

Abstract
Mating-type switching in the fission yeast Schizosaccharomyces pombe is initiated by a strand-specific imprint located at the mating-type (mat1) locus. We show that the imprint corresponds to a single-strand DNA break (SSB), which is site- but not sequence-specific. We identified three novel cis-acting elements, involved in the formation and stability of the SSB. One of these elements is essential for a replication fork pause next to mat1 and interacts in vivo with the Swi1 protein. Another element is essential for maintaining the SSB during cell cycle progression. These results suggest that the DNA break appears during the S-phase and is actively protected against repair. Consequently, during the following round of replication, a polar double-strand break is formed. We show that when the replication fork encounters the SSB, the leading-strand DNA polymerase is able to synthesize DNA to the edge of the SSB, creating a blunt-ended recombination intermediate.
Keywords: fission yeast, imprinting, mating type, recombination, replication
Introduction
In the fission yeast Schizosaccharomyces pombe, a stable single-strand DNA lesion triggers programmed gene conversion at the mating-type locus. This process restricts mating-type interconversion to one of the two sister chromatids during DNA replication. Following chromosome segregation at mitosis, the sister cells express two different and complementary mating-type alleles (Arcangioli and Thon, 2003). Extensive pedigree analysis at the single-cell level demonstrated that two consecutive divisions are required to produce one switched cell among four related cousins. Furthermore, the sister of the switched cell is competent for switching during the next division, forming a chain of recurrent switching (Miyata and Miyata, 1981; Egel and Eie, 1987; Klar, 1987, 1990). It was proposed that an imprinting event segregates with a specific strand of DNA at the mating-type locus (mat1) (Klar, 1987). The mating type of the cell is determined by the allele present at the mat1 locus on the right arm of chromosome II: mat1P in P cells and mat1M in M cells (Beach et al, 1982). The mat1 allele can be replaced efficiently by genetic information contained in one of the two silent donor cassettes mat2P and mat3M (Beach, 1983; Kelly et al, 1988). A noticeable feature of the mating-type loci is that they are flanked by homologous sequences (Figure 1A). The H1 homology box (59 bp) is located on the centromere-distal side of the cassettes and the H2 homology box (135 bp) on the centromere-proximal side (Kelly et al, 1988). Both sequences are thought to be essential for base pairing during the initiation and resolution steps of the gene conversion process required for mating-type switching.
Figure 1.
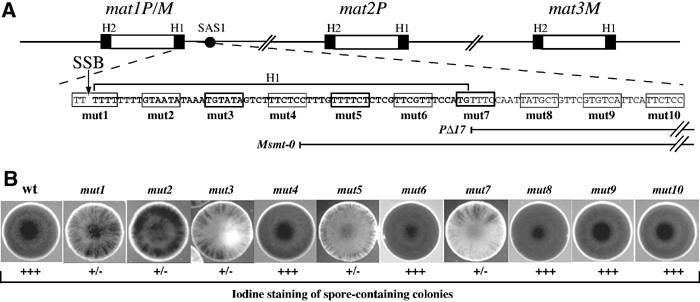
Structure of the mating-type region and phenotypes of the cis-acting mutations. (A) Schematic representation of the mating-type loci on the right arm of chromosome II. The black boxes indicate the H1 and H2 homology sequences and the SAS1 element is shown as a black circle. The H1 sequence is in bold and an arrow indicates the position of the SSB. The positions of the PstI (CTGCAG) substitutions are indicated by rectangles and numbered (mut1–mut10) and the Msmt-0 and PΔ17 deletions are shown. (B) The switching efficiencies for the mutant strains (mut1 to mut10) were analyzed by exposing the colonies to iodine vapors. The PstI substitutions at positions 1, 2, 3, 5 and 7 reduce (+/−) the switching efficiencies.
Recent molecular studies suggest that the imprint at mat1 is either a single-strand DNA break (Arcangioli, 1998) or an alkali-labile DNA modification (Dalgaard and Klar, 1999). The discovery of this novel type of single-strand DNA lesion (called SSB) has triggered new investigations of the mating-type interconversion process. The SSB was mapped to the upper strand at the junction of the mat1 allele and the H1 homology box. This SSB fulfills all of the imprinting criteria described previously for mating-type switching (Crouse, 1960; Klar, 1987). Interestingly, the position of the break at mat1 differs by three nucleotides between the mat1P and mat1M alleles (Nielsen and Egel, 1989; Arcangioli, 1998). The enzyme or process involved in the SSB formation at mat1 is unknown. The SSB is stable throughout the entire length of the cell cycle and is transiently converted to a polar double-strand break (DSB) during the S-phase. The DSB appears on the distal side of mat1, indicating that the replication fork is approaching mat1 from the H1 side (Arcangioli, 1998). Consequently, it was proposed that the leading-strand replication complex is stalled at or near the SSB. The broken chromatid can be healed by a gene-conversion event, initiated at the H1 homology sequence also present at the opposite donor loci. Once DNA repair synthesis starts, it proceeds into the silent locus ending after the H2 homology region (Arcangioli and de Lahondes, 2000). Several recent experiments indicate that the polarity of replication at mat1 is achieved by a replication termination site (RTS1) located about 700 bp centromere-proximal to the H2 box, thus forcing the locus to be replicated from the H1 side, a necessary condition for SSB formation. Another element called MPS1 (for mat1 pause site I) was also described around the H1 box as important in SSB pathway formation (Dalgaard and Klar, 1999, 2000). We showed that the SSB is reformed at every generation on the newly synthesized upper DNA strand (Arcangioli, 2000). Taken together, these experiments demonstrate that following DNA replication, the SSB is found on the upper, neo-synthesized lagging strand.
Functional studies of the region centromere-distal to mat1 revealed the presence of two cis-acting elements, SAS1 and SAS2 (for switching-activating sites), located 140 and 60 bp from the break at mat1 (Arcangioli and Klar, 1991). The switching-activating protein Sap1 interacts with SAS1 (Arcangioli et al, 1994) and is essential for growth, independently of mating-type switching, and participates in chromosome morphogenesis (de Lahondès et al, 2003). Individual deletions of SAS1 or SAS2 reduced the level of the break and double deletions abolished break formation. Similar deletions in M or P strains have been named Msmt-0 or PΔ17, respectively (see Figure 1; Arcangioli and Klar, 1991; Styrkarsdottir et al, 1993). Three trans-acting gene products (Swi1p, Swi3p and Swi7p) were identified as necessary for the initial formation of the lesion, since their mutations cause a reduced steady-state level of the SSB. Swi1 (Egel et al, 1984) has sequence identity in many species and was named topoisomerase 1-associated factor 1 (Tof1) in Saccharomyces cerevisiae, where it was shown to regulate DNA damage responses during the S-phase (Park and Sternglanz, 1999; Dalgaard and Klar, 2000; Foss, 2001). swi7 encodes the catalytic subunit of the DNA polymerase α (Singh and Klar, 1993). The replication-pausing activities of both RTS1 and MPS1 are strongly reduced in swi1 or swi3 mutants, whereas the activity of at least MPS1 is not affected in swi7 or Msmt-0 mutants. It was therefore proposed that swi1 and swi3 act upstream of swi7 and the cis-acing SAS1 and SAS2 elements in SSB pathway formation (Dalgaard and Klar, 2000, 2001).
To advance our understanding of the process of SSB formation and stability, we undertook a functional analysis of the DNA sequence at the SSB site and within its vicinity. To preserve the distances between already known and novel elements, we introduced substitution mutations. Several new sequences located within the H1 homology box were characterized as essential for the SSB steady-state level, and consequently for mating-type switching. Using several molecular approaches (two-dimensional (2D) gel electrophoresis, genomic sequencing and chromatin immunoprecipitation), we were able to ascribe specific activities to these novel elements and propose a model for SSB formation and maintenance. Significantly, we found that during DNA replication, the leading-strand DNA polymerase synthesizes DNA until the last 5′ nucleotide of the broken DNA, forming a blunt-ended recombination intermediate.
Results
Identification of novel cis-acting elements necessary for efficient mating-type switching
Systematic linker-scanning mutagenesis was performed within the H1 sequence distal to mat1. Each mutation consists of a 6 bp PstI substitution located within every 10 bp, starting from the SSB and extending 100 bp from the break site, and are named mut1–mut10 (Figure 1A). As the mutated DNA fragments contain the mat1P allele, following homologous recombination, each mutant initially expresses the P mating type. The mutants were first analyzed by the iodine-staining assay, an indirect measure of the mating-type switching efficiency. The starch reaction with iodine vapors stains spore-containing colonies black, whereas slow-switching mutants exhibit streaky iodine-staining patterns and colonies unable to switch the mating type appear yellowish. The position of the mutations (mut1–mut10) and their effects on iodine staining are shown in Figures 1A and B. None of the mutations completely abolished mating-type switching. Five mutations (mut1, mut2, mut3, mut5 and mut7) exhibit variable streaky staining, indicative of reduced switching efficiencies, whereas the five other mutations appear to have no effect (mut4, mut6, mut8, mut9 and mut10). The mut7 mutation pinpoints the cis-acting SAS2 element, grossly mapped by deletional analysis (Arcangioli and Klar, 1991). Interestingly, the first three mutations (mut1, mut2 and mut3), upon re-streaking, produce a noticeable proportion of black colonies by iodine staining. As this staining was stable and similar to wild-type levels, we hypothesized that these were spontaneous revertants of the mutation. To quantify the rate of reversion, we counted the percentage of colonies that were half black and half streaky. As shown in Figure 2A, the efficiency of reversion decreased as a function of distance from the DNA lesion (mut1>mut2>mut3). The other mutations were also tested and showed no detectable reversion of the PstI site (data not shown). To demonstrate that reversion indeed occurred, we prepared DNA from two independent clones from each mutant and amplified the DNA sequence flanking the SSB, using primers specific for the P or M alleles at mat1 (Figure 2B). Amplified DNA fragments were assayed for the presence of the PstI mutation by enzymatic digestion. From each of the three mutants, we isolated DNA resistant to PstI digestion, which correlated with the rate of reversion obtained by iodine staining (mut1>mut2>mut3), and DNA sequences were identical to the wild type. Interestingly, mut1 never gave rise to PstI at the M allele (see Discussion).
Figure 2.
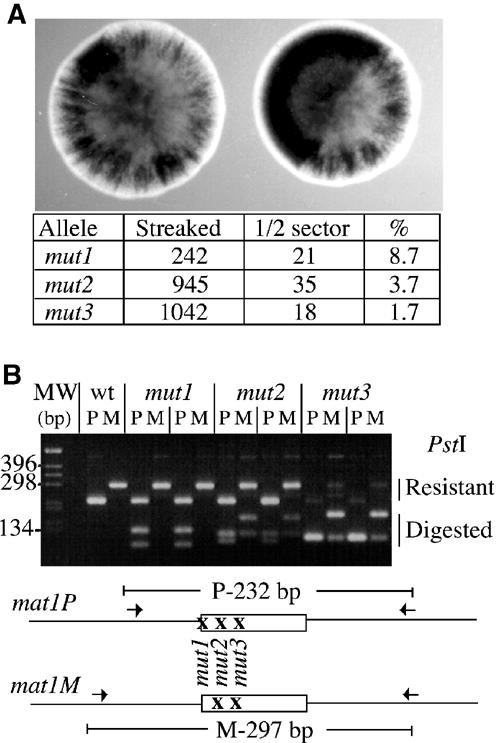
Reversion of the mut1, mut2 and mut3 substitutions. (A) Reversion rates for strains (mut1, mut2 and mut3) per cell division were calculated from the number of half-black sectors per streaky colonies. (B) DNA from two independent streaky colonies in each strain was isolated, and amplified with two different sets of primers specific for mat1P (P) or mat1M (M), and analyzed for the presence of the PstI site. The mat1M and mat1P PCR products from wild-type DNA are resistant to PstI. Upon growth, the mut1 mutant gives rise to a mat1M allele, completely resistant to PstI digestion. The mut2 and mut3 give rise to a mixed population of resistant and digested DNA for both P and M alleles. The differential migration of the PstI-digested PCR products is due to the position of the PstI sites.
Effects of the mutations on the SSB steady-state levels and position
As the reduced mat1 switching efficiency can be due to reduced SSB formation, we analyzed the level of the break for each mutant strain. Genomic DNA from each strain was prepared using the traditional DNA extraction procedure, which converts the SSB to a DSB by shearing the fragile site (Arcangioli, 1998). We observed the three mat1 HindIII DNA fragments of 12.6 kbp, together with the two sheared products mat1-distal and mat1-proximal of 7.2 and 5.4 kbp, respectively (Figure 3A). Strains mut3, mut5 and mut7 exhibit reduced levels of the mat1 cut fragments, indicating reduced steady-state levels of the break, consistent with reduced switching efficiencies. However, mut1 and mut2 mutations, which exhibit a mild reduction of the switching phenotype, showed wild-type levels of the break. Importantly, when the genomic DNA was subsequently digested with PstI, we never observed transfer of the mutated sequence into the silent mat2P and mat3M loci (data not shown). The phenotypes observed (iodine staining and break level) were not due to the insertion of the LEU2 gene, since several mutations were introduced without LEU2 to confirm the mutant phenotypes (Arcangioli and Klar (1991) and data not shown). We used the genomic sequencing methodology to map the position of the SSB in each mutant. Genomic DNA was prepared from wild-type and mutant (mut3, mut5 and mut7) strains using low-melting agarose plugs, which protect the fragile site from shearing (Arcangioli, 1998). For each mutant, the SSB is present on the upper strand, and in the same position as the wild-type break, but at reduced levels (Figure 3B). As previously shown, two mat1-distal upper DNA strands, of 233 and 236 nucleotides, were observed, corresponding to the SSB at mat1M and mat1P, respectively (Nielsen and Egel, 1989; Arcangioli, 1998). To avoid the high rate of reversion observed in mut1, we used a stable mat1M strain with a deletion of the mat2P and mat3M silent cassettes (strain SP714). This strain is viable and contains wild-type levels of the SSB (Klar and Miglio, 1986). The same PstI mutation, as previously used for the P allele (mut1), was introduced into the M allele, in this stable donorless background (named mut1-3). Figure 3C shows that mut1-3 exhibits a SSB on the upper strand, at the same position and level as compared to the parental SP714 strain. From this result, we conclude that the SSB is site-specific and sequence independent.
Figure 3.
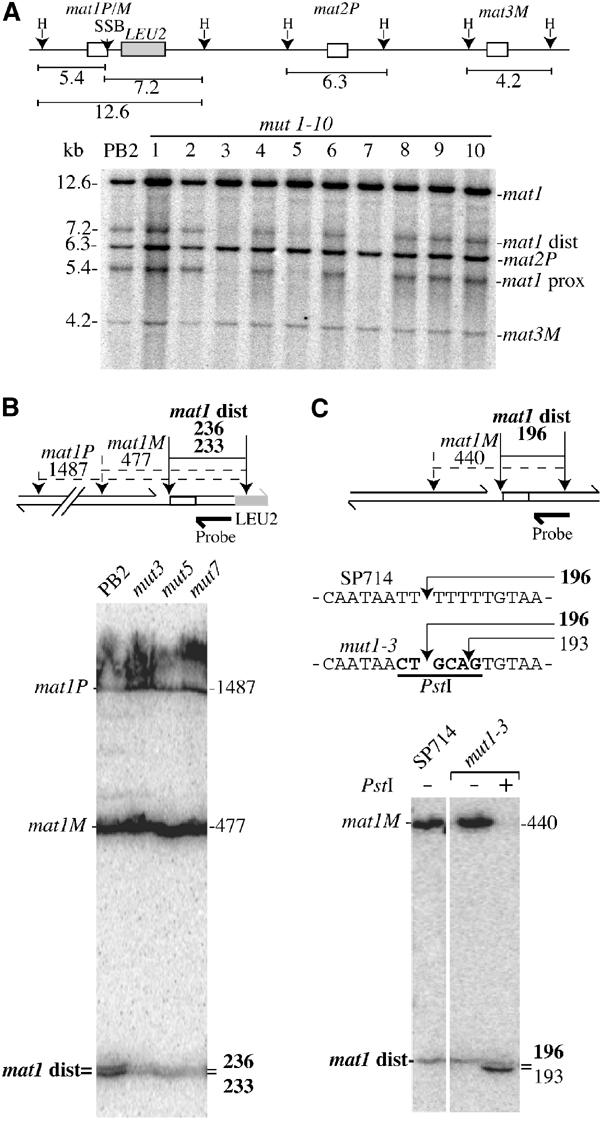
Level and position of the SSB in mutant strains. (A) Upper panel: Schematic representation of the mating-type region and size (in kbp) of the HindIII (H) DNA fragments. Genomic DNA from the wild-type (PB2) and each mutant strain (mut1 to mut10) was prepared by the classical method, digested by HindIII and analyzed by Southern blot (lower panel). The probe used is a mat1P HindIII radiolabeled fragment. The SSB present in 20–30% of mat1 molecules is transformed to a DSB during DNA purification. Therefore, the 12.6 kbp mat1 fragment is broken into two subfragments (mat1-proximal, 5.4 kbp, and mat1-distal, 7.2 kbp). The probe also recognizes the mat2P (6.3 kbp) and mat3M (4.2 kbp) HindIII DNA fragments. (B) Upper panel: Positions and distances for the SSB (arrow) and the SspI restriction site polymorphisms (dotted arrows) in the mat1P and M loci. Note the position of the LEU2 gene, which displaces the SspI site by 37 nucleotides. The probe used is a mat1-distal single-stranded radiolabeled fragment. The sizes and names of the DNA fragments are shown. Lower panel: Genomic sequencing analysis of the SSB in wild-type (PB2), mut3, mut5 and mut7 strains. The sizes and names of the labeled DNA fragments are indicated. (C) Position of the SSB in the stable mat1M donorless strains, SP714 and mut1-3 (absence of LEU2). Upper panel: Positions and distances between the SSB (arrow) and the SspI sites (dotted arrows), located on both sides of the SSB. Sequences of the upper strand containing the SSB in the wild-type strain (SP714), and in the mutant strain mut1-3, containing the PstI substitution (in bold), are indicated. Lower panel: Genomic DNA was prepared, digested with SspI and analyzed as above, with the sizes and names of the DNA fragments indicated. Upon PstI digestion of the mutant DNA, the mat1M DNA fragment (440 nucleotides) was cleaved, liberating a 193 DNA fragment (no SSB and cut by PstI), which migrates just below the wild-type 196 mat1-distal fragment (with the SSB and not cut by PstI).
mut3 reduced Swi1p interaction at mat1 and the replication pause at MPS1
The replication fork pause MPS1 appears to be a prerequisite for SSB formation and was reported to be fully active in the Msmt-0 mutant strain, containing a 262 bp deletion, that removes all of the cis-acting elements starting from the mut4 position and extends beyond SAS1 (Figure 1A). We investigated whether the mut3 mutation affects the MPS1 pause site by 2D gel electrophoresis (Brewer and Fangman, 1988). mut3 exhibits a reduced pausing efficiency compared to the wild-type and the two other mutant strains mut5 and mut7 (Figure 4A). As swi1 was previously shown to be important for MPS1 function, we tested whether Swi1p interacts in vivo with this DNA element by chromatin immunoprecipitation (ChrIP). We constructed a strain containing an HA tag of Swi1 (AHR1, see Materials and methods). This strain, which exhibits wild-type levels of mating-type switching, SSB formation and MPS1 activity (data not shown), was combined with mut3. Primer pairs flanking mat1 were used for quantitative real-time PCR (Figure 4B) and semiquantitative PCR analyzed by agarose gel electrophoresis (data not shown) of DNA prepared from HA-immunoprecipitated material. The H1 sequence containing the MPS1 element was strongly associated with Swi1 and this interaction was almost abolished in the mut3-containing strain (Figure 4B). Collectively, these data indicate that Swi1 interacts physically (directly or indirectly) with the sequence mutated at mut3 and that this interaction is required for DNA-replication pausing. A weaker Swi1 interaction was observed with the other side of mat1, containing the RTS1 element in the wild-type background. However, the strain containing the mut3 mutation, about 2 kbp away from RTS1, enhances the Swi1 interaction with RTS1 in a reproducible manner (Figure 4B and Materials and methods). This result suggests that Swi1 interacts with these two replication fork pause elements in a mutually exclusive manner.
Figure 4.
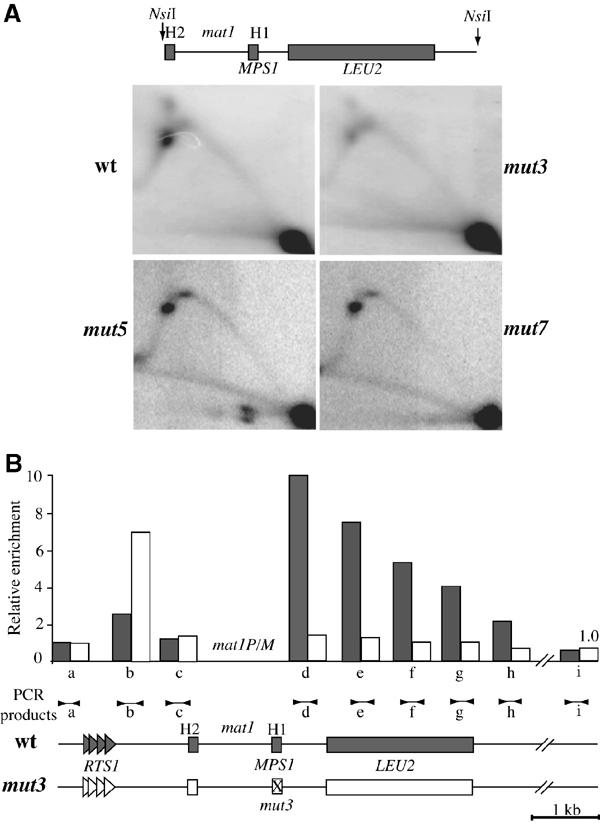
mut3 strongly reduces MPS1 activity and Swi1 interaction. (A) Analysis of replication intermediates in the wild-type, mut3, mut5 and mut7 strains by 2D gel electrophoresis. Schematic representation of the mat1-NsiI restriction fragment. The quantification of the hybridization signals indicates that the MPS1 spot is 10-fold lower for mut3 (data not shown). (B) Chromatin immunoprecipitation analysis of Swi1p with the flanking mat1 sequences. The lower panel shows the wild-type (AHR1) and mut3-containing strain (AHR2) in gray and white, respectively. The positions of RTS1 and MPS1 are indicated on both sides of mat1. Note that mat1 was not analyzed since the same sequences are present at the two donor loci. Genomic DNA from wt (AHR1) and mut3 (AHR2) strains was isolated from crude extracts and the anti-HA (Swi1) immunoprecipitated chromatin was assayed by quantitative PCR (Materials and methods). Each sample was assayed in triplicate, giving a standard deviation between 5 and 10% for the quantitation of the DNA sample. The positions of PCR products (a–i) are shown above the schematic representation of the mat1 region. Following quantification, the relative enrichment for each PCR product was plotted, with the highest enrichment set at 10. The gray and white bars in the histogram indicate wt and mut3, respectively.
Mut7 is essential to maintain the SSB during cell cycle progression
The high level and stability of the SSB suggests that the break is actively protected against repair during cell cycle progression and that its recombinational repair occurs within the same phase as its formation, namely the S-phase (Arcangioli 1998). To elucidate whether mut3, mut5 and mut7 substitutions affect the formation or maintenance of the SSB, we followed the level of the break during cell cycle progression. Small G2 cells were enriched from an asynchronous cell population, by lactose gradient centrifugation, and transferred to fresh media allowing cells to continue cycling in a synchronized fashion (Fantes and Nurse, 1978). Cell samples from the synchronized population were taken at 20 min intervals, and genomic DNA was prepared and analyzed by genomic sequencing. Figure 5A compares SSB levels among the wild-type and mutant strains during cell cycle progression. Like wild type, mut3 exhibits constant levels of the SSB in all phases of the cell cycle, albeit with lower levels (Figure 5C). We obtained a similar result with a swi1-deleted strain (data not shown). In contrast, mut7 shows variable levels of the SSB, which peaked at the 100 min time point and then diminished. This peak overlaps with the first replication period (S1), as measured by septation (Figures 5B and C). A second increase of the SSB was observed at the beginning of the second septation peak (200 min, at S2). A modest increase of the SSB level was also observed for mut5 during the S-phase. A block and release experiment using hydroxyurea (HU), an inhibitor of ribonucleotide reductase, gives similar results (data not shown). These data indicate that the mut7 mutation reveals an important cis-acting element (SAS2) necessary for maintaining the SSB during cell cycle progression, and strongly support the hypothesis that the SSB is formed during DNA replication.
Figure 5.
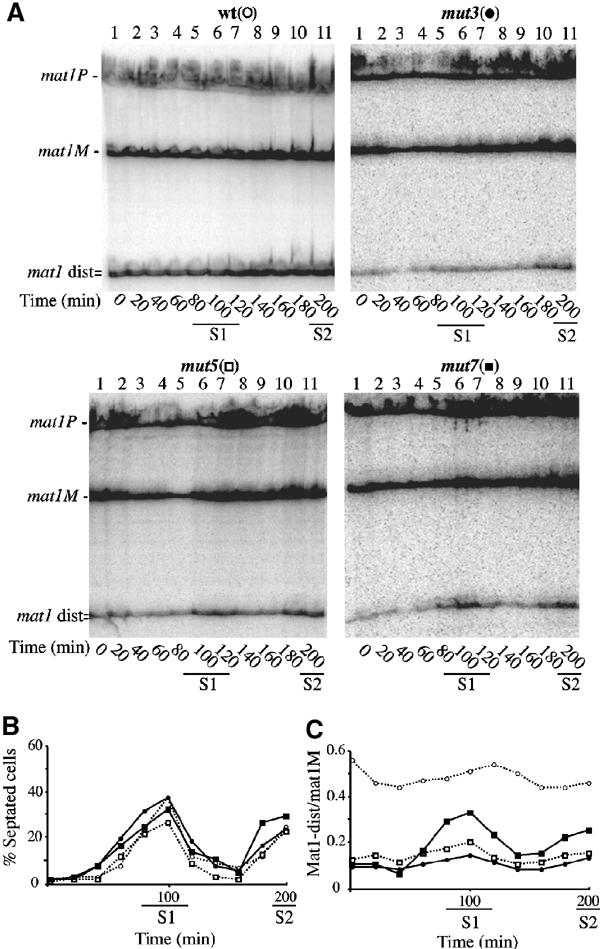
Stability of the SSB during cell cycle progression. Wild-type (○), mut3 (•), mut5 (□) and mut7 (▪) strains were synchronized by lactose gradients (enriching for small G2 cells) and samples from each culture were taken every 20 min. (A) Purified DNA was analyzed by genomic sequencing as in Figure 3B. (B) Cell cycle progression for each culture was followed by measuring the proportion of septated cells (as well as cell density, not shown) as a function of time for almost two generations (same strain symbols as used in (A)). Septum formation coincides with DNA replication period (indicated by S1 and S2) in S. pombe. (C) The intensity of the mat1M and mat1-distal bands was quantified using a PhosphoImager and the relative intensity of mat1-distal/mat1M, for each time point, was plotted (same strain symbols as used in (A)). The mat1-distal DNA fragment varied slightly during cell cycle progression for the wild-type, mut3 and mut5 strains, but peaked for mut7 around the 100 and 200 min time points (same symbols). These results are representative of three independent experiments.
Leading-strand polymerase synthesizes DNA to the edge of the SSB 5′ end
Maintaining the SSB at mat1 is thought to be necessary for DSB formation during the next round of replication. However, the molecular nature of the broken-end intermediate depends on the ability of the leading-strand machinery to synthesize DNA to the 5′ edge of the DNA template. To enrich for replication intermediates, we used a strain containing the cdc25-22 conditional mutant allele to synchronize the cell population. To reduce rapid disappearance of the 3′ end of the leading strand during gene conversion, the strain used was also donorless and mat1M stable. Cell samples from the synchronized population were taken at various intervals and genomic DNA was analyzed by genomic sequencing to map the position of the 3′ end of the leading strand stalled at mat1 (Figure 6A). Figure 6B shows the position and kinetics of the appearance and disappearance of a novel band overlapping the S-phase, peaking at the 70–90 min time points. Another band was present in all lanes and corresponds to the upper strand containing the SSB (data not shown). To estimate the size of the transitory band, we used the presence of the PstI sites in the mut1-3 and mut1-4 strains (mat1M, donorless background) as size markers (Figure 6F, right panel). Upon PstI digestion, mut1-4 and mut1-3 produce lower-strand DNA fragments, of wild-type sequence, with a one-nucleotide resolution between them (196 and 197 nucleotides, respectively). If the leading DNA polymerase stalls at the 5′ end of the mat1M cut template, it will produce a fragment of 196 nucleotides, migrating at the same position as the mut1-4 fragment. Figure 6B shows that the leading-strand DNA intermediate co-migrates with the mut1-4 size marker.
Figure 6.
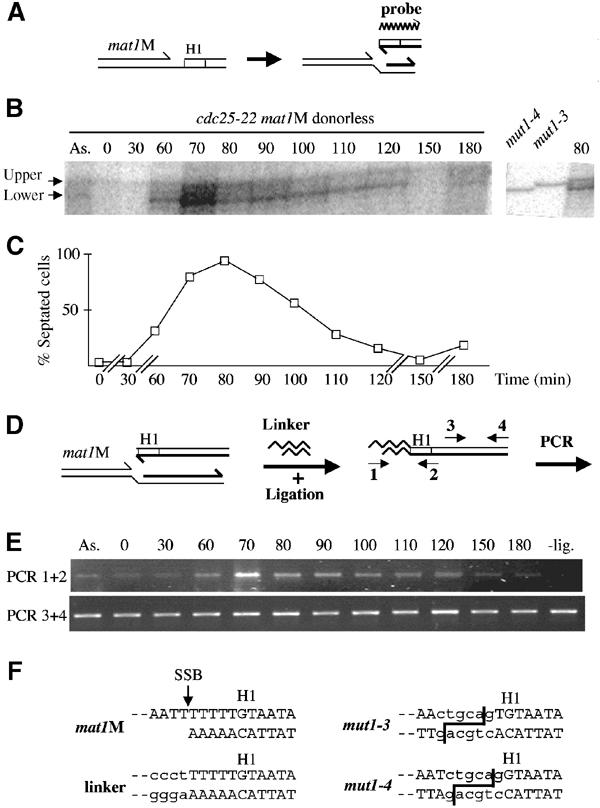
Mapping of the stalled leading-strand 3′ end at mat1. (A) Schematic representation of the stalled leading strand at mat1 and polarity of the probe. (B) Autoradiogram of purified genomic DNA from a cdc25-22 mat1M donorless synchronized cell culture, digested with SspI and analyzed on a genomic sequencing gel. Left panel: Kinetics of the stalled leading strand. ‘As.' represents the asynchronous cell population and the time (min) after release from the temperature block is indicated. The transitory band is labeled as ‘lower' and the constant band as ‘upper'. This upper band is due to low contamination of the labeled probe by the template used in the labeling reaction (data not shown). Right panel: Migration of the mat1M-distal lower strand of mut1-3 and mut1-4 digested by SspI and PstI, together with the 80 min time point. The position of the PstI sites are shown in (F), right panel. (C) Cell cycle progression was followed by measuring the proportion of septated cells (and cell density, not shown) as a function of time for almost two generations. Time is indicated in minutes (min). (D) Schematic representation of the LM-PCR method. Two pairs of primers were used: (1–2) and (3–4). (E) PCR products using primers (1–2) or (3–4) were amplified for 32 or 26 cycles, respectively, and analyzed on agarose gels. The time is indicated in minutes after temperature block and release. A ‘- lig' control experiment was also performed using the 70-min time point in the absence of ligase. (F) Sequence of the SSB in a mat1M donorless strain. The left panel shows the identical sequence obtained from eight independent clones of the LM-PCR products (using primers 1–2). An arrow indicates the position of the SSB and the linker sequence is written in lower-case letters. Right panel: PstI sequences are indicated in lower-case letters and the cleavage sites are shown for mut1-3 and mut1-4 mutations.
To validate the formation of a transitory blunt end, we performed ligation-mediated PCR (LM-PCR). The genomic DNA was ligated with phosphorylated, blunt-ended linkers. The ligation efficiency was analyzed by PCR, using nested PCR primers (1–2) to increase the specificity of the amplification and mat1-distal primers (3–4) as a control (Figure 6D). As anticipated from the previous result, PCR products were observed from the DNA isolated during the DNA-replication period, concomitant with the presence of the stalled nascent leading strand (Figure 6E). The PCR product was cloned and sequenced. Figure 6F (left panel) shows the sequences of the amplified DNA, demonstrating the conserved and blunt-ended nature of the recombination intermediate.
Discussion
Several novel cis-acting elements in the distal mat1 sequence have been identified. These elements indicate that several molecular steps are required for efficient SSB formation and maintenance for mating-type switching (Figure 7).
Figure 7.
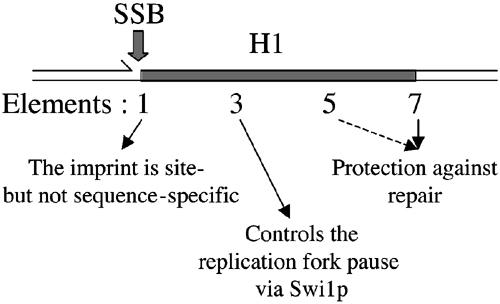
Role of the cis-acting elements in the H1 sequence. The positions of the SSB and the cis-acting elements are shown and their different roles in SSB formation and maintenance are indicated.
SSB is site- but not sequence-specific
We found that our PstI substitutions at the SSB site (mut1) did not change the position, or the efficiency of break formation, indicating distance-dependent and sequence-independent regulation of the SSB position. As compared to mat1M, the break at mat1P is shifted by three nucleotides (Nielsen and Egel, 1989), but the sequences to the right-hand side, containing H1, are identical for both mat1P and mat1M. Therefore, we should also reconsider the importance of sequence information within the P or M alleles. Future work is needed to understand how the position of the break is determined.
Replication fork progresses to the edge of the SSB 5′ end
Due to the presence of the mat1P DNA sequence in the initial DNA fragment used for homologous recombination, we propose that the removal of the PstI mutation for mut1 in the M allele is an obligatory step for initiating gene conversion. This proposal, together with leading-strand progression to the end of the break site, indicates that four heterologous nucleotides (within the PstI sequence) are made upon strand invasion with the donor. These few bases might be removed soon after strand invasion by editing functions (polymerase or flap endonuclease activities), to allow priming of repair synthesis during gene conversion (Paques and Haber, 1997; Palmer et al, 2003). An additional argument for the 3′ end running to the edge of the SSB site comes from the (partial) persistence of the two distal PstI substitutions (mut2 and mut3) at mat1M upon switching. In these two mutants, only eight or 18 nucleotides within the 3′ end of the invading strand are perfectly homologous with the donor information. Removal of these heterologous sequences might require mismatch and/or excision repair factors, as shown in S. cerevisiae (Saparbaev et al, 1996; Sugawara et al, 1997). The efficient loss of the PstI mutations, within a 20 bp window (mut1–mut3), followed a gradient, suggesting that the repair might be functionally linked to the initiation of DNA synthesis during gene conversion. This indicates that only 20 bp of perfect homology is needed for strand invasion at mat1.
To our knowledge, this is the first in vivo demonstration of a leading-strand polymerase reaching a DNA end. This is a recurrent question regarding telomere replication at native ends of linear chromosomes. It is thought that leading-strand synthesis continues to the very end of the template molecule, but this was never formally demonstrated. The ability of the replication machinery to synthesize DNA to the very last nucleotide of a broken template seems a safe attempt to minimize the loss of DNA information. This raises important questions regarding how a replication fork (helicases or polymerases) senses and responds to oncoming damage. For example, is the entire replication fork maintained or collapsed before mat1 switching? Is lagging and leading-strand synthesis transiently uncoupled, allowing the lagging-strand synthesis to proceed further, as recently proposed in E. coli (Pages and Fuchs, 2003)? Perhaps the novel cis-acting elements described here prevent the replication fork from collapsing, and/or assist in recruiting the homologous recombination machinery for gene conversion during mating-type switching.
Identification of the mat1 replication pause site
Previous work has shown that the mat1 pause site 1 (MPS1) activity requires the function of both swi1 and swi3, and might be one of the early steps in mating-type switching (Dalgaard and Klar, 2000). Significantly, we found that Swi1 interacts, directly or indirectly, with a short sequence present in H1 and that the mut3 mutation almost abolishes this interaction. Furthermore, the mut3 site is necessary for MPS1 function, whereas the two other elements defined by mut5 and mut7 play a minor role (Figure 4A). The pause acts upstream of the SSB formation, and protection or maintenance functions, revealed for the first time by mut5 and mut7, act downstream of the SSB formation. Consequently, Swi1 is more likely involved in the formation and not protection of the SSB (see below). It was not possible to determine whether the mut3 site is sufficient for Swi1 function, since the protein is also required for the mat1-proximal replication-termination function (RTS1), consistent with the cumulative effect of iodine staining for the double-mutant strain mut3 Δswi1 (data not shown). It is noteworthy that the modest Swi1 interaction observed with RTS1 in the wild-type background seems stimulated in the mut3-containing strain. Previous observations indicated that the close proximity of MPS1 and RTS1 interferes with their functions (Dalgaard and Klar, 2001), but the role of RTS1 was only described in an Msmt-0 mutant strain or on a plasmid, where SSB formation and switching are prevented. We imagine that the two replication fork barriers (RFBs), on either side of mat1, communicate with each other, such that Swi1 can interact with only one of the two sites, or that Swi1 interacts with both elements during the S-phase, but this interaction remains stable at MPS1 and is only transitory during the S-phase at RTS1.
Finally, it was proposed that RTS1 optimizes imprinting by controlling the direction of replication at mat1 (Dalgaard and Klar, 2000). An additional function for RTS1 might be to block any fork of replication running from the centromere-proximal side of mat1, especially during the mating-type switching process, which lasts about 30 min (at 24°C) and overlaps the replication period (Arcangioli and de Lahondes, 2000). This model is consistent with the proposed function of S. cerevisiae Tof1 (Swi1 in S. pombe) in the Mec1 (Rad3/ATR in S. pombe/humans) pathway, which is necessary to activate and maintain the S/M checkpoint (Foss, 2001). Recently, Tof1 was found to interact physically in vivo, with the replication machinery and stalled forks, using a global approach (Katou et al, 2003). Yet, we still do not know when and how Swi1 is recruited to the mut3 site in H1 and whether Swi1 is required to stop replication progression and/or to stabilize the stalled fork at mat1.
A model for chromosomal imprinting
A central question regarding programmed DNA rearrangements is how the recombination machinery is targeted to specific chromatin sites at the right time (Haber, 1998; Gellert, 2002). All of the double cis-acting mutant combinations tested exhibited a synergistic mating-type switching reduction, but never abolished switching, as observed by iodine staining (data not shown). This reinforces the idea that these cis-acting sites participate in concert for the initial process of mating-type switching. This feature is reminiscent of the ordered recruitment of transcription factors, allowing site-specific transcriptional initiation (Stargell and Struhl, 1996), in which some factors are able to first interact with DNA sequences in the presence of nucleosomes, thus permitting subsequent assembly of an enhanceosome (Merika and Thanos, 2001). The affinity of transcription regulators to specific DNA sequences is not sufficient, and only concerted binding leads to active enhanceosome assembly. To extend the previous imprinting model (Dalgaard and Klar, 2000), perhaps the Swi1-dependent replication fork pause allows Sap1 (de Lahondès et al, 2003) to interact with the SAS1 element, and this in turn facilitates further assembly of several, as yet, unidentified regulatory proteins to other cis-acting elements (mut5 and mut7 sites). This particular DNA–protein assembly might offer a multifunctional surface providing ordered, efficient and concerted DNA break formation on the neo-synthesized lagging strand, with the end result being that, during the S-phase, only one of the two sister chromatids is cleaved. Analysis of the chromatin nature at mat1 will allow us to support this model. The apparent active protection of the SSB, revealed by the mut7 mutation, suggested that the imprinting complex is a stable chromosomal structure of mat1, masking the SSB from repair. Consequently, during the subsequent DNA-replication period, two major events could be envisioned. The first event, associated with the leading-strand machinery, creates the blunt-ended DNA intermediate, channeling the homologous recombination machinery into mating-type switching. The second event, associated with the lagging-strand machinery, could reproduce the entire process of imprinting complex assembly, including SSB formation and its subsequent protection. This hypothetical scenario is proposed to explain the asymmetric pattern of mating-type switching in cell lineages of fission yeast. Finally, our results also raise interesting questions concerning how single-stranded lesions are maintained/protected and how repair occurs within different genomic regions/contexts.
The replication/recombination connection
The connection between replication fork progression and recombination is accumulating (Rothstein et al, 2000). It was traditionally proposed that both programmed and accidental pausing might lead to the formation of DSBs. The results described above suggest that DNA-replication fork pauses and single-strand DNA lesions are tightly associated. Perhaps other programmed recombination events and systems possess a similar association. For example, DNA lesions have been detected in cell lines competent for somatic hypermutation or class switch recombination of immunoglobulin genes. These lesions are reported to be single stranded or double stranded, during the late S-phase (Papavasiliou and Schatz, 2000; Kong and Maizels, 2001; Haber, 2001). Another example is the RFB, found in the rDNA repeats of all eukaryotic organisms. In S. cerevisiae, the RFB has been associated with recombination hotspot activity and single-strand DNA lesions (Keil and Roeder, 1984; Brewer and Fangman, 1988; Weitao et al, 2003). Fragile chromosomal sites in higher eukaryotes are also induced by conditions that partially inhibit DNA replication. Furthermore, it was proposed that stalled replication forks, escaping the ATR replication checkpoint, induce common fragile-site expression (Casper et al, 2002), which may directly contribute to chromosome instability and tumor cell biology (Richards, 2001). Similarly, Mec1, the homolog of mammalian ATR in S. cerevisiae, prevents chromosomal breakage in replication-slow zones (Cha and Kleckner, 2002). Clearly, the progression of the replication fork along the DNA must translate the information required for chromosomal plasticity in a wide variety of biological systems.
>Materials and methods
Strains
The mat1P DNA fragments containing the PstI substitutions were introduced into the mat1 locus by homologous recombination into the SP162 strain (h90 leu1-32 ura4-D8) using the lithium transformation procedure (Moreno et al, 1991), producing mut1–mut10 mutant strains. Using the same procedure, the mat1M DNA fragments containing the PstI substitutions were introduced by homologous recombination into the SP714 donorless strain (mat1M leu1-32 ura4-D8 ade6-M210 mat2,3::LEU2), producing strains mut1-3 and mut1-4, respectively. Each construction has been sequenced.
A C-terminal HA tag of swi1 within the chromosome was introduced using a PCR-based gene-targeting system (Bahler et al, 1998). The swi1-HA PCR product was transformed into either the mat1::LEU2 derivative of SP837 (Arcangioli and Klar, 1991), producing strain AHR1, or mut3, producing strain AHR2. All primer sequences are available upon request.
Genomic DNA preparation and analysis
DNA was isolated by a classical method (Moreno et al, 1991), digested with the HindIII enzyme and analyzed by Southern blots. The probe was made using a 10.4 kbp mat1P HindIII fragment. Alternatively, DNA was prepared and digested (as indicated) in agarose plugs and resolved on an 8% denaturing PAGE (Arcangioli 1998). DNA was electroblotted on to a Hybond-N+ membrane (Amersham) for 1 h at 300 mA and revealed with a single-stranded probe immediately distal to H1. Single-stranded DNA probes, specific for the mat1-distal upper and lower strands, were prepared by primer extension and purified using denaturing PAGE conditions.
2D gels
2D gel analysis of replication intermediates was carried out as described previously (Brewer and Fangman, 1988). DNA was prepared and digested with NsiI in agarose plugs (Arcangioli, 1998). Enriched fractions for replication intermediates were obtained using BND cellulose columns. Gels were hybridized with a 2.6 kbp mat1-distal specific fragment, containing LEU2 from S. cerevisiae.
Synchronization conditions
Strains were grown in YES media, at 33°C, until exponential phase. Synchronization was achieved using lactose gradients, as described previously (Arcangioli, 2000), except that two consecutive lactose gradient centrifugations were performed. Small G2 cells (5% of the cells) from the initial culture were collected and incubated in prewarmed YES media at 33°C. The cdc25-22 temperature-sensitive allele blocks the cell cycle at the G2/M transition at the nonpermissive temperature, allowing synchronous progression through the cell cycle, upon release at the permissive temperature. DNA was prepared and digested with restriction enzymes in agarose plugs. All synchronization experiments were performed at least twice.
ChrIP analysis
Exponential cultures of AHR1 (wt) and AHR2 (mut3) grown in YES at 33°C were crosslinked with formaldehyde for 12 min and sonicated such that the majority of sheared fragments were between 0.5 and 1.0 kbp. Whole-cell extraction, immunoprecipitation and DNA extraction of the immunoprecipitated material were performed as described (Meluh and Broach, 1999). The Anti-HA Affinity Matrix from Roche (anti-HA covalently bound to agarose beads) was used for the immunoprecipitation reactions. Samples were assayed by quantitative PCR with a LightCycler (ABI PRISM 7000 Sequence Detection System from Perkin-Elmer) according to the manufacturer's recommendations. Relative enrichments were obtained by first normalizing the total input DNA and ChrIP DNA to the furthest distal sequence, 5 kbp from the SSB, amplified by primer pair (i), as indicated in Figure 4B. The input normalized value was divided by the ChrIP normalized value, giving the full relative in vivo enrichment. ChrIP experiments were repeated at least twice, with independent DNA preparations. Primers for all PCR analyses were synthesized by GENSET SA guaranteed oligos, and chosen using the Primer Express™ Version 2.0 software (Perkin-Elmer). Primer pair sequences used for strain construction and ChrIP are available upon request.
Ligation-mediated PCR
The oligonucleotides BEL5A 5′OH TGACCTTTTGTAGGACAAGGTACCGGT TGTGAAGAAAGCCAATACCCT 3′OH and BEL5B 5′P AGGGTATTGGCTTTC 3′OH (Eurogentec) were hybridized, creating a blunt-ended linker for the LM-PCR reactions. DNA from the same time points obtained from the synchronized cdc25-22 culture were prepared in agarose plugs, melted at 70°C for 10 min, and centrifuged. The supernatant was treated with RNAse (Roche) and β Agarase I (NEB). Genomic DNA at 5 ng/μl was mixed with the hybridized oligonucleotides at 0.2 μM and incubated with T4 DNA ligase and buffer (NEB) for at least 3 h at 16°C. The ligation product was directly used at a 1/20 dilution for ‘Hot Start' PCR: 95°C (5 min), then 32 cycles at 94°C (30 s)/60°C (1 s)/72°C (30 s). The PCR product was digested and cloned into the pUC18 vector. Eight independent clones were sequenced and found to have identical, wild-type mat1M information.
Acknowledgments
We are grateful to G-F Richard and J Weitzman for reading the manuscript and contributing helpful comments. This work was supported by fellowships from the Ministère de l'Education National, de la Recherche et de la Technologie (MENRT) and La Ligue Contre le Cancer to AK, the European Molecular Biology Organization (EMBO) and the Fondation pour la Recherche Medical (FRM) to AH, as well as by grants from the Human Frontiers in Science Program (HFSP) and Association pour la Recherche sur le Cancer (ARC) to BA.
References
- Arcangioli B (1998) A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast. EMBO J 17: 4503–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B (2000) Fate of mat1 DNA strands during mating-type switching in fission yeast. EMBO Rep 1: 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B, Copeland TD, Klar AJS (1994) Sap1, a protein that binds to sequences required for mating-type switching, is essential for viability in Schizosaccharomyces pombe. Mol Cell Biol 14: 2058–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B, de Lahondes R (2000) Fission yeast switches mating-type by a replication-recombination coupled process. EMBO J 19: 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B, Klar AJS (1991) A novel switch-activating site (SAS1) and its cognate binding factor (Sap1) required for efficient mat1 switching in Schizosaccharomyces pombe. EMBO J 10: 3025–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangioli B, Thon G (2003) Mating-types cassettes: structure, switching and silencing. In Molecular Biology of Schizosaccharomyces pombe, Egel R (ed), pp 129–147. Berlin: Springer Verlag [Google Scholar]
- Bahler J, Wu J-Q, Longtine MS, Shah NG, McKenzie A III, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14: 943–951 [DOI] [PubMed] [Google Scholar]
- Beach DH (1983) Cell type switching by DNA transposition in fission yeast. Nature 305: 682–688 [Google Scholar]
- Beach D, Nurse P, Egel R (1982) Molecular rearrangement of mating-type genes in fission yeast. Nature 296: 682–683 [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL (1988) A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 55: 637–643 [DOI] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW (2002) ATR regulates fragile site stability. Cell 111: 779–789 [DOI] [PubMed] [Google Scholar]
- Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602–606 [DOI] [PubMed] [Google Scholar]
- Crouse HV (1960) The controlling element in sex chromosome behavior in Sciara. Genetics 45: 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgaard JZ, Klar AJS (1999) Orientation of DNA replication establishes mating-type switching pattern in S. pombe. Nature 400: 181–184 [DOI] [PubMed] [Google Scholar]
- Dalgaard JZ, Klar AJS (2000) Swi1 and Swi3 perform imprinting, pausing and termination of DNA replication in S. pombe. Cell 102: 745–751 [DOI] [PubMed] [Google Scholar]
- Dalgaard JZ, Klar AJS (2001) A DNA replication-arrest site RTS1 regulates imprinting by determining the direction of replication at mat1 in S. pombe. Genes Dev 15: 2060–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lahondès R, Ribes V, Arcangioli B (2003) The fission yeast Sap1 protein is essential for chromosome stability. Euk Cell 2: 910–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egel R, Beach DH, Klar AJS (1984) Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc Natl Acad Sci USA 81: 3481–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egel R, Eie B (1987) Cell lineage asymmetry in Schizosaccharomyces pombe: unilateral transmission of high-frequency state of mating-type switching in diploid pedigree. Curr Genet 12: 429–433 [Google Scholar]
- Fantes PA, Nurse P (1978) Control of the timing of cell division in fission yeast. Cell size mutants reveal a second control pathway. Exp Cell Res 115: 317–329 [DOI] [PubMed] [Google Scholar]
- Foss EJ (2001) Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics 157: 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M (2002) V(D)J recombination: RAG proteins, repair factors, and regulation. Annu Rev Biochem 71: 101–132 [DOI] [PubMed] [Google Scholar]
- Haber JE (1998) Mating-type gene switching in Saccharomyces cerevisiae. Annu Rev Genet 32: 561–599 [DOI] [PubMed] [Google Scholar]
- Haber JE (2001) Hypermutation: give us a break. Nat Immunol 2: 902–903 [DOI] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424: 1078–1083 [DOI] [PubMed] [Google Scholar]
- Keil RL, Roeder GS (1984) Cis-acting recombination-stimulating activity in a fragment of the ribosome DNA of S. cerevisiae. Cell 39: 377–386 [DOI] [PubMed] [Google Scholar]
- Kelly M, Burke J, Smith M, Klar A, Beach D (1988) Four mating-type genes control sexual differentiation in the fission yeast. EMBO J 7: 1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar AJS (1987) Differential parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature 326: 466–470 [DOI] [PubMed] [Google Scholar]
- Klar AJS (1990) The developmental fate of fission yeast cells is determined by the pattern of inheritance of parental and grandparental DNA strands. EMBO J 9: 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klar AJS, Miglio LM (1986) Initiation of meiotic recombination by double-strand DNA breaks in Schizosaccharomyces pombe. Cell 46: 725–731 [DOI] [PubMed] [Google Scholar]
- Kong Q, Maizels N (2001) DNA breaks in hypermutating immunoglobulin genes: evidence for a break-and-repair pathway of somatic hypermutation. Genetics 158: 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meluh PB, Broach JR (1999) Immunological analysis of yeast chromatin. Methods Enzymol 304: 414–430 [DOI] [PubMed] [Google Scholar]
- Merika M, Thanos D (2001) Enhanceosomes. Curr Opin Genet Dev 2: 205–208 [DOI] [PubMed] [Google Scholar]
- Miyata H, Miyata M (1981) Mode of conjugation in homothallic cells of Schizosaccharomyces pombe. J Gen Appl Microbiol 27: 365–369 [Google Scholar]
- Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- Nielsen O, Egel R (1989) Mapping of the double-strand breaks at the mating-type locus in fission yeast by genomic sequencing. EMBO J 8: 269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages V, Fuchs RP (2003) Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science 300: 1300–1303 [DOI] [PubMed] [Google Scholar]
- Palmer S, Schildkraut E, Lazarin R, Nguyen J, Nickoloff JA (2003) Gene conversion tracts in Saccharomyces cerevisiae can be extremely short and highly directional. Nucleic Acids Res 31: 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavasiliou FN, Schatz GG (2000) Cell-cycle-regulated DNA double-strand breaks in somatic hypermutation of immunoglobulin genes. Nature 80: 216–221 [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE (1997) Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol 17: 6765–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Sternglanz R (1999) Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast 15: 35–41 [DOI] [PubMed] [Google Scholar]
- Richards R (2001) Fragile and unstable chromosomes in cancer: causes and consequences. Trends Genet 17: 339–346 [DOI] [PubMed] [Google Scholar]
- Rothstein R, Michel B, Gangloff S (2000) Replication fork pausing and recombination or ‘gimme a break'. Genes Dev 14: 1–10 [PubMed] [Google Scholar]
- Saparbaev M, Prakash L, Prakash S (1996) Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1–RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 142: 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Klar AJS (1993) DNA polymerase α is essential for mating-type switching in fission yeast. Nature 361: 271–276 [DOI] [PubMed] [Google Scholar]
- Stargell LA, Struhl K (1996) Mechanisms of transcriptional activation in vivo: two steps forward. Trends Genet 8: 311–315 [DOI] [PubMed] [Google Scholar]
- Styrkarsdottir U, Egel R, Nielsen O (1993) The smt-0 mutation which abolishes mating-type switching in fission yeast is a deletion. Curr Genet 23: 184–186 [DOI] [PubMed] [Google Scholar]
- Sugawara N, Pâques F, Colaiácovo M, Haber JE (1997) Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci USA 94: 9214–9219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitao T, Budd M, Hoopes LL, Campbell JL (2003) Dna2 helicase/nuclease causes replicative fork stalling and double-strand breaks in the ribosomal DNA of Saccharomyces cerevisiae. J Biol Chem 278: 22513–22522 [DOI] [PubMed] [Google Scholar]