

Abstract
Cell movement is driven by the coordinated regulation of cytoskeletal reorganization through Rho GTPases downstream of integrin and growth-factor receptor signaling. We have reported that mDia, a target protein of Rho, interacts with Src and DIP. Here we show that DIP binds to p190RhoGAP and Vav2, and that DIP is phosphorylated by Src and mediates the phosphorylation of p190RhoGAP and Vav2 upon EGF stimulation. When endogenous DIP was inhibited by expressing dominant-negative mutants of DIP or siRNA, phosphorylation of p190RhoGAP and Vav2 upon EGF stimulation was diminished, and EGF-induced actin organization, distribution of p190RhoGAP and Vav2, and cell movement were affected. Therefore, DIP seems to transfer the complex of the three proteins from cytosol to beneath the membrane, and the three proteins, in turn, can be phosphorylated by Src. DIP inactivated Rho and activated Rac following EGF stimulation in the membrane fraction. Thus, DIP acts as a regulatory molecule causing Src kinase-dependent feedback modulation of Rho GTPases downstream of Rho-mDia upon EGF stimulation, and plays an important role in cell motility.
Keywords: DIP, mDia, p190RhoGAP, Rac, Rho, Src, Vav2
Introduction
Cell migration is critical for many processes, including embryogenesis, inflammatory response, wound healing and tumor metastasis (Horwitz and Parsons, 1999), and is believed to be regulated, in part, by integrin- and growth factor-mediated signaling events (Miranti and Brugge, 2002; Yamada and Even-Ram, 2002). However, the mechanisms controlling migration remain incompletely understood. Effective cell migration involves dynamic changes in cell shape that reflect the underlying rearrangement of the cytoskeleton. These events are controlled by a variety of processes such as switching on and off the activity of Rho family GTPases (Etienne-Manneville and Hall, 2002). Many target and regulatory molecules of Rho GTPases have been identified (Bishop and Hall, 2000). However, much remains to be learned regarding the details of their spatiotemporal regulation during morphological changes and cell migration. Recent progress in the field of Rho GTPase research leads to the idea that Rho family GTPases regulate each other's activities (Caron, 2003).
Two major Rho GTPase target molecules, Rho kinase (ROCK) and mDia, appear to participate in Rho-mediated effects on stress fiber formation (Ishizaki et al, 1996; Matsui et al, 1996; Watanabe et al, 1997). Rho kinase induces the pre-existing actin filament bundling through acto-myosin contraction (Amano et al, 1996) while mDia triggers de novo actin polymerization (Watanabe et al, 1999; Tominaga et al, 2000). Rho kinase is known to contribute to another major function of Rho, focal adhesion formation through engagement or clustering of integrin (Tominaga et al, 1993, 1998; Hotchin and Hall, 1995). Rho kinase also causes retraction of focal adhesions by strong acto-myosin contraction (Amano et al, 1996), leading to tail detachment in migrating cells.
We recently identified two novel pathways involving mDia, a Rho-mDia-Src kinase pathway (Tominaga et al, 2000) and an mDia-DIP pathway (Satoh and Tominaga, 2001), in which DIP (mDia interacting protein) binds to mDia through its fyn-like SH3 domain and colocalizes with mDia and Src kinase at focal adhesions. Others have recently reported that the Rho-Dia-Src pathway is also important for endosome motility (Gasman et al, 2003). These findings suggest that mDia-related proteins, Src kinase and DIP, play inter-related roles in cell motility. Here we report that upon EGF stimulation, DIP is phosphorylated by Src kinase, and binds and mediates the phosphorylation of p190RhoGAP and Vav2. As a result, DIP regulates Rho and Rac activity negatively and positively, respectively, downstream of Rho-mDia, in a Src kinase-dependent manner.
Results
DIP phosphorylation is induced by both Src kinase and EGF, and DIP co-precipitates with p190RhoGAP and Vav2
Previous studies have suggested the importance of interactions among mDia, Src kinase and DIP in the turnover of focal adhesions (Tominaga et al, 2000; Satoh and Tominaga, 2001). To further examine these interactions, we adopted a biochemical approach. COS7 cells were transfected with DIP alone or DIP and vSrc cDNAs. DIP was immunoprecipitated following stimulation with several growth factors, and immunoblotted with antiphosphotyrosine (PY) antibody (Figure 1A, left). EGF or PDGF treatment produced tyrosine phosphorylation of DIP in cells expressing DIP alone. Surprisingly, DIP was even more strongly tyrosine phosphorylated when coexpressed with vSrc, suggesting that Src is important for DIP phosphorylation. Furthermore, we noticed that in addition to DIP (∼80 kDa), two proteins of ∼200 and ∼100 kDa were phosphorylated by EGF stimulation and these two proteins also appeared to be more prominently phosphorylated in cells expressed with vSrc (Figure 1A). We prepared a membrane fraction (see Materials and methods) for IP experiments because considerable amounts of mDia and Src could be detected in a RIPA-insoluble fraction (data not shown). We also observed clear tyrosine phosphorylation of these two proteins and DIP in the membrane fractions following EGF treatment (Figure 1A, right). Therefore, we focused on the analysis of the membrane fractions in the following experiments. By screening with antibodies against candidate proteins, we identified the tyrosine-phosphorylated ∼200 and ∼100 kDa proteins as p190RhoGAP and Vav2, respectively, in untransfected cells (Figure 1B). We also excluded the possibility that the ∼200 kDa protein was the EGF receptor (data not shown). These data indicate that EGF stimulation induced tyrosine phosphorylation of endogenous DIP, p190RhoGAP and Vav2 (Figures 1B, upper, and C). Like mDia and Src, DIP/HA, p190RhoGAP and Vav2 were all distributed predominantly in the RIPA-insoluble fraction without EGF stimulation in serum-starved cells overexpressing DIP/HA (Figure 1C). DIP could be immunoprecipitated with p190RhoGAP (Figure 1D, upper) and Vav2 (Figure 1D, lower) using cells overexpressing DIP with p190RhoGAP or Vav2. Phosphorylation of p190RhoGAP, DIP and Vav2 again depended on EGF stimulation (Figure 1D). The time course of DIP phosphorylation upon EGF stimulation was examined using membrane fractions of DIP/HA-expressing cells. p190RhoGAP and Src were co-precipitated with DIP, and phosphorylated together with DIP at 5 and 10 min after EGF application (Figure 1E).
Figure 1.

DIP is tyrosine phosphorylated by vSrc and growth factors, and co-precipitated with phosphorylated forms of p190RhoGAP and Vav2 by EGF stimulation. (A) Phosphorylation of DIP by vSrc, EGF and PDGF in COS7 cells. Cells were stimulated with either 15% serum, 100 ng/ml EGF or 10 ng/ml PDGF for 10 min. Two other phosphorylated proteins (arrowheads and an asterisk), detected only by EGF stimulation in the left panel, are more clearly shown when membrane fraction was used for IP (right). (B) Co-precipitation of endogenous DIP with endogenous p190RhoGAP, Vav2 and cSrc, and phosphorylation of the four proteins by EGF stimulation in COS7 cells. Transferred membrane was first immunoblotted with anti-PY (upper) and reprobed with anti-DIP, anti-p190, anti-Vav2 and anti-Src antibodies (lower panels). IgG was used as a control for IP. (C) Distribution of DIP, p190RhoGAP, Vav2 and cSrc in COS7 cells transfected with DIP/HA cDNA. RIPA-soluble fraction (lane 1) was immunoprecipitated with anti-HA antibody, and RIPA-insoluble fraction was boiled with SDS sample buffer and loaded with the IP samples (lane 2). Transferred membrane was blotted with anti-p190, anti-Vav2, anti-Src and anti-HA (for DIP) antibodies. (D) DIP is co-immunoprecipitated with p190RhoGAP or Vav2 in COS7 cells overexpressing p190RhoGAP/Myc or Vav2/Myc with DIP/HA. Membrane fractions were precipitated with anti-Myc antibody and immunoblotted with anti-PY, anti-HA and anti-Myc antibodies. (E) Time course of DIP phosphorylation by EGF stimulation in serum-starved DIP/HA-expressing COS7 cells. From the membrane fraction, DIP was immunoprecipitated with anti-HA antibody and phosphorylation was detected with anti-PY antibody (upper). The same membrane was used for IB with anti-HA, anti-p190 and anti-Src antibodies (lower panels). (F) Phosphorylation of DIP, p190RhoGAP and Vav2 was lost in serum-starved src/fyn−/− cells expressing DIP/HA and Vav2/Myc. The membrane fraction of the cells was immunoprecipitated with anti-HA antibody (lanes 2 and 4). Tyrosine phosphorylation was detected with anti-PY antibody (upper). The existence of DIP, p190RhoGAP or Vav2 in IP samples was confirmed using anti-HA, anti-p190 or anti-Myc antibody, respectively (lower panels). src/fyn−/− cells expressing DIP/HA, Vav2/Myc and cSrc were treated as above (lanes 1 and 3). Note that phosphorylation of DIP, p190RhoGAP and Vav2 occurred only in cSrc-expressing cells (lane 3). (G) Cell migration assay using stable cell lines. Vector alone (MOCK) or DIP/pcDNA4/HisMax was stably transfected in src/fyn−/− or src/fyn+/+ cells. The number of cells that moved through the transwell was counted after 2 and 4 h. Quantitative analyses from three independent experiments. #P<0.01. The expression level of DIP in each cell line is shown at the bottom.
To confirm that these phosphorylation events were induced by Src kinase downstream of EGF, we used the src/fyn−/− cells expressing DIP/HA. We could not detect any phosphorylation of DIP, p190RhoGAP or Vav2 upon EGF stimulation using these cells (Figure 1F, lane 4, upper). However, phosphorylation of the three proteins was recovered by expressing cSrc in the src/fyn−/− cells (Figure 1F, lane 3, upper), indicating that Src kinase is required for EGF-induced phosphorylation of the three proteins. Binding of DIP to p190RhoGAP and Vav2 was detected in the membrane fraction without EGF stimulation both in untransfected cells (Figure 1B, lower three panels) and overexpressing cells (Figures 1C, right, and 1F, lower panels). To further examine the significance of the interaction between DIP and Src in vivo, we carried out a cell migration assay using src/fyn−/− and src/fyn+/+ cells with or without DIP expression (Figure 1G). In this assay, cells expressing both DIP and Src exhibited increased cell motility at 2 and 4 h, while DIP expression without Src did not induce such effects. Thus, Src is necessary for DIP-induced cell motility.
DIP phosphorylation is necessary for Src kinase-mediated p190RhoGAP and Vav2 activation
How do Src kinase, p190RhoGAP and Vav2 interact? To address this question, we first determined the region of DIP phosphorylated by EGF stimulation. DIP consists of three major regions: SH3, proline rich (Pro) and leucine rich (LR) (Figure 2A, left). Using several deletion mutants of DIP, we found that the Pro region is necessary for EGF-dependent DIP phosphorylation in vivo (Figure 2A, right). The importance of the Pro region in EGF-dependent DIP phosphorylation was further confirmed by an in vitro Src kinase assay using GST fusion proteins (Figure 2B). Following EGF stimulation, only the Pro+ region was strongly phosphorylated by immunoprecipitated Src (Figure 2B, far right). Next, in order to identify the DIP regions required for interaction with the other three proteins, we carried out an in vitro pull-down assay by mixing the GST fusion proteins (Figure 2B) with 35S-labeled in vitro translated proteins of p190RhoGAP, Vav2 and Src (Figure 2C, upper). All three fusion proteins bound mainly to a Pro+ construct, although p190RhoGAP appeared to bind to other regions of DIP (Figure 2C, lower). There are only two tyrosine residues in the Pro+ construct, one in the Pro region (227 aa) and another in connection between the SH3 and Pro regions (161 aa) (Figure 2B, left). To determine which tyrosine of DIP is phosphorylated by Src upon EGF stimulation, we mutated each tyrosine to phenylalanine and the resultant mutants were assayed for phosphorylation. Vav2/Myc was overexpressed in this experiment because endogenous Vav2 was found not to be abundant in COS7 cells (Figure 1B). Phosphorylation was preserved in Y227FDIP (Figure 2D, left). In contrast, tyrosine phosphorylation of p190RhoGAP and Vav2 as well as DIP was almost completely abolished in the cells expressing a mutant lacking the Pro region (ΔPro), a Y161FDIP mutant or a Y161F/Y227F mutant (Figure 2D, left). The finding that slight phosphorylation of p190RhoGAP left in the Y161F mutant disappeared in the Y161F/Y227F mutant suggests that Y227 is also involved in the DIP-mediated phosphorylation. ΔPro failed to bind Vav2 (Figure 2D, right), consistent with the result shown in Figure 2C. On the other hand, whereas tyrosine phosphorylation of p190RhoGAP was diminished in cells expressing ΔPro, Y161FDIP or Y161F/Y227F (Figure 2D, left), binding of these three mutants to p190RhoGAP was preserved (Figure 2D, right). As suggested in Figure 2C, other regions of DIP might be necessary for binding to p190RhoGAP. These findings suggest that Y161 is predominantly involved in DIP phosphorylation upon EGF stimulation, and raise the possibility that Src kinase phosphorylates Y161 and Y227 of DIP upon binding to its Pro region. This mechanism might account for the reported observations that activation of p190RhoGAP and Vav2 occurred through Src kinase-induced tyrosine phosphorylation (Chang et al, 1995; Marignani and Carpenter, 2001). The Src-dependent mechanism in migration assay was confirmed by the result that an Src kinase inhibitor, PP1, blocked DIP-induced cell migration in COS7 cells (Figure 2E). No significant changes in migration between GFP- and DIP/GFP-expressing cells could be due to considerable endogenous expression of DIP in COS7 cells (Satoh and Tominaga, 2001). The importance of the Pro region of DIP in cell migration was again confirmed (Figure 2E).
Figure 2.
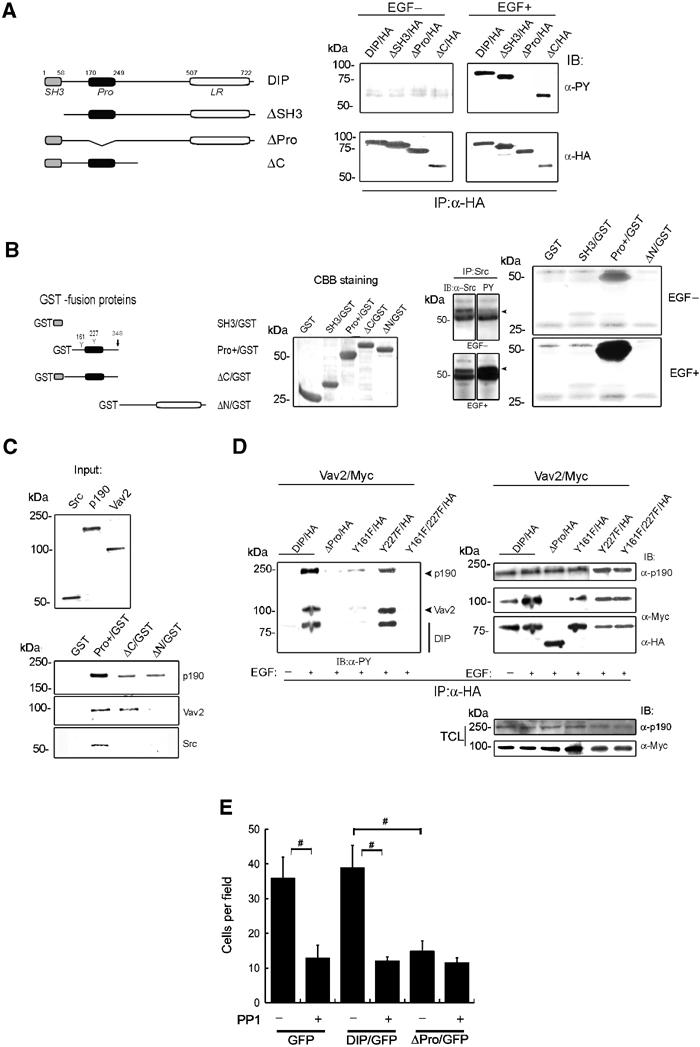
DIP phosphorylation in its proline-rich (Pro) region by Src kinase is necessary for activation of p190RhoGAP and Vav2. (A) Phosphorylation of DIP needs its Pro region in vivo. A construct design of DIP cDNAs (left). COS7 cells were transfected with each HA-tagged DIP cDNA. The RIPA-soluble fraction was immunoprecipitated with anti-HA antibody. IP samples were subjected to IB with anti-PY (right, upper panels) or anti-HA (right, lower panels) antibody. Note that only the mutant DIP lacking a proline-rich region (ΔPro/HA) lost tyrosine phosphorylation by EGF (right, upper), although all the constructs of DIP were expressed in COS7 cells (right, lower). (B) In vitro Src kinase (IVK) assay using GST-fusion proteins. A construct design of the GST-fusion proteins (left) and their expression by CBB staining (second left). Pro+/GST indicates a region (159–348 aa) including two tyrosine residues (Y) in the connection between SH3 and Pro regions and in a Pro region. NP-40-soluble fraction from cSrc-transfected COS7 cells was subjected to IP. Amounts of immunoprecipitated Src and its phosphorylation are shown (arrowheads in the third panel). Using these IP-Src, IVK assay was performed (the fourth panel). (C) In vitro pull-down assay using GST-fusion proteins. The assay was performed by mixing the GST-fusion proteins (B) and in vitro translated (IVT) 35S-labelled proteins. 1/10 input of the three IVT proteins (upper). (D) Phosphorylation of p190RhoGAP and Vav2 was affected by DIP mutants in COS7 cells transfected with DIP/HA or DIP mutants/HA cDNAs along with Vav2/Myc cDNA. Membrane fractions of the cells were subjected to IP with anti-HA antibody and IBs with anti-PY, anti-Myc and anti-HA antibodies. In all, 1/10 samples of TCL were used for determining the expression level (right lower two panels). (E) Cell migration assay using COS7 cells transfected with pEGFP or DIP/pEGFP or ΔPro/pEGFP. Before applying to the transwell chamber, cells were treated with or without PP1 (50 μM) for 30 min. Cells moving through the transwell were counted after 3 h. #P<0.01.
Activation of DIP regulates Rho and Rac activities
p190RhoGAP is known as GTPase-activating protein (GAP) for Rho, whereas Vav2 is reported as guanine nucleotide exchange factor (GEF) for Rac. These events regulating small GTPase activity have been proposed to depend on the phosphorylation of p190RhoGAP and Vav2 (Chang et al, 1995; Marignani and Carpenter, 2001). Therefore, we looked at the activities of Rho and Rac with or without EGF stimulation, using a pull-down assay to determine the physiological significance of the tyrosine phosphorylation of DIP. We used HEK293 cells for these experiments because Rho and Rac were expressed more abundantly in the membrane fraction of the cells compared with that in the RIPA-soluble fraction (Figure 3A and data not shown). Time-course analyses of endogenous Rho and Rac activities induced by DIP upon EGF stimulation revealed that DIP expression decreased Rho activity and increased Rac activity at 2–10 min after EGF stimulation (Figure 3B). These time courses parallel those of DIP and p190RhoGAP phosphorylation (Figure 1E). Therefore, the following studies were performed at 7.5 min after EGF stimulation. We transfected wild-type RhoA or Rac1 together with DIP cDNAs for Rho or Rac activation assay, respectively, because we needed to prepare large amounts of membrane fractions. The DIP-dependent phosphorylation of p190RhoGAP and Vav2 was not affected by overexpression of RhoA or Rac1 (Figure 3C, lower fifth–eighth panels). Rho activity was reduced upon EGF stimulation in cells expressing wild-type DIP, but not in cells expressing the dominant-negative ΔPro (Figure 3C, left). On the other hand, EGF stimulation increased Rac activity in cells expressing DIP but not in cells expressing ΔPro (Figure 3C, right). Basal Rac activity in ΔPro-transfected cells was always reduced compared with that in MOCK cells, perhaps due to loss of interaction of ΔPro with Vav2 (Figure 2D). When we examined the effect of DIP overexpression on Cdc42 activity using GST-PAK-CRIB, we could not detect any effects of DIP (data not shown). These results suggest that DIP tyrosine phosphorylation leads to Rho inactivation and Rac activation through phosphorylation of p190RhpGAP and Vav2.
Figure 3.

Regulation of Rho inactivation and Rac activation by DIP in HEK293 cells. (A) Endogenous active Rho exists mainly in the membrane fraction (M) but not in the RIPA-soluble fraction (S). (B) Time course of DIP effects on endogenous Rho and Rac activities in cells transfected with DIP/HA cDNA. Active RhoA or Rac1 in the membrane fraction was determined. In all, 1/10 of the samples were used for input (asterisks). (C) Effects of DIP expression on Rho and Rac activities with or without 7.5 min EGF stimulation in cells expressing DIP/HA or ΔPro/HA with RhoA/HA (left) or Rac1/HA (right). In all, 1/10 of the samples was used for input (asterisks) and blotted with anti-HA antibody. DIP phosphorylation was confirmed using anti-PY antibody. The phosphorylation and expression of P190RhoGAP and Vav2 were confirmed using DIP/HA immunoprecipitated samples (lower fifth to eighth panels). Quantitative analyses from three independent experiments (bottom). #P<0.01. (D) DIP-specific siRNA blocked the effect of EGF on Rho and Rac activities. The assays were carried out using the same preparations with or without EGF stimulation for 7.5 min. Quantitative analyses from three independent experiments (bottom). #P<0.01. Asterisks indicate input. (E) Rho and Rac activities modified by DIP were reversed by co-expression of p190RhoGAP mutant (p190(30-1)) (left) and Vav2 mutant (Vav2-R425G, right), respectively. Asterisks indicate input.
In addition, we confirmed the effect of endogenous DIP on Rho and Rac activities upon EGF stimulation by blocking endogenous human DIP expression with siRNA (87% reduction in 72 h, n=8) (Figure 3D, fourth panel). In cells where DIP expression was inhibited, Rho inactivation and Rac activation upon EGF stimulation were blocked (Figure 3D) as in the ΔPro-transfected cells (Figure 3C). To further confirm the involvement of p190RhoGAP and Vav2 downstream of DIP, we used a RhoGAP-deficient p190 (p190 (30-1)) (Brouns et al, 2001) and a Vav2 mutant (Vav2-R425G), reported not to activate Rac (Tamas et al, 2003). Both mutants inhibited the effects of DIP on Rho inactivation and Rac activation (Figure 3E). These results strongly suggest that Src kinase-induced activation of DIP with EGF stimulation regulates Rho negatively and Rac positively through p190RhoGAP and Vav2 activation, respectively.
Rho-mDia-Src pathway activates DIP
DIP is a protein downstream of mDia and Src kinase. Therefore, in order to examine the interaction of the Rho-mDia-Src pathway with DIP, we applied LPA to activate Rho in NIH3T3 cells. As found with EGF stimulation, p190RhoGAP could always be co-precipitated with DIP regardless of LPA stimulation (Figures 4A (left), 1B and 2D). Also, similar to EGF stimulation, LPA stimulation induced tyrosine phosphorylation of both DIP and p190RhoGAP (Figure 4A, lane 1+, right), although phosphorylation of p190RhoGAP was very weak. A tyrosine-phosphorylated protein intermediate in size between p190 and DIP (asterisk in lane 1+) was found to be FAK (data not shown). This result is consistent with the previous report that FAK binds Src and is phosphorylated in response to LPA (Clark and Brugge, 1995), suggesting that enough stimulation of Rho occurred by LPA. DIP phosphorylation disappeared with coexpression of an mDia lacking the FH1 region (mDiaΔFH1), to which Src cannot bind (Figure 4A, lane 2+, right) (Tominaga et al, 2000). DIP phosphorylation was stronger with EGF treatment than LPA treatment, and the phosphorylation was profoundly enhanced by concomitant treatment with EGF and LPA (Figure 4B). PP1 inhibited the DIP phosphorylation induced by not only EGF but also LPA (Figure 4B). The result that LPA induced an outcome similar to that caused by EGF might represent a novel Rho-mDia-Src-DIP pathway. Active mDia with (ΔGBDmDia) or without (ΔGBDmDiaΔFH1) its FH1 domain (an Src-binding domain) (Tominaga et al, 2000) was used for Rho and Rac activation assay to stimulate only the mDia-Src pathway (Figure 4C). A combination of ΔGBDmDia with DIP inactivated Rho and activated Rac (lane 5). These changes were reversed with a combination of ΔGBDmDia with ΔPro (lane 6). A combination of ΔGBDmDiaΔFH1 with wild-type DIP (lane 7) produced similar results observed in control (lane 1). These findings strongly support the existence of the Rho-mDia-Src-DIP pathway.
Figure 4.

Rho-mDia-Src pathway activates DIP in NIH3T3 cells. (A) LPA phosphorylates DIP through a Rho-mDia-Src pathway in cells transfected with 0.5 μg of mDia1/pEFm or mDia1ΔFH1/pEFm together with 1.5 μg of DIP/pcDNA3 and 1.5 μg of cSrc/pcDNA3. After stimulation with 10 μM LPA for 10 min, the membrane fraction was immunoprecipitated with anti-HA antibody, and immunoblotted with anti-HA (left), anti-p190 (left) and anti-PY (right) antibodies. mDia expression in TCL was confirmed using anti-Myc antibody (left lower). (B) LPA-induced phosphorylation occurs in an Src-dependent manner in cells overexpressing DIP/HA. Cells were stimulated with EGF alone, LPA alone, or EGF plus LPA. DIP phosphorylation in the membrane fraction was determined by HA-IP (lower) and PY-IB (upper). DIP phosphorylation by either stimulus was abolished by 30 min pretreatment with PP1 (50 μM) (right three lanes). (C) Rho and Rac activities induced by active mDia are affected by DIP in cells transfected with cDNAs, a combination of dominant active mDia (ΔGBDmDia1/pEFm, 0.3 μg), dominant active mDia lacking the FH domain (ΔGBDmDiaΔFH1/pEFm, 0.3 μg), DIP/HA/pcDNA3 (0.5 μg) and/or ΔPro/HA/pcDNA3 (0.5 μg), and 0.3 μg of RhoA/HA/pEF-BOS or Rac1/HA/pEF-BOS. Membrane fraction was used for Rho and Rac activation assay. The relative values of Rho and Rac activities normalized to lane 1 are shown (fold). The lower three panels indicate the amounts of mDia and DIP, and the DIP phosphorylation level in TCL.
Change of stress fiber formation following EGF stimulation
It has been reported that in normal fibroblasts, stress fiber abundance decreases upon EGF stimulation and begins to recover within 10 min. However, such recovery was not observed in cells overexpressing p190RhoGAP, even 30 min after EGF application (Chang et al, 1995). Therefore, we examined whether DIP could modulate stress fiber formation upon EGF stimulation in NIH3T3 cells. Untransfected fibroblasts (no GFP signal) lost stress fibers at 2 min after EGF stimulation then began to regain them at 5–10 min, with complete recovery at 20 min, similar to previous reports (Figure 5) (Chang et al, 1995). In contrast, in cells expressing DIP/GFP, stress fibers did not recover until 40 min and membrane ruffling was observed at 5–10 min (Figure 5A). Cells expressing ΔPro already showed clear stress fiber recovery at 5 min compared with control cells (Figure 5B). These data suggest that DIP plays a critical role in the connection between Src and p190RhoGAP (Arthur et al, 2000; Arthur and Burridge, 2001). The effect of siRNA specific to mouse DIP (78% reduction in 72 h, n=5) (Figure 7C) on this stress fiber recovery was examined (Figure 5C). DIP-specific siRNA-treated cells began to regain stress fibers 5 min after EGF stimulation, similar to ΔPro-expressing cells (Figure 5B).
Figure 5.
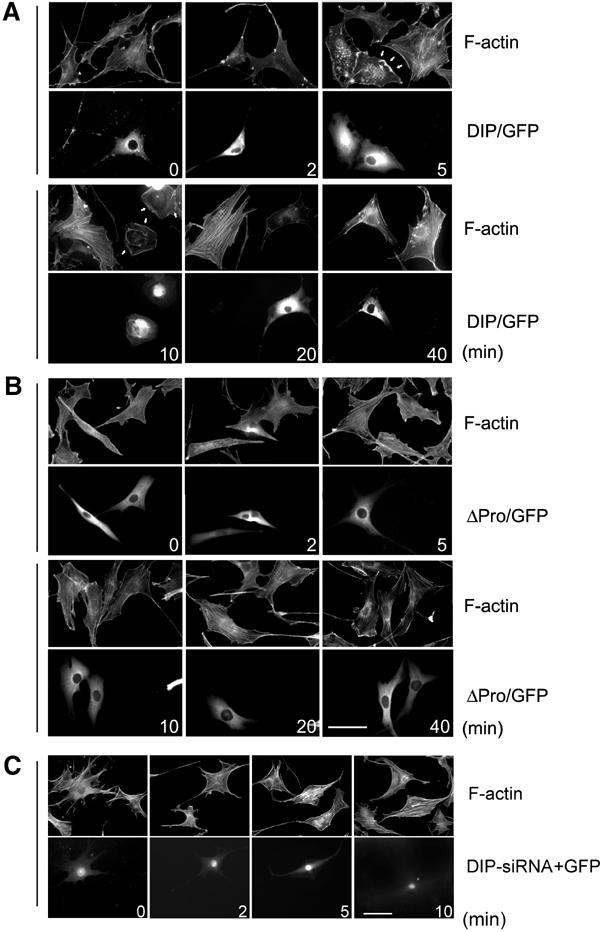
DIP regulates actin organization downstream of EGF stimulation. (A, B) F-actin staining in NIH3T3 cells expressing DIP/GFP or ΔPro/GFP. Bar, 50 μm. (C) F-actin staining in NIH3T3 cells transfected with DIP-specific siRNA with pEGFP. The siRNA-treated cells are indicated by GFP expression (lower panels).
Figure 7.
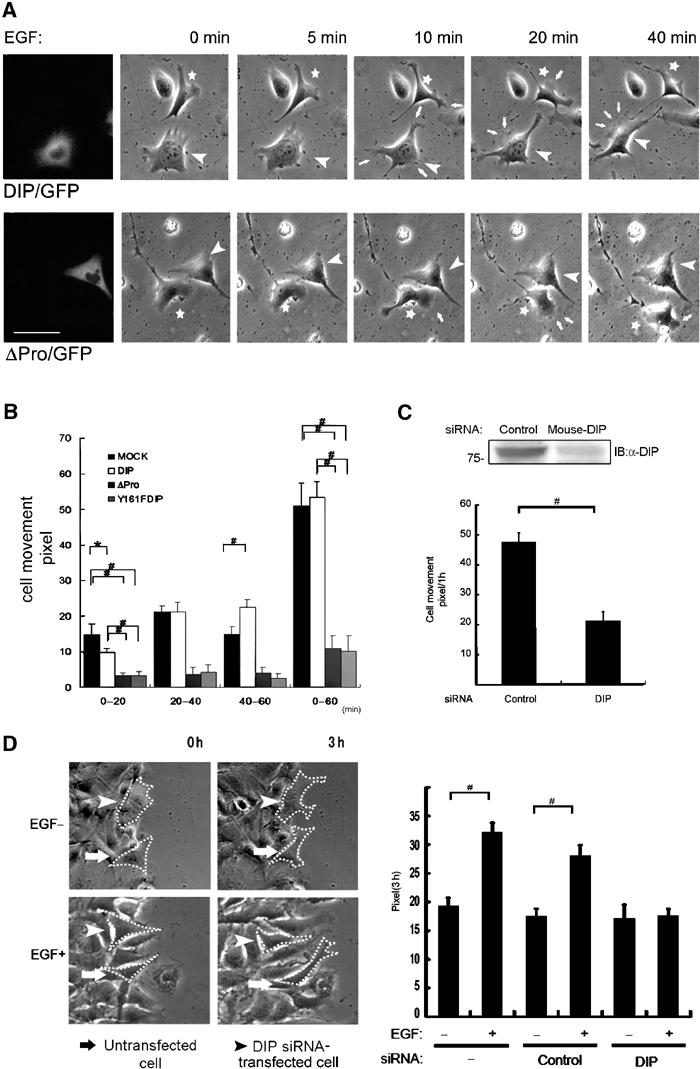
DIP regulates cell motility (time-lapse study). (A) Morphology and motility of src/fyn+/+ fibroblasts upon 100 ng/ml EGF stimulation. A representative cell with (arrowhead) or without (asterisk) DIP/GFP expression (upper) and with (arrowhead) or without (asterisk) ΔPro/GFP expression (lower). Arrows indicate membrane ruffling. DIP/GFP- and ΔPro/GFP-expressing cells are shown in the far left panels and in the other panels (arrowheads). Bar, 25 μm. (B) Quantitative analyses of movement of src/fyn+/+ fibroblasts after EGF treatment. *P<0.05; #P<0.01. (C) Quantitative analyses of movement of siRNA-treated src/fyn+/+ fibroblasts. DIP expression after siRNA treatment is shown (upper). Data indicate cell movements during the 60-min period following EGF stimulation. n=5 for each group. #P<0.01. (D) Wound-healing assay of siRNA-treated NIH3T3 cells. The left panels show the representative cell movements. Dot lines indicate cell margins. Note that DIP-specific siRNA-treated cells did not move regardless of EGF treatment, whereas untransfected cells moved well 3 h after EGF stimulation (left lower). The right panel shows quantitative analyses of cell movements. #P<0.01.
Endogenous DIP determines the distribution of p190RhoGAP and Vav2, and their phosphorylation is dependent on DIP
To confirm the importance of endogenous DIP in the findings described above, we examined the phosphorylation of p190RhoGAP and Vav2 using DIP-specific siRNA (Figure 6A) in HeLa cells. We prepared membrane fraction from cells with or without EGF stimulation. Only p190RhoGAP was immunoprecipitated to distinguish it from EGFR close in size. Interestingly, in addition to DIP, p190RhoGAP and Vav2 were diminished in amounts in membrane fractions from cells treated with the DIP-specific siRNA but not from cells treated with control siRNA (Figure 6A, left). Phosphorylation of p190RhoGAP and Vav2 was also strongly reduced in EGF-stimulated cells treated with the DIP-specific siRNA (Figure 6A, right). To find a reason for the apparent depletion of p190RhoGAP and Vav2 in the membrane fraction of the DIP-specific siRNA-treated cells, we examined the amounts of p190RhoGAP, Vav2 and Src in total cell lysates (TCL), as well as RIPA-soluble and membrane fractions. We noticed that the distribution of p190RhoGAP and Vav2 but not Src was changed from the membrane fraction to the RIPA-soluble fraction following the DIP-specific siRNA treatment (Figure 6B). These results suggest that DIP might recruit p190RhoGAP and Vav2 to the plasma membrane. We also examined the changes of the cell morphology and the localization of p190RhoGAP and Vav2 with DIP after EGF application in siRNA-treated cells (Figure 6C). In control siRNA-treated cells, DIP was co-localized well with p190RhoGAP (panels (a–c)) or Vav2 (panels (f–h)), especially at the cell periphery and membrane ruffles. In contrast, membrane ruffles were not observed, and p190RhoGAP and Vav2 were localized mainly in the cytosole or at the peri-nucleus in the DIP-specific siRNA-treated cells (panels (e) and (j)).
Figure 6.

Endogenous DIP is important for phosphorylation of p190RhoGAP and Vav2, and for their distribution in HeLa cells. (A) Cells were treated with control or DIP-specific siRNA (DIP/siRNA) for 72 h. The membrane fraction of the cells was subjected to IBs with anti-DIP, anti-Vav2 (left) or anti-PY (right) antibody. Only for p190RhoGAP detection, the membrane fraction was first immunoprecipitated with anti-p190 antibody, and then the IP samples were immunoblotted with anti-p190 (left) or anti-PY (right) antibody. (B) Distribution of p190RhoGAP and Vav2 was changed from membrane fraction to RIPA-soluble fraction with DIP/siRNA treatment. TCL, RIPA-soluble and membrane fractions were obtained from cells treated with siRNA. Endogenous DIP, p190RhoGAP, Vav2 and cSrc were detected by IB. (C) Cell morphology and distribution of p190RhoGAP (a–e) and Vav2 (f–j) were changed with DIP/siRNA treatment. Cells were treated with siRNA for 72 h and stimulated with EGF. Merge images are shown in (c) and (h). Exposure time was 500 ms except for the DIP in (d) and (i) (1000 ms). (D) Co-localization of Vav2 with DIP but not with ΔPro at membrane ruffles. Cells expressing Vav2/GFP and DIP/DsRed or ΔPro/DsRed were stimulated with EGF. Note that membrane ruffles and co-localization of Vav2 with DIP (upper) were lost in a ΔPro-expressing cell (lower).
To further confirm the interaction of DIP with Vav2 in living cells, we overexpressed DIP/DsRed and Vav2/pEGFP. DIP was co-localized well with Vav2 at membrane ruffles (Figure 6D, upper). On the other hand, no such co-localization was observed when Vav2 was overexpressed with ΔPro/DsRed (Figure 6D, lower), similar to the DIP-specific siRNA-treated cells (Figure 6C).
DIP regulates cell movement and cell shape
To investigate the involvement of DIP in cell movement before and after EGF stimulation, we performed time-lapse recording of cells expressing DIP. We chose src/fyn+/+ fibroblasts because these cells moved well and membrane ruffling was easily observed, and because endogenous DIP is not abundant. DIP-expressing cells typically stayed at the same place, without extension for at least 10 min following EGF stimulation, but exhibited membrane ruffles compared with control cells (asterisk) (Figures 7A, upper, and B, and Movie 1 in Supplementary data). Then movement of the cells was recovered and even progressed to a greater-than-normal level after 40–60 min (Figure 7B and Movie 1 in Supplementary data). Cells expressing ΔPro/GFP appeared triangular in shape without membrane ruffles and did not move at all, even upon EGF stimulation (Figure 7A, lower, and Movie 2 in Supplementary data). The shape and movement of cells expressing the Y161FDIP/GFP were similar to those of ΔPro/GFP-expressing cells (data not shown and Figure 7B). Together with the results shown in Figure 5, these data suggest that in the period shortly after EGF stimulation, DIP increases membrane ruffles and inhibits motility through loss of stress fibers and focal contacts formation in DIP overexpressing cells compared with control cells. These changes might occur through activation of Vav2 and p190RhoGAP by DIP. Stress fiber formation and focal contacts were restored at 20 min after EGF stimulation, leading to a restoration of cell movement (Figures 7A and B). On the other hand, ΔPro overexpression led to an abundance of stress fibers and loss of membrane ruffles (Figures 5 and 7A, lower), suggesting that ΔPro cannot activate p190RhoGAP and Vav2. To further confirm the physiological significance of DIP, we investigated cell movement using mouse DIP-specific siRNA (Figure 7C, upper). Cell movement was significantly inhibited by knockdown of endogenous DIP with the DIP-specific siRNA compared with control siRNA-treated cells (Figure 7C, lower), consistent with the results obtained using ΔPro or Y161F DIP (Figure 7B).
Wound-healing assay was performed in NIH3T3 cells using siRNA (Figure 7D). EGF-induced cell movement following wounding in DIP-specific siRNA-treated cells was significantly diminished compared with untransfected and control siRNA-treated cells.
Discussion
Proper cell movement requires that cytoskeletal rearrangement occurs through the coordinated spatiotemporal regulation of Rho GTPases (Horwitz and Parsons, 1999). In this study, we have demonstrated a critical function of DIP in the regulation of these proteins. DIP was found to bind to Src kinase, p190RhoGAP and Vav2 in RIPA-soluble fraction and transfer them to the membrane fraction. Under conditions where the three proteins bind together beneath the plasma membrane, DIP is phosphorylated by Src following EGF stimulation; then the phosphorylated DIP, in turn, inactivates Rho by mediating the Src-induced phosphorylation of p190RhoGAP. In addition, phosphorylated DIP activates Rac by mediating the Src-induced phosphorylation of Vav2. LPA alone can induce weak DIP phosphorylation in the absence of EGF while simultaneous application of EGF and LPA causes synergistic effects on DIP phosphorylation (Figure 4). We hypothesize that this synergistic interaction works in native cells as well, because it is well known that integrin activation causes activation of RhoGTPases and that integrins and their matrix protein ligands are close collaborators with growth factors (Miranti and Brugge, 2002; Yamada and Even-Ram, 2002). Furthermore, our time-lapse analysis clearly demonstrated that DIP plays a pivotal role in cell motility (Figure 7). In addition, several changes—in DIP phosphorylation, Rho and Rac activities, and stress fiber formation and membrane ruffling—occur in a similar time period after EGF stimulation, further supporting that DIP is a key molecule causing Rac activation and feedback inactivation of Rho through phosphorylation of p190RhoGAP and Vav2. These effects can be achieved by endogenous DIP since phosphorylation of p190RhoGAP and Vav2, regulation of Rho and Rac activities, and cell motility in time-lapse and wound-healing assays were reduced in cells treated with DIP-specific siRNA (Figures 3, 5 and 7). Thus, cell motility could be finely tuned through a Rho-mDia-Src-DIP pathway in a collaborative fashion.
Several previous reported lines of evidence have shown that p190RhoGAP activated by Src causes cytoskeletal disruption (Arthur et al, 2000; Arthur and Burridge, 2001). Our data make it clear that DIP plays a critical role in the connection between Src and p190RhoGAP, thereby permitting the actions of p190RhoGAP on the cytoskeleton. EGF stimulation has been reported to cause a loss of stress fiber formation through p190RhoGAP activation. Our study shows that EGF-induced activation of DIP could represent the underlying mechanism for this phenomenon. Vav2 has been found to be activated by growth factors such as EGF (Moores et al, 2000). In our experiments, EGF-induced DIP activation caused Vav2 phosphorylation, Rac activation and membrane ruffling. These data also suggest that DIP activates Rac via Vav2 phosphorylation. mDia has been reported to activate Rac by Cas (Tsuji et al, 2002), probably through a Src-Cas-Crk-DOCK180-Rac pathway (Kiyokawa et al, 1998). The pathway that we have identified seems to be a more direct means for Rac activation by mDia, although we cannot prove which pathway is more predominant in native, moving cells.
Cell movement is a final outcome of various steps such as protrusion in the front and retraction in the tail of cells (Nobes and Hall, 1999). In the front of cells, many regulatory molecules appear to be involved in cell protrusion. As most of the data shown in this study were obtained in the 5–10 min period following EGF stimulation, DIP might be one of the key molecules regulating the initial steps of cell movement after stimulation of integrin and growth factor receptors. The fact that cell motility was rather reduced during 0–20 min after EGF stimulation might be explained by a scenario in which overexpressed DIP acts predominantly on p190RhoGAP, leading to Rho and thereby ROCK inactivation. As a result, new stress fiber formation, retraction in the cell tail and new focal adhesion formation in cell front were inhibited, causing prevention of cell movement (Figure 8). Time-lapse and staining studies showed that cells regained motility 20–40 min after EGF stimulation, probably through diminished negative feedback of Rho regulation via p190RhoGAP, which began to induce new focal adhesion formation in the cell front and stress fiber formation (Figure 8).
Figure 8.

Proposed model for DIP function in the activation of p190RhoGAP and Vav2. DIP binds to p190RhoGAP and Vav2, thereby making a complex and transferring them beneath the membrane. Upon EGF stimulation, cSrc phosphorylates DIP. Then, phosphorylation of p190RhoGAP and Vav2 is induced presumably through conformational change of the complex by cSrc. Phosphorylated p190RhoGAP inactivates Rho, and in turn the inactivated Rho blocks retraction (in the cell tail) and new focal adhesion formation (in the cell front) through Rho kinase (ROCK) inhibition, preventing cell movement. On the other hand, phosphorylated Vav2 activates Rac, and in turn the activated Rac causes protrusion in the cell front.
Recently, it has been proposed that Rho activity level determines focal complex turnover and contractility through mDia and Rho kinase activation, respectively, by p190RhoGAP (Small and Kaverina, 2003). This model was proposed on the basis of a report describing the regulation of Rho activity by p190RhoGAP (Arthur and Burridge, 2001). In addition, Rho GTPases have been thought to be regulated by each other, themselves, their target molecules and/or their regulatory molecules. The present study clearly supports these proposals by clarifying a novel function of DIP, namely the activation of Rac through Vav2 and feed-back inactivation of Rho through p190RhoGAP.
Materials and methods
Materials and plasmids
The following antibodies were used: mAb antiphosphotyrosine (4G10) (Upstate Biotechnology), mAbs anti-rat-HA (3F10) (for immunoprecipitation (IP)) and anti-mouse-HA (12CA5) (for immunoblotting (IB)) (Roche), mAbs anti-p190 (G19020), anti-Rac1 (R56220) and anti-FAK (BD Transduction Laboratories), mAbs anti-Myc (9E10) and anti-RhoA (26C4) and polyclonal anti-goat Vav2 (C-19 or P-18) (Santa Cruz Biotechnology), mAb anti-Src (327) (Oncogene Research Products), mAb anti-HisG (R940-25) (Invitrogen). Anti-DIP polyclonal antibodies (055 for immunostaining and A1 for IP) were made by ourselves, as described previously (Satoh and Tominaga, 2001). Alexa Fluor 594-conjugated phalloidin (Molecular Probes) was used for F-actin. For IP, Protein A- and G-sepharose 4B were used (Zymed Laboratories Inc.). All chemicals and reagents except for PP1 (from Biomed Research Laboratories) were obtained from Sigma.
pcDNA3-cSrc and kinase-dead Src were provided by M Hamaguchi, and vSrc cDNA (provided by S Hanafusa) was subcloned into pcDNA3. Rat wt and mutant p190RhoGAP cDNAs (provided by J Settleman) were subcloned into Myc-tagged pcDNA3. Human Vav2 cDNA (provided by J Brugge) was subcloned into Myc-tagged pcDNA3. GST-RBD-Rhotekin, GST-PAK-CRIB, and pEF-BOS-HA-Rho and -Rac1 cDNAs were provided by M Negishi.
Point mutations were introduced by using oligonucleotide-directed mutagenesis, and all constructs were verified by DNA sequencing. GST-fusion proteins were made according to the manufacturer's instructions (Amersham Biosciences).
Cell culture and transfection
COS7, NIH3T3 fibroblast, src/fyn−/−, src/fyn+/+, HEK293 and HeLa cells were cultured in DME containing 10% FBS, penicillin, streptomycin and L-glutamine. Zeocin (Invitrogen; 400 and 250 μg/ml) were added to src/fyn−/− and src/fyn+/+ cells (provided by C Lowell) stably expressing DIP/pcDNA4/HisMax or pcDNA4/HisMax (as MOCK) (Invitrogen), respectively. The cells were cultured in DME containing 10% FBS and L-glutamine.
Cells were plated 1 day before transfection with the indicated amount of cDNAs (total 2 μg/60-mm dish or 4 μg/100-mm dish) in OPTI-MEM using LipofectAMINE plus reagent (Invitrogen) according to the manufacturer's protocol. When transfected with two kinds of cDNAs to the same cells, the ratios of cDNAs were kept one to one unless otherwise stated. After 3–4 h incubation, the medium was changed with serum-starved DME for 18–20 h before the experiments.
Immunoprecipitation, immunoblotting analyses and preparation of membrane fraction
IP and IB were performed by using the membrane fraction of cells as described previously (Tominaga et al, 1998). In brief, cells were plated with 50–60% of confluency. The next day, cDNA was transfected and serum starved for 20 h. After that, cells were stimulated with growth factors. In the case of EGF treatment, 100 ng/ml EGF was applied for 10 min unless otherwise stated. For IP from the membrane fraction, the cells were washed twice with ice-cold PBS containing 0.1 μM sodium orthovanadate (Na3VO4) and resuspended in TNE buffer (10 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, complete EDTA-free protease inhibitor cocktail (PIC) (Roche), 1 μM Na3VO4). Samples were centrifuged for 15 min at 100 000 g. The pellets were resuspended in TNE buffer with 1% Nonidet P-40 (NP-40) and sonicated for 30 s. Following centrifugation at 100 000 g for 30 min, the supernatants were adjusted protein concentrations and subjected to IPs. In the indicated experiment, IP was performed using soluble fraction after lysis with RIPA buffer (TNE buffer with 1% Triton X-100, 0.5% sodium deoxycholate and 0.1% SDS). For IB, protein-blotted PVDF membranes were sometimes reprobed with other antibodies.
Cell migration assay
Cells were serum starved in DME+0.1% BSA for 16–20 h, trypsinized and suspended in DME+0.1% BSA. After incubation for 30 min in CO2 incubator, cells (1 × 105) in a 0.1 ml were added to the upper surface of a transwell chamber (6.4 mm in diameter, 8 μm in pore size; Becton Dickinson), and the lower chamber was filled with 0.6 ml of conditional medium obtained from the supernatant of culture with HT1080 cells (provided by K Itoh) (Yoshioka et al, 2003) in MEM+0.005% vitamin C+0.1% BSA for 24 h. After the indicated period of culture in a CO2 incubator, the cells on the upper surface of the membrane of a transwell chamber were removed and the migrating cells on the lower membrane surface were fixed, stained with crystal violet and counted in five different regions in the field. In the experiments using COS7 cells, cells were transfected with GFP-tagged cDNA. GFP signal on the lower membrane was calculated in the same way as above.
Pull-down and in vitro kinase assay
A pull-down assay using GST-fusion proteins with 35S-labeled in vitro translated proteins was performed as described previously (Satoh and Tominaga, 2001). The measurement of RhoA and Rac1 activities was performed as previously described (Katoh et al, 2000), with a minor modification as follows. HEK293 cells were transfected with 1.5 μg of HA-DIP/pcDNA3 or HA-ΔPro/pcDNA3 and 0.5 μg of HA-RhoA/pEF-BOS or HA-Rac1/pEF-BOS as above. After adjusting protein concentration, the membrane fraction was incubated with 20 μg of GST-RBD or GST-PBD beads in ice-cold cell lysis buffer (50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 2 mM MgCl2, 1% NP-40, 10% glycerol, 1 mM dithiothreitol, PIC) for 30 min at 4°C. After washing with the cell lysis buffer, the bound GTP-RhoA or GTP-Rac1 was determined by IB with anti-RhoA or anti-Rac1 antibody for endogenous protein, or anti-HA antibody for overexpressed protein. In all, 1/10 of membrane fractions were used for IB or IP with anti-HA or anti-PY antibody to confirm the protein amount (input) and phosphorylation.
An in vitro kinase (IVK) assay was performed as described previously (Tominaga et al, 1998). Briefly, COS7 cells were transfected with cSrc cDNA. Following the lysis of cells with 1% NP40-TNE buffer and centrifugation, cSrc was immunoprecipitated from NP40-soluble fraction. IVK assay was performed by mixing the IP-Src with GST-DIP fragments in the presence of [γ32]P-ATP for 10 min.
siRNA preparation and transfection
siRNAs specific to human and mouse DIP were designed (5′-GUGGCAACUGGUACAACCUdU-3′ and 5′-AGGUUGUACCAGUUGCCACdT-3′ and 5′-ACCUUGUCUCGAAGAGGCAdU-3′ and 5′-UGCCUCUUCGAGACAAGGU-3′, respectively), and constructed using the Silencer™ siRNA Construction Kit (Ambion Inc., TX, USA). A control siRNA specific to GAPDH was made. The siRNAs were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells plated at 40–50% were transfected with 50 nM siRNAs with or without 0.2 μg of pEGFP, cultured for 72 h and subjected to the experiments. For IPs, cells were serum starved for 20 h before experiments. For Rho and Rac activation assays, cells were treated with siRNA for 48 h, transfected with HA-RhoA/pEF-BOS or HA-Rac1/pEF-BOS, serum starved for 20 h and subjected to the assays.
Immunofluorescence study
Cells were plated on cover glasses, transfected and serum starved for 16–20 h before the immunostainings, as previously described (Satoh and Tominaga, 2001). In brief, cells were fixed with 4% paraformaldehyde for 20 min and incubated with primary antibodies for 90 min. Then, cells were incubated with secondary antibodies (Alexa Fluor 488- or 594-labeled anti-IgGs (Molecular Probes)). Images were obtained using an Olympus fluorescent microscope with a cooled CCD camera (ORCA-ER, Hamamatsu Photonics) and IP-Lab Image software (Solution Systems, Tokyo).
Time-lapse microscopy
src/fyn+/+ cells were plated on FN-coated 35-mm dishes at a density of 5 × 104, transfected with 0.5 μg of pEGFP-DIP or -DIP mutants cDNAs and serum starved in DME+0.1% BSA for 16–20 h. A dish was transferred to the chamber (Olympus) for maintaining the cells in 5% CO2 and 37°C on the microscopy, and live differential interference contrast and fluorescence images of the cells were monitored every 2 min using IP-Lab Image software. Nuclear translocation was quantified and expressed as amount of pixels.
Wound-healing assay
NIH3T3 cells were transfected with siRNA and 0.1 μg of pEGFP as above, seeded on FN-coated coverslips in 24-well plates after 48 h, and serum starved after 4 h of plating. After 20 h, the monolayer was wounded. Cells were washed once, fresh serum-free mediun was added, and cells were allowed to invade the wound for 3 h under observation with time-lapse microscopy. Nuclear translocation was quantified as the amount of pixels.
Statistical analysis
Values are expressed as mean±s.e. One-way ANOVA and an unpaired t-test were carried out for statistical analysis.
Supplementary Material
Movie 1 in Supplementary data
Movie 2 in Supplementary data
Acknowledgments
We thank M Negishi (Kyoto University), J Settleman (Harvard Medical School), J Brugge (Harvard Medical School), S Hanafusa (Osaka Bioscience Institute), M Hamaguchi (Nagoya University), C Lowell (UCSF), and K Yoshioka and K Itoh (Osaka Medical Center for Cancer & Cardiovascular Diseases) for providing materials; MJ Caterina (Johns Hopkins University) for a critical reading of the manuscript; and H Konishi (Mie University) for technical assistance. This work was supported by grants from a Grant-in-Aid for Scientific Research on Priority Areas (to TT) of Japan.
References
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271: 20246–20249 [DOI] [PubMed] [Google Scholar]
- Arthur WT, Burridge K (2001) RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell 12: 2711–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur WT, Petch LA, Burridge K (2000) Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol 10: 719–722 [DOI] [PubMed] [Google Scholar]
- Bishop AL, Hall A (2000) Rho GTPases and their effector proteins. Biochem J 348 (Part 2): 241–255 [PMC free article] [PubMed] [Google Scholar]
- Brouns MR, Matheson SF, Settleman J (2001) p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol 3: 361–367 [DOI] [PubMed] [Google Scholar]
- Caron E (2003) Rac signallings: a radical view. Nat Cell Biol 5: 185–187 [DOI] [PubMed] [Google Scholar]
- Chang JH, Gill S, Settleman J, Parsons SJ (1995) c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol 130: 355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Brugge JS (1995) Integrin and signal transduction pathways: the road taken. Science 268: 233–239 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420: 629–635 [DOI] [PubMed] [Google Scholar]
- Gasman S, Kalaidzidis Y, Zerial M (2003) RhoD regulates endosome dynamics through Diaohanous-related formin and Src tyrosine kinase. Nat Cell Biol 5: 195–204 [DOI] [PubMed] [Google Scholar]
- Horwitz AR, Parsons JT (1999) Cell migration—movin' on. Science 286: 1102–1103 [DOI] [PubMed] [Google Scholar]
- Hotchin NA, Hall A (1995) The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J Cell Biol 131: 1857–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S (1996) The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J 15: 1885–1893 [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Yasui H, Yamaguchi Y, Aoki J, Fujita H, Mori K, Negishi M (2000) Small GTPase RhoG is a key regulator for neurite outgrowth in PC12 cells. Mol Cell Biol 20: 7378–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T, Matsuda M (1998) Activation of Rac1 by a Crk SH3-binding protein, DOCK180. Genes Dev 12: 3331–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marignani PA, Carpenter CL (2001) Vav2 is required for cell spreading. J Cell Biol 154: 177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito T, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K (1996) Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J 15: 2208–2216 [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 4: E83–E90 [DOI] [PubMed] [Google Scholar]
- Moores SL, Selfors LM, Fredericks J, Breit T, Fujikawa K, Alt FW, Brugge JS, Swat W (2000) Vav family proteins couple to diverse cell surface receptors. Mol Cell Biol 20: 6364–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144: 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh S, Tominaga T (2001) mDia-interacting protein acts downstream of Rho-mDia and modifies Src activation and stress fiber formation. J Biol Chem 276: 39290–39294 [DOI] [PubMed] [Google Scholar]
- Small JV, Kaverina I (2003) Microtubules meet substrate adhesions to arrange cell polarity. Curr Opin Cell Biol 15: 40–47 [DOI] [PubMed] [Google Scholar]
- Tamas P, Solti Z, Bauer P, Illes A, Sipeki S, Bauer A, Farago A, Downward J, Buday L (2003) Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J Biol Chem 278: 5163–5173 [DOI] [PubMed] [Google Scholar]
- Tominaga T, Ishizaki T, Narumiya S, Barber DL (1998) p160ROCK mediates RhoA activation of Na–H exchange. EMBO J 17: 4712–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, Alberts AS (2000) Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell 5: 13–25 [DOI] [PubMed] [Google Scholar]
- Tominaga T, Sugie K, Hirata M, Morii N, Fukata J, Uchida A, Imura H, Narumiya S (1993) Inhibition of PMA-induced, LFA-1-dependent lymphocyte aggregation by ADP ribosylation of the small molecular weight GTP binding protein, rho. J Cell Biol 120: 1529–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Ishizaki T, Okamoto M, Higashida C, Kimura K, Furuyashiki T, Arakawa Y, Birge RB, Nakamoto T, Hirai H, Narumiya S (2002) ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J Cell Biol 157: 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S (1999) Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol 1: 136–143 [DOI] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 16: 3044–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Even-Ram S (2002) Integrin regulation of growth factor receptors. Nat Cell Biol 4: E75–E76 [DOI] [PubMed] [Google Scholar]
- Yoshioka K, Foletta V, Bernard O, Itoh K (2003) A role for LIM kinase in cancer invasion. Proc Natl Acad Sci USA 100: 7247–7252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1 in Supplementary data
Movie 2 in Supplementary data