 1988;21:144–146. doi: 10.1055/s-2007-1014666. [DOI] [PubMed] [Google Scholar]
- 45.Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist–a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/S0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 46.Parsons CG, Danysz W, Quack G. Memantine and the amino-alkyl-cyclohexane MRZ 2/579 are moderate affinity uncompetitive NMDA receptor antagonists–in vitro characterisation. Amino Acids. 2000;19:157–166. doi: 10.1007/s007260070044. [DOI] [PubMed] [Google Scholar]
- 47.Palmer GC. Neuroprotection by NMDA receptor antagonists in a variety of neuropathologies. Curr Drug Targets. 2001;2:241–271. doi: 10.2174/1389450013348335. [DOI] [PubMed] [Google Scholar]
- 48.Finucane TE. Memantine for patients with Alzheimer disease. JAMA. 2004;291:1695. doi: 10.1001/jama.291.14.1695-a. [DOI] [PubMed] [Google Scholar]
- 49.Hirsch CH. Memantine was better than placebo in Alzheimer disease already being treated with donepezil. ACP J Club. 2004;141:38. [PubMed] [Google Scholar]
- 50.Tariot PN, Farlow M, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 51.Blanpied TA, Boeckman FA, Aizenman E, et al. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 52.Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Danysz W, Parsons CG, Kornhuber J, et al. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents–preclinical studies. Neurosci Biobehav Rev. 1997;21:455–468. doi: 10.1016/S0149-7634(96)00037-1. [DOI] [PubMed] [Google Scholar]
- 54.Bossi SR, Simpson JR, Isacson O. Age dependence of striatal neuronal death caused by mitochondrial dysfunction. NeuroReport. 1993;4:73–76. doi: 10.1097/00001756-199301000-00019. [DOI] [PubMed] [Google Scholar]
- 55.Guyot MC, Hantraye P, Dolan R. Quantifiable bradykinesia, gait abnormalities and Huntington’s disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience. 1997;79:45–56. doi: 10.1016/S0306-4522(96)00602-1. [DOI] [PubMed] [Google Scholar]
- 56.Lee ST, Chu K, Park JE, et al. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006;1118:199–207. doi: 10.1016/j.brainres.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 57.Ouary S, Bizat N, Altairac S, et al. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: implication for neuroprotection studies. Neuroscience. 2000;97:521–530. doi: 10.1016/S0306-4522(00)00020-8. [DOI] [PubMed] [Google Scholar]
- 58.Teunissen CE, Markerink-van Ittersum M, de Bruijn C, et al. Evaluation of 3-nitrotyrosine as a marker for 3-nitropropionic acid-induced oxidative stress in Lewis and Wistar rats and strain-specific whole brain spheroid cultures. Brain Res. 2002;931:5–20. doi: 10.1016/S0006-8993(01)03331-5. [DOI] [PubMed] [Google Scholar]
- 59.Nasr P, Gursahani HI, Pang Z, et al. Influence of cytosolic and mitochondrial Ca(2+), ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem Int. 2003;43:89–99. doi: 10.1016/S0197-0186(02)00229-2. [DOI] [PubMed] [Google Scholar]
- 60.Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim GW, Copin JC, Kawase M, et al. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000;20:119–129. doi: 10.1097/00004647-200001000-00016. [DOI] [PubMed] [Google Scholar]
- 62.Karanian DA, Baude AS, Brown QB, et al. 3-Nitropropionic acid toxicity in hippocampus: protection through N-methyl-D-aspartate receptor antagonism. Hippocampus. 2006;16:834–842. doi: 10.1002/hipo.20214. [DOI] [PubMed] [Google Scholar]
- 63.Tozzi A, Costa C, Di Filippo M, et al. Memantine reduces neuronal dysfunction triggered by in vitro ischemia and 3-nitropropionic acid. Exp Neurol. 2007;207:218–226. doi: 10.1016/j.expneurol.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 64.Ikonomidou C, Stefovska V, Turski L. Neuronal death enhanced by N-methyl-D-aspartate antagonists. Proc Natl Acad Sci USA. 2000;97:12885–12890. doi: 10.1073/pnas.220412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frankiewicz T, Parsons CG. Chronic memantine does not block 3-nitropropionic acid-delayed ischaemic tolerance in rat hippocampal slices ex vivo. Neurotox Res. 2004;5:617–622. doi: 10.1007/BF03033181. [DOI] [PubMed] [Google Scholar]
- 66.Stefani A, Chen Q, Hernandez J, et al. Physiological and molecular properties of AMPA/Kainate receptors expressed by striatal medium spiny neurons. Dev Neurosci. 1998;20:242–252. doi: 10.1159/000017318. [DOI] [PubMed] [Google Scholar]
- 67.Le Page F, Schreiber V, Dherin C, et al. Poly(ADP-ribose) polymerase-1(PARP-1) is required in murine cell lines for base excision repair of oxidative DNA damage in absence of DNA polymerase beta. J Biol Chem. 2003;278:18471–18477. doi: 10.1074/jbc.M212905200. [DOI] [PubMed] [Google Scholar]
- 68.Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin, and other cytotoxic anticancer drugs: a cautionary note. Cancer Res. 1989;49:5870–5878. [PubMed] [Google Scholar]
- 69.Lazebnik YA, Kaufmann SH, Desnoyers S, et al. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 70.Shah GM, Shah RG, Poirier GG, et al. Different cleavage pattern for poly(ADP-ribose) polymerase during necrosis and apoptosis in HL-60 cells. Biochem Biophys Res Commun. 1996;229:838–844. doi: 10.1006/bbrc.1996.1889. [DOI] [PubMed] [Google Scholar]
- 71.Gobeil S, Boucher CC, Nadeau D, et al. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): implication of lysosomal proteases. Cell Death Differ. 2001;8:588–594. doi: 10.1038/sj.cdd.4400851. [DOI] [PubMed] [Google Scholar]
- 72.Andreassen OA, Ferrante RJ, Hughes DB, et al. Malonate and 3-nitropropionic acid neurotoxicity are reduced in transgenic mice expressing a caspase-1 dominant-negative mutant. J Neurochem. 2000;75:847–852. doi: 10.1046/j.1471-4159.2000.0750847.x. [DOI] [PubMed] [Google Scholar]
- 73.McCracken E, Dewar D, Hunter AJ. White matter damage following systemic injection of the mitochondrial inhibitor 3-nitropropionic acid in rat. Brain Res. 2001;892:329–335. doi: 10.1016/S0006-8993(00)03266-2. [DOI] [PubMed] [Google Scholar]
- 74.Schulz JB, Matthews RT, Beal MF. Role of nitric oxide in neurodegenerative diseases. Curr Opin Neurol. 1995;8:480–486. doi: 10.1097/00019052-199512000-00016. [DOI] [PubMed] [Google Scholar]
- 75.Schulz JB, Matthews RT, Jenkins BG, et al. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995a;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Behrens MI, Koh J, Canzoniero LM, et al. 3-Nitropropionic acid induces apoptosis in cultured striatal and cortical neurons. NeuroReport. 1995b;6:545–548. doi: 10.1097/00001756-199502000-00034. [DOI] [PubMed] [Google Scholar]
- 77.Muir KW, Lees KR. Clinical experience with excitatory amino acid antagonist drugs. Stroke. 1995;26:503–513. doi: 10.1161/01.str.26.3.503. [DOI] [PubMed] [Google Scholar]
- 78.Davis SM, Albers GW, Diener HC, et al. Termination of acute stroke studies involving selfotel treatment ASSIST steering committed. Lancet. 1997;349:32. doi: 10.1016/S0140-6736(05)62166-6. [DOI] [PubMed] [Google Scholar]
- 79.Ferger B, Eberhardt O, Teismann P, et al. Malonate-induced generation of reactive oxygen species in rat striatum depends on dopamine release but not on NMDA receptor activation. J Neurochem. 1999;73:1329–1332. doi: 10.1046/j.1471-4159.1999.0731329.x. [DOI] [PubMed] [Google Scholar]
- 80.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 81.Bloom FE. In: The pharmacological basis of therapeutics. Goodman Gilman A, Rall TW, Nies AS, Taylor P, editors. Pergamon; New York: 1990. pp. 244–268. [Google Scholar]