

Abstract
Immunoglobulin (Ig) G4-related lung disease is a fibroinflammatory entity that presents in protean ways. Diagnostically, IgG4-related lung disease requires a high clinical index of suspicion complemented by elevated serum IgG4 levels and/or biopsy that shows the characteristic pathological features. The disease is almost always responsive to systemic corticosteroids. However, relapse is common following their discontinuation. The authors present three cases to highlight the diverse clinical features, and to illustrate the diagnostic and therapeutic approaches to this disease.
Keywords: Corticosteroids, IgG4-related lung disease, Immunoglobulins
Abstract
La maladie pulmonaire liée aux immunoglobulines (Ig) G4 est une entité fibro-inflammatoire de caractère protéiforme. Sur le plan diagnostique, elle exige un fort indice de présomption clinique, conjugué à des taux d’IgG4 sérique élevés ou à une biopsie qui en révèle les caractéristiques pathologiques. La maladie répond presque toujours aux corticoïdes systémiques. Cependant, les récidives sont fréquentes après la fin du traitement. Les auteurs présentent trois cas qui font ressortir les caractéristiques cliniques diversifiées de la maladie et en illustrent les approches diagnostiques et thérapeutiques.
Learning Objectives:
To recognize the protean clinical manifestations of immunoglobulin (Ig) G4-related lung disease (IgG4RLD).
To understand the importance of serum Igs (and IgG subclass phenoytping) in the diagnosis of this disease entity.
CanMEDS Competency: Medical Expert
Pre-test:
What are the characteristic histological features of IgG4RLD?
How is the diagnosis of IgG4RLD classified?
What is the standard treatment for IgG4RLD?
IgG4RLD is a fibroinflammatory entity that has diverse clinical manifestations (1). Histopathologically, this disease is characterized by dense infiltration of IgG4-positive plasma cells and lymphocytes in affected organs. The first description of IgG4-related disease was with autoimmune pancreatitis (2,3). Since then, IgG4-related disease has been described in other organs, including the biliary tract, pancreas, liver and lungs (4). IgG4RLD shares the same histopathological features with its pancreatic prototype. Apart from dense infiltration by IgG-positive plasma cells and lymphocytes, storiform fibrosis and obliterative phlebitis are also common findings of this disease (Figure 1) (1). Extrapulmonary manifestations of IgG4RLD are common and include autoimmune pancreatitis, periaortitis, interstitial nephritis, chronic sclerosing sialadenitis and prostatitis (5). Radiographically, there are four major subtypes of IgG4RLD that are based on thoracic computed tomography (CT) appearance (6): solid nodular type (solitary nodular lesions that include a mass); round-shaped ground-glass opacity (GGO) type, characterized by multiple round-shaped GGOs; alveolar interstitial type showing honeycombing, bronchiectasis or diffuse GGOs; and bronchovascular type showing thickening of bronchovascular bundles and interlobular septa. Diagnosis is based on symptoms, biochemistry (serum IgG4 level >1.35 g/L or the ratio of IgG4 to total IgG >40%) (7); and characteristic histopathological findings classified into definite (symptoms and biochemical and histopathology), probable (symptoms and histopathology only) or possible (symptoms and biochemical markers only) disease (6). Disease presentation can be variable and, thus, requires a high index of suspicion. In the present article, we describe three patients with IgG4RLD to illustrate the protean manifestations of this disease in the lungs.
Figure 1).
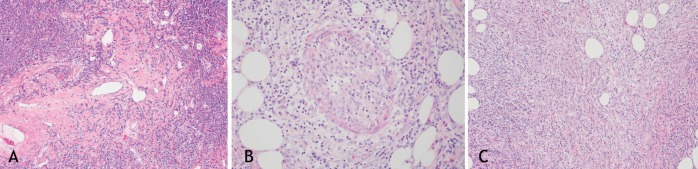
A An illustration of central sclerosis in a case of immunoglobulin (Ig)G4 disease involving the lung (hematoxylin and eosin stain, original magnification ×10). B Illustration of phlebosclerosis in a case of IgG4 disease involving the mediastinum and mesentery (hematoxylin and eosin stain, original magnification ×20). C Illustration of storiform fibrosis in a case of IgG4 disease involving the mediastinum and mesentery (hematoxylin and eosin stain, original magnification ×20)
CASE PRESENTATIONS
Case 1
A 38-year-old female nonsmoker presented to medical attention with progressive onset of dyspnea and cough productive of white sputum. Despite multiple courses of antibiotics, her symptoms progressed. Her medical history was significant for type II diabetes mellitus, microcytic anemia, hypothyroidism, xerostomia, xerophthalmia and salivary gland enlargement. Schirmer’s test was positive, suggestive of Sjogren’s syndrome, but antibodies against SSa/Ro and SSb/La antigens were negative. Bronchoscopy was unremarkable. A right heart catheterization revealed pulmonary hypertension with a mean pulmonary arterial pressure of 46 mmHg with a normal pulmonary capillary wedge pressure of 11 mmHg. She was started on bosentan and tadalafil but her dyspnea progressively increased to a modified Medical Research Council dyspnea grade of 4 (ie, ‘breathless when dressing’). She also became hypoxemic and required supplemental domiciliary oxygen. Her pulmonary function tests showed moderate airflow limitation (forced expiratory volume in 1 s [FEV1], 1.74 L [66% predicted]; and forced vital capacity [FVC], 2.5 L [82% of predicted]) and severe impairment of gas exchange (diffusing capacity for carbon monoxide [DLco] 28% of predicted). A thoracic CT scan showed subpleural bullae, and diffuse cystic and interstitial fibrotic changes (Figure 2). Serum IgG levels were elevated, as were the levels of all four subclasses of IgG. An open lung biopsy showed dense IgG4-positive plasma cells, lymphocytic infiltration and storiform fibrosis. Owing to worsening anemia (hemoglobin <70 g/L), a bone marrow biopsy was performed that showed IgG4-positive plasma cell hypertrophy. The patient was treated with four cycles of cyclophosphamide, vincristine, prednisone and rituximab, which improved her breathlessness (modified Medical Research Council dyspnea grade III, ‘stopping for breath after walking 100 yards or less’).
Figure 2).
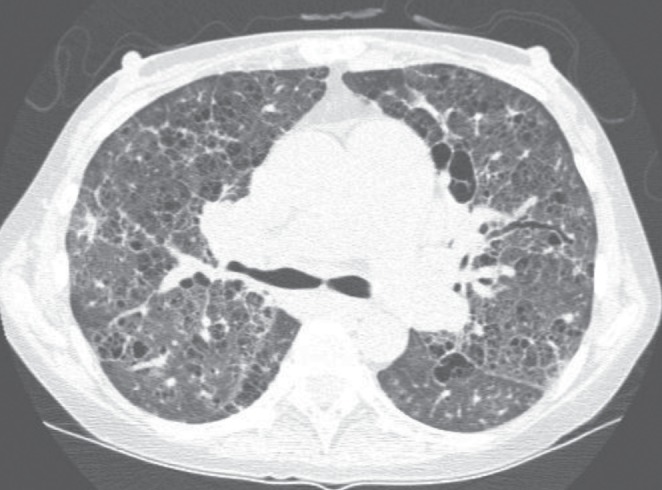
A representative section of a noncontrast computed tomography scan of the chest in patient 1. Diffuse cystic and reticular changes are apparent in addition to septal thickening throughout the pulmonary parenchyma. Bronchiectasis is also apparent
Case 2
A 56-year old woman presented with a three-month history of cough productive of white phlegm. She had no dyspnea or wheeze. She was a lifetime nonsmoker with a medical history of type II diabetes mellitus and hypercholesterolemia. Her physical examination and lung function measurements were normal (FEV1 94% of predicted; FVC 84% of predicted and DLco 94% of predicted). A plain frontal chest radiograph showed a pulmonary nodule. A CT scan of the chest demonstrated multiple pulmonary nodules (Figure 3A). Sampling of the lesion demonstrated inflammatory pseudotumour rich in plasma cells (Figure 3B). Prednisone 20 mg daily for 10 days was prescribed and resulted in marked improvement in the patient’s cough. The cough recurred following corticosteroid discontinuation, necessitating another short course of oral steroid therapy, which abated her symptoms. There has been no progression of the lesion on follow-up CT scan eight months later. Serum IgG levels were obtained before the initiation of prednisone, which showed elevated levels of IgG and IgG4 subclasses. With steroid treatment, the IgG4 subclass measurements normalized.
Figure 3).
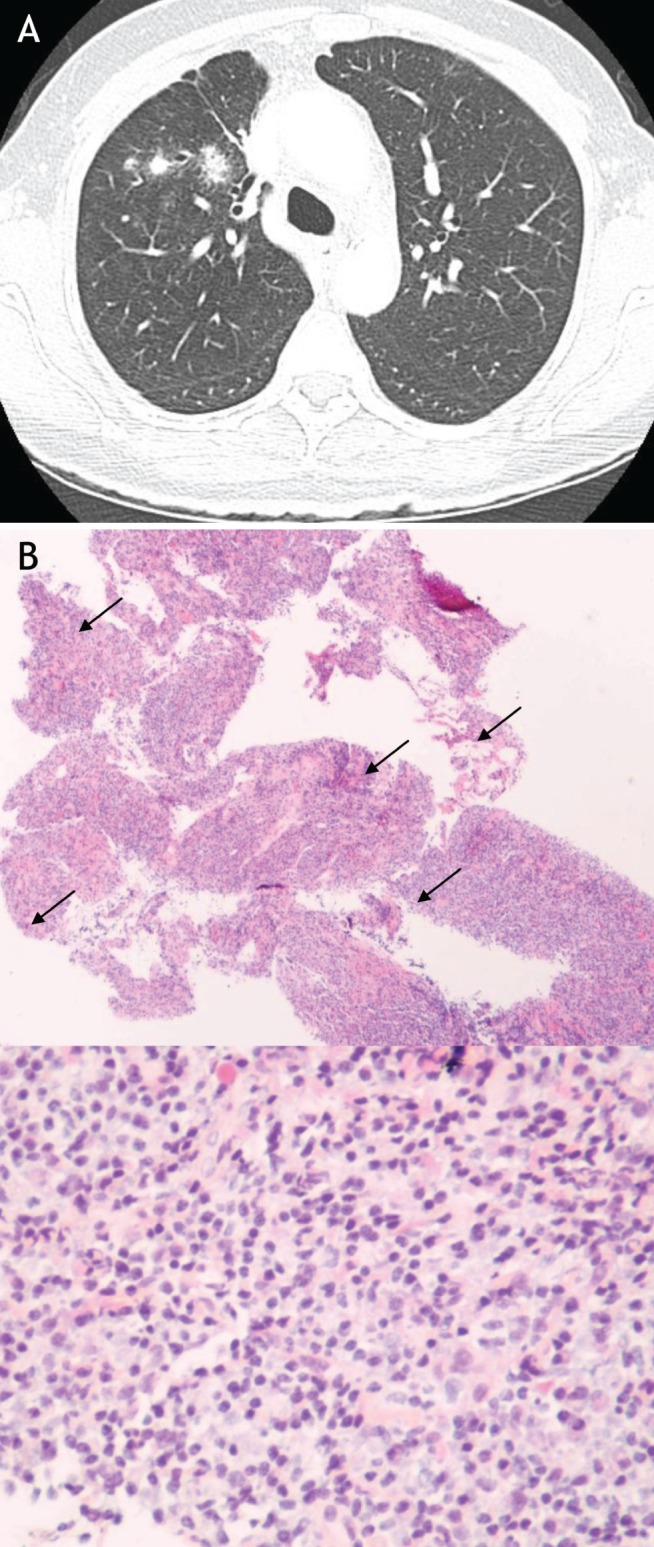
A Representative section of a thoracic computed tomography scan of patient in case 2. Multiple pulmonary nodules on original computed tomography. B Histopathology of hematoxylin and eosin-stained pulmonary biopsy specimen at low- (40×) and high-power (100×) magnification. The biopsy was taken from the largest pulmonary nodule and demonstrates the characteristic fibroinflammatory infiltrate of an inflammatory pseudotumour. The high-power section illustrates the presence of numerous plasma cells (examples indicated by arrows)
Case 3
A 62-year-old man presented with a one-year history of nasal congestion, cough, dyspnea, hearing difficulty and anosmia. He experienced recurrent infections of the upper respiratory tract. He was a non-smoker with a medical history of hypertension, hyperthyroidism treated with radioactive iodine, and left-sided spontaneous pneumothorax that did not require chest tube drainage. Physical examination revealed left cervical lymphadenopathy but no nasal obstruction or sinus tenderness. Chest radiography showed mild bilateral apical pleural thickening. Spirometry was within normal limits but a bronchoprovocation challenge was positive for hyper-reactive airways (FEV1 92% of predicted; FVC 101% of predicted; DLco 98% of predicted; a methacholine challenge test with a provocating dose producing a 20% fall of FEV1 was 0.59 mg). Due to the history of recurrent infections, an Ig panel was ordered, which showed polyclonal elevation of IgG, predominantly of the IgG4 subclass (23.4 g/L, reference range 0.05 g/L to 1.25 g/L). A biopsy of the nasopharynx showed reactive lymphoid hyperplasia. He was started on a tapering course of prednisone and his symptoms completely resolved.
DISCUSSION
In the present article, we described three patients with IgG4RLD, each presenting with a different phenotype. In case 1, our patient had symptoms, serum IgG4 levels and histopathology consistent with a definite diagnosis of IgG4RLD. In case 2, our patient had possible IgG4RLD because she had symptoms consistent with the disease as well as elevated serum IgG4 levels. Her serum IgG4 level returned to normal after initiation of oral corticosteroids. A study by Ghazale et al (8) demonstrated that serum IgG4 levels had a sensitivity of 67% to 95% in detecting this syndrome. In case 3, elevated serum IgG4 levels along with reactive lymphoid aggregates in the nasopharynx suggested a definite case of IgG4RLD with upper respiratory tract involvement. His IgG4 subclass showed marked elevation and, despite treatment with prednisone, remained elevated at 2.82 g/L (reference range 0.052 g/L to 1.25 g/L). In all three patients, corticosteroid treatments resulted in short-term therapeutic responses. The patient in case 1 was treated with a full chemotherapeutic regimen, and this, to our knowledge, is the first reported case of such treatment in a definite case of IgG4RLD.
Several pathogenic mechanisms had been proposed to explain the disparate presentation and response to corticosteroids in IgG4RD. Some have suggested that IgG4RD is a disease of autoimmunity (9). Based on human leukocyte antigen haplotypes/genotypes studies, association between autoantibodies directed against lactoferrin and carbonic anhydrase II and susceptibility to IgG4RLD have been implicated. A related theory has proposed that Helicobacter pylori infection triggers autoimmune antibody production through molecular mimicry in genetically susceptible individuals (10). Others have proposed that IgG4RLDs are a consequence of an imbalance between T-helper (Th) 2 and regulatory T (Treg) cell immune responses. Th2-mediated IgG4 production is believed to be stimulated by Treg cells under the influence of interleukin-10. A recent study indicates that Treg cells can suppress allergy in a phenomenon known as immune deviation. Normally, Treg cell response is directed against T cell epitopes of the respective antigen, suppressing antigen-specific T cell proliferation and reducing the production of Th-1- and Th-2-type cytokines. In IgG4RLD, overexpression of interleukin-10 and transforming growth factor-β have been suggested to play an important role in skewing the immune response toward a fibroproliferative state (11). A review of the literature (Table 1) identifies a number of case reports and case series that have described the many facets of this disease, although there is yet insufficient information to comment on the disease’s epidemiology. Similarly, there is a paucity of information on management and prognosis of patients affected by IgG4RLD. Because the manifestation and severity of IgG4RLD are disparate and protean, management is guided by patient symptoms and physiological impairment of organs. Typically, systemic corticosteroids are the first line of treatment. Although there is no universal consensus on the dose or the duration, experts suggest 0.5 mg/kg/day to 1 mg/kg/day for two to four weeks and then in tapering doses over three months. As illustrated by our cases, most cases of IgG4RLD are partially or totally responsive to corticosteroids, although the rates of recurrence are high following their discontinuation. In nonresponsive or frequently recurring cases, other immunosuppressives are suggested, as illustrated in case 1. However, none of these therapies, including corticosteroids, has been validated in randomized controlled trials. More recently, some have used rituximab to deplete B cells and have reported excellent response rates (1). The role of surgery is uncertain. In cases of isolated lung lesions refractory to immunosuppressive therapy, a surgical option may be considered if patients have significant symptoms and/or demonstrate significant organ impairment. Future studies will further inform our understanding of the pathogenesis, natural progression, treatment and prognosis.
TABLE 1.
Immunoglobulin G4 (IgG4)-related lung disease reported in the literature*
| Author (ref), year | Total cases, n | Mean age at Dx, years | n, sex | Clinical features | Dx findings reported | Extrapulmonary involvement | Outcome |
| Matsui et al (12), 2012 | 25 | 61 | 18 M, 7 F | Cough (n=5), dyspnea (n=1) | IgG4/IgG positive cells >40%, elevated serum IgG4 levels, | AIP (n=3), nephritis (n=3), RPF (n=1), hypophysitis (n=1) | All responded to steroids, one relapse after 3 years |
| Matsui et al (13), 2012 | 18 | 62 | 14 M, 4 F | Mediastinal lymphadenopathy, thickening of perilymphatic hilum | High serum IgG4, biopsy with abundant IgG4+ plasma cell infiltration | AIP (n=12), CSS (n=9), RPF (n=3), lacrimal gland (n=5), bile duct (n=2), renal (n=3), prostate (n=3) | Good response to corticosteroids |
| Inoue et al (5), 2009 | 13 | 70 | 9 M, 4 F | Cough (n=7), fever (n=4), dyspnea (n=2) on exertion; solid nodules (n=4); GGOs (n=2), alveolar interstitial pattern (n=2), bronchovascular lesions (n=5) | Bx of involved organs showed lymphoplasmacytic infiltration, irregular fibrosis, obliterative vasculitis | AIP (n=3), CSS (n=3), periaortitis (n=2), renal (n=3), prostate (n=1) | NR |
| Yamashita et al (14), 2008 | 3 | 72 | 3 M | Dyspnea (n=1); pleural effusion (n=1); multifocal consolidation (n=1) | Lymphoplasmacytic infiltration, prominent vascular involvement, IgG4/IgG >40% | CSS (1), lacrimal gland (1) | Resolved with steroid (n=1); resected with no recurrence; diuresed with no recurrence |
| Suzuki et al, (15), 2011 | 2 | NR | NR | NR | NR | Pericardial effusion (n=1), CSS (n=1), RPF (n=1) | Steroids effective; complicated by NTM |
| Single case reports (16–30) | 17 | 63 Others NR | 10 M, 2 F Others NR | Dry cough, dyspnea, fever; atopic asthma; lung mass | Most with biopsy proven organ involvement with lymphoplasmacytic infiltration | Ocular inflammation, acquired hemophilia A, pericarditis, lacrimal gland, CSS, renal, prostate | Prednisone generally effective |
| Total | 78 |
Search criteria: Pubmed and Google scholar. (“immunoglobulin g”[MeSH Terms] OR “immunoglobulin g”[All Fields] OR “igg4”[All Fields]) AND related[All Fields] AND (“disease”[MeSH Terms] OR “disease”[All Fields]) AND (“lung”[MeSH Terms] OR “lung”[All Fields]). AIP Autoimmune pancreatitis; Bx Biopsy; CSS Chronic sclerosing sialadenitis; Dx Diagnosis; F Female; GGO Ground-glass opacities; M Male; NR Not reported; NTM Nontuberculous mycobacterium; ref Reference; RPF Retroperitoneal fibrosis
Post-test
-
What are the characteristic histological features of IgG4RLD?
IgG4RLD is characterized by dense infiltration of IgG4-positive plasma cells and lymphocytes. In more severe cases, storiform fibrosis and obliterative phlebitis are also notable in the affected lung tissue (Figure 1).
-
How is the diagnosis of IgG4RLD classified?
Diagnosis is based on symptoms, biochemistry and histopathological findings, and is classified into definite (if patients have symptoms and biochemical and histopathology evidence), probable (if patients have symptoms and histopathology evidence only) or possible (if patients have symptoms and biochemical markers of disease only).
-
What is the standard treatment for IgG4RLD?
IgG4RLD is responsive in most cases to systemic corticosteroids. However, the disease may recur following cessation of systemic corticosteroids.
REFERENCES
- 1.Stone JH, Zen Y, Deshpande V. IgG4-related disease. New Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 2.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. New Engl J Med. 2001;344:732–8. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982–4. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 4.Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES. Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. Int J Rheumatol. 2012;23:2960. doi: 10.1155/2012/232960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue D, Zen Y, Abo H, Gabata T, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology. 2009;251:260–70. doi: 10.1148/radiol.2511080965. [DOI] [PubMed] [Google Scholar]
- 6.Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 7.Masaki Y, Kurose N, Yamamoto M, et al. Cutoff values of serum IgG4 and histopathological IgG4+ plasma cells for diagnosis of patients with IgG4-related disease. Int J Rheumatol. 2012;2012:580814. doi: 10.1155/2012/580814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghazale A, Chari ST, Smyrk TC, et al. Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol. 2007;102:1646–53. doi: 10.1111/j.1572-0241.2007.01264.x. [DOI] [PubMed] [Google Scholar]
- 9.Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology. 2000;118:573–81. doi: 10.1016/s0016-5085(00)70264-2. [DOI] [PubMed] [Google Scholar]
- 10.Kountouras J, Zavos C, Chatzopoulos D. Autoimmune pancreatitis, Helicobacter pylori infection, and apoptosis: A proposed relationship. Pancreas. 2005;30:192–3. doi: 10.1097/01.mpa.0000151576.91790.f8. [DOI] [PubMed] [Google Scholar]
- 11.Koyabu M, Uchida K, Miyoshi H, et al. Analysis of regulatory T cells and IgG4-positive plasma cells among patients of IgG4-related sclerosing cholangitis and autoimmune liver diseases. J Gastroenterol. 2010;45:732–41. doi: 10.1007/s00535-010-0199-3. [DOI] [PubMed] [Google Scholar]
- 12.Matsui S, Taki H, Shinoda K, et al. Respiratory involvement in IgG4-related Mikulicz’s disease. Mod Rheumatol. 2012;22:31–9. doi: 10.1007/s10165-011-0504-x. [DOI] [PubMed] [Google Scholar]
- 13.Matsui S, Hebisawa A, Sakai F, et al. Immunoglobulin G4-related lung disease: Clinicoradiological and pathological features. Respirology. 2013;18:480–7. doi: 10.1111/resp.12016. [DOI] [PubMed] [Google Scholar]
- 14.Yamashita K, Haga H, Kobashi Y, Miyagawa-Hayashino A, Yoshizawa A, Manabe T. Lung involvement in IgG4-related lymphoplasmacytic vasculitis and interstitial fibrosis: Report of 3 cases and review of the literature. Am J Surg Pathol. 2008;32:1620–6. doi: 10.1097/PAS.0b013e318172622f. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki N, Saeki T, Shimaoka Y, et al. [Two cases of IgG4-related disease with pleural effusion] Nihon Kokyuki Gakkai zasshi. 2011;49:97–102. [PubMed] [Google Scholar]
- 16.Vandenbos F, Benchetrit M, Tieulie N, Ambrosetti D, Chanalet S, Burel-Vandenbos F. [Diagnosis of IgG4-related systemic disease in a patient with an ocular tumor associated with lung nodules] Revue de pneumologie clinique. 2012;68:370–3. doi: 10.1016/j.pneumo.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Sugino K, Gocho K, Ishida F, et al. Acquired hemophilia A associated with IgG4-related lung disease in a patient with autoimmune pancreatitis. Intern Med. 2012;51:3151–4. doi: 10.2169/internalmedicine.51.8133. [DOI] [PubMed] [Google Scholar]
- 18.Odaka M, Mori S, Asano H, Yamashita M, Kamiya N, Morikawa T. Thoracoscopic resection for a pulmonary nodule with the infiltrate of IgG4-positive plasma cells. Asian J Endosc Surg. 2012;5:176–8. doi: 10.1111/j.1758-5910.2012.00149.x. [DOI] [PubMed] [Google Scholar]
- 19.Sekiguchi H, Horie R, Utz JP, Ryu JH. IgG4-related systemic disease presenting with lung entrapment and constrictive pericarditis. Chest. 2012;142:781–3. doi: 10.1378/chest.11-2608. [DOI] [PubMed] [Google Scholar]
- 20.Zhang YJ, Zhao JX, Liu R, Liu XY. [A case report of IgG4-related sclerosing disease with lung involvement] Beijing da xue xue bao Yi xue ban. 2012;44:311–5. [PubMed] [Google Scholar]
- 21.Chapman EM, Gown A, Mazziotta R, Churg A. Pulmonary hyalinizing granuloma with associated elevation in serum and tissue IgG4 occurring in a patient with a history of sarcoidosis. Am J Surg Pathol. 2012;36:774–8. doi: 10.1097/PAS.0b013e318248713d. [DOI] [PubMed] [Google Scholar]
- 22.Umeda M, Fujikawa K, Origuchi T, et al. A case of IgG4-related pulmonary disease with rapid improvement. Mod Rheumatol. 2012;22:919–23. doi: 10.1007/s10165-012-0600-6. [DOI] [PubMed] [Google Scholar]
- 23.Nakazawa S, Shimizu K, Nakano T, et al. An immunoglobulin G4-related disease mimicking postoperative lung cancer recurrence. Mod Rheumatol. 2012;22:787–90. doi: 10.1007/s10165-011-0580-y. [DOI] [PubMed] [Google Scholar]
- 24.Sprangers B, Lioen P, Meijers B, et al. The many faces of Merlin: IgG4-associated pulmonary-renal disease. Chest. 2011;140:791–4. doi: 10.1378/chest.10-3086. [DOI] [PubMed] [Google Scholar]
- 25.Toyoshima M, Chida K, Kono M, et al. IgG4-related lung disease in a worker occupationally exposed to asbestos. Intern Med. 2010;49:1175–8. doi: 10.2169/internalmedicine.49.3314. [DOI] [PubMed] [Google Scholar]
- 26.Fujiu K, Sakuma H, Miyamoto H, Yamaguchi B. Immunoglobulin G4-related inflammatory pseudotumor of the lung. General Thorac Cardiovasc Surg. 2010;58:144–8. doi: 10.1007/s11748-009-0491-4. [DOI] [PubMed] [Google Scholar]
- 27.Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: A clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33:1886–93. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 28.Takato H, Yasui M, Ichikawa Y, et al. Nonspecific interstitial pneumonia with abundant IgG4-positive cells infiltration, which was thought as pulmonary involvement of IgG4-related autoimmune disease. Intern Med. 2008;47:291–4. doi: 10.2169/internalmedicine.47.0411. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi H, Shimokawaji T, Kanoh S, Motoyoshi K, Aida S. IgG4-positive pulmonary disease. J Thorac Imag. 2007;22:360–2. doi: 10.1097/RTI.0b013e31813fab9f. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Sun YC, Feng RE, Liu XF, Liu GJ, Liu HG. [IgG4-related lung disease: A case report and review of the literature] Chin J Tuberculos Respir Dis. 2012;35:752–7. [PubMed] [Google Scholar]