

Abstract
The crystal structure of the recombinant collagen-binding domain of Yersinia adhesin YadA from Yersinia enterocolitica serotype O:3 was solved at 1.55 Å resolution. The trimeric structure is composed of head and neck regions, and the collagen binding head region is a novel nine-coiled left-handed parallel β-roll. Before the β-roll, the polypeptide loops from one monomer to the rest, and after the β-roll the neck region does the same, making the transition from the globular head region to the narrower stalk domain. This creates an intrinsically stable ‘lock nut' structure. The trimeric form of YadA is required for collagen binding, and mutagenesis of its surface residues allowed identification of a putative collagen-binding surface. Furthermore, a new structure–sequence motif for YadA β-roll was used to identify putative YadA-head-like domains in a variety of human and plant pathogens. Such domains may therefore be a common bacterial strategy for avoiding host response.
Keywords: adhesin, collagen binding, MAD phasing, X-ray crystallography, YadA
Introduction
Collagens are the major component of extracellular matrix (ECM) and, in addition to providing mechanical strength and rigidity to tissues, they are involved in a number of cellular processes such as cell attachment, haemostasis, differentiation and bacterial adhesion (for a review, see Juliano and Haskill, 1993; Foster and Höök, 1998). Collagen-binding proteins are therefore required for many essential processes, but only a few structurally different classes of the collagen-binding proteins have been extensively investigated. These include matrix metalloproteinases (reviewed in Bode et al, 1999), the structurally related I domains from the α-1 subunit of integrin (Lee et al, 1995; Emsley et al, 1997) and domain A3 of the von Willebrand factor (Huizinga et al, 1997), and the extracellular domain of matrix protein BM-40 (Hohenester et al, 1996). Furthermore, only two bacterial collagen-binding domain structures are known, the Staphylococcus aureus collagen-binding domain (CBD; Symersky et al, 1997) and the CBD of Clostridium histolyticum class I collagenase (Wilson et al, 2003).
The Yersinia adhesin YadA is a homotrimeric collagen-binding protein expressed at +37°C on the outer membrane of Yersinia enterocolitica and Yersinia pseudotuberculosis. It forms a fibrillar matrix on the bacterial surface (Hoiczyk et al, 2000). YadA is encoded by the 70 kb Yersinia virulence plasmid pYV, and the monomeric YadA from Y. enterocolitica serotype O:3 has a relative molecular mass of about 44 kDa (Skurnik et al, 1984; Skurnik and Wolf-Watz, 1989), as estimated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS–PAGE). The native form of YadA is very stable, as the Yersinia bacteria could still bind to type I collagen, that is, had the active form of YadA on the outer surface, even after 20 min incubation at +80°C or after different protease treatments (Emödy et al, 1989). Moreover, neither treatment with 1 M urea nor changing the pH from 5.0 to 10.0 released bound collagen from Y. enterocolitica bacteria. Trimeric YadA can even be seen in SDS–PAGE after boiling in sample buffer (Skurnik et al, 1984; Tamm et al, 1993). YadA belongs to the family of lollipop-shaped adhesins, with a globular head domain, a rigid stalk domain and a C-terminal membrane anchor (Hoiczyk et al, 2000), and recently features of YadA were identified suggesting that it is an autotransporter (Roggenkamp et al, 2003). YadA and its ability to bind collagen are essential for virulence in Y. enterocolitica; loss of this ability leads to avirulence in mice (Tamm et al, 1993; Roggenkamp et al, 1995). In contrast, YadA does not appear to be needed for virulence in Y. pseudotuberculosis (Han and Miller, 1997), and in the third human pathogenic Yersinia species, Y. pestis, YadA is not expressed at all due to a frame shift in the yadA gene (Rosqvist et al, 1988; Skurnik and Wolf-Watz, 1989).
Y. enterocolitica causes several different types of diseases ranging from mild diarrhoea to septicaemia and mesenteric lymphadenitis with reactive arthritis, iritis and erythema nodosum as sequelae (Cover and Aber, 1989). The first step of infection requires invasion of the intestinal mucosa by the bacteria, initiated by the chromosomally encoded proteins Invasin and Ail (Miller and Falkow, 1988; Isberg and Tran van Nhieu, 1995). After invasion, YadA is the major adhesin during infection; it is needed for adherence of bacteria to epithelial cells (Heesemann and Grüter, 1987) and to ECM proteins collagen, laminin and fibronectin (Emödy et al, 1989; Schulze-Koops et al, 1992, 1993; Tertti et al, 1992; Flügel et al, 1994). YadA also protects bacteria by being responsible for autoagglutination, serum resistance, complement inactivation and phagocytosis resistance (Skurnik et al, 1984; Balligand et al, 1985; Piltz et al, 1992; China et al, 1993).
The functional mapping of biological properties to the YadA polypeptide sequence has been extensive. Carboxy (C)-terminal deletion mutants were not expressed on the bacterial surface and failed to oligomerise, suggesting that the C-terminal membrane anchor is responsible for oligomerisation (Tamm et al, 1993; Roggenkamp et al, 2003). The head domain of YadA appears to be the collagen-binding region, based on binding studies with different deletion mutants (for a review, see El Tahir and Skurnik, 2001). Deletion of amino acids 29–81 led to a loss of adhesion to neutrophils, but autoagglutination and binding to collagen or laminin were not affected (Roggenkamp et al, 1996). The point mutations H156Y and H159Y specifically abrogated binding of YadA to ECM (Roggenkamp et al, 1995). Moreover, the collagen-binding ability and autoagglutination of YadA was totally lost by deletion of residues 83–104 (Tamm et al, 1993). Finally, the NSVAIGXXS motif, repeated eight times in the head domain, has been found to be essential in YadA collagen interaction, because mutation of the middle residues in most of the motifs abolishes collagen binding totally (El Tahir et al, 2000). Recently, further deletion mutant studies have demonstrated that both the head domain and the neck are needed for collagen binding and, furthermore, the neck region is required for autoagglutination (Roggenkamp et al, 2003).
To understand the basis of collagen binding to YadA as well as virulence of Y. enterocolitica, we have solved the first YadA crystal structure of the globular collagen-binding head domain. The novel structure explains these properties and the extreme stability of YadA.
Results
The structure of YadA26–241 was solved utilising seleno-methionyl labelled double mutant Yad26–241-2M (I130M, I157M) and multiple anomalous dispersion (MAD). The mutant crystals grew from similar conditions to native ones (Nummelin et al, 2002) in space group R32. (The native data had been indexed in R3 (Nummelin et al, 2002) in case the crystals were merohedrally twinned, but it became clear that the two-fold is crystallographic, and so all data were rescaled to R32 (Table I).) The electron density quality was good throughout the structure (Figure 1), but 37 residues were disordered in the final model (see below). The stereochemistry of the molecule is good (Table I). However, only 81% of residues are in the most favourable areas of the Ramachandran plot instead of greater than 90% as is usual in well-defined structures of this resolution. This can be explained by the fact that the structure contains 17 of the less common type II β-turns (see below). In 16 of these, a non-Gly residue occupies the i+2 position (Figure 1) in an αL conformation, which is usually permitted only for Gly (Rose et al, 1985).
Table 1.
Data collection, structure determination and refinement
| Data collection | ||||
|---|---|---|---|---|
| Crystal | Native (R32) | SeMet (R32) | SeMet | SeMet |
| Unit cell (a=b, c) Å | 67.05, 221.95 | 65.17, 230.49 | ||
| Wavelength (Å) | 0.9102 | 0.9793 (peak) | 0.9795 (inflection) | 0.9184 (remote) |
| Resolution range (Å) | 20.0–1.55 (1.61–1.55) | 20.0–2.0 (2.07–2.00) | 30.0–2.0 (2.07–2.00) | 30.0–2.0 (2.07–2.00) |
| Total observations | 355 936 | 635 275 | 229 637 | 277 509 |
| Unique reflections | 32 335 (2683) | 24 672 (2454) | 24 671 (2466) | 24 726 (2466) |
| Data completeness (%) | 99.8 (99.4) | 99.9 (100.0) | 99.8 (99.9) | 99.8 (99.9) |
| Rmerge (%) | 4.4 (42.9) | 8.1 (30.0) | 8.5 (34.9) | 9.6 (38.9) |
| I/σ | 15.3 (4.5) | 23.8 (5.4) | 7.6 (1.6) | 7.4 (1.6) |
| Phase determination | ||||
| Anomalous scatterer | 2 Se sites | |||
| Figure of merit | 0.28 | |||
| Phasing power | 0.80 | |||
| Refinement statistics | ||||
| Resolution range (Å) | 20–1.55 | 20–2.00 | ||
| No. of reflections Rwork/Rfree | 23 731/2569 | 21 795/2265 | ||
| Atoms (protein/water) | 1309/105 | 1228/77 | ||
| Rfree (%) | 20.4 | 22.9 | ||
| Rwork (%) | 19.5 | 19.7 | ||
| R.m.s.d bond length (Å) | 0.004 | 0.006 | ||
| R.m.s.d. bond angle (o) | 1.384 | 1.435 | ||
| Mean B-value (Å2) main/side chain | 14.9/17.9 | 16.6/19.9 | ||
| Missing residues | 26–31, 52–61, 221–241 | 26–31, 51–61, 221–241 | ||
| Alternate conformations | 10 | None | ||
| Ramachandran plot | ||||
| Residues in most favoured regions (%) | 81.0 | |||
| Residues in additional allowed regions (%) | 17.6 |
Figure 1.
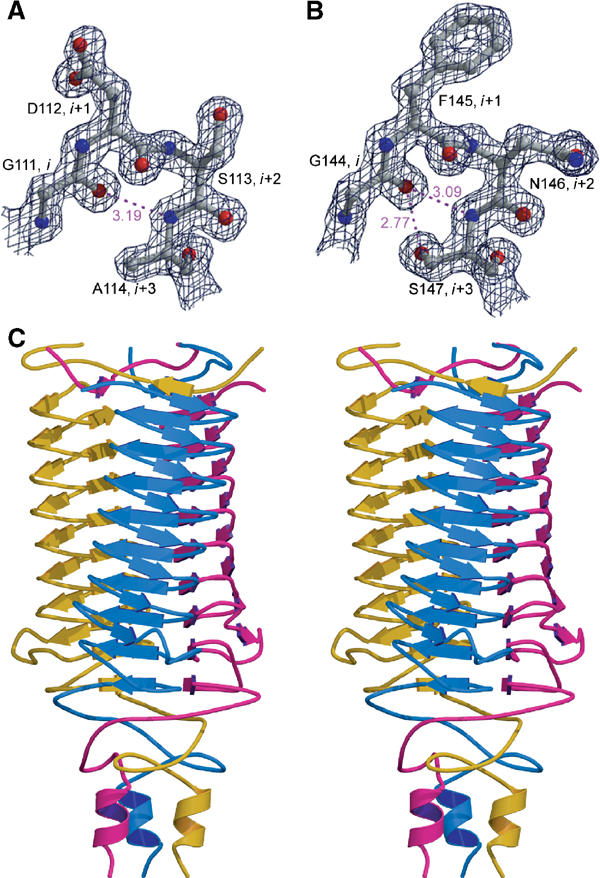
Electron density of (2Fo-Fc) map contoured at 1.5σ for turns T1 (A) and T2 (B) showing S113 and N146 in i+2 positions in left-handed α-helical conformation. Hydrogen bonding from G111 and G144 to i+3 is illustrated in violet with the distance between atoms in Å. The figure was prepared using BOBSCRIPT (Esnouf, 1999) and Raster3D. (C) Stereo picture of trimeric YadA head domain. The N-termini are up and the C-termini are down. The strands are drawn as arrows, helices as ribbons and the different monomers are in different colours.
The head domain
The YadA head domain is a tight trimer about 53 Å long and 37 Å wide (Figure 1C). The monomers consist of two different regions identified from the sequence, the head (26–194) and the neck (195–219; Hoiczyk et al, 2000). Amino (N)-terminal residues 26–31 and loop 52–61 are missing in the final model, as well as the start of the stalk domain (221–241). The head region is composed almost solely of β-sheets making a novel nine coiled left-handed parallel β-roll (LPBR), surrounded by a partly disordered (N)-terminal random coil and a C-terminal neck region, which consists of a random coil and a short helix at the start of the stalk domain. There are no stabilising disulphide bridges to account for the stability of the trimer.
The YadA LPBR structure is composed of a 14-residue repeat motif, of the form turn (T1)—three-residue inner strand (IS)–turn (T2) and a three-residue outer strand (OS; Figure 2). The variation in the coil length, from 13 to 16 residues, is caused by a variation in the T1 length, so that the longest one is at the C-terminus of the LPBR. The other turn, T2, is always the same length. The hydrophobic interior of the trimer is composed of the inner strands, which constitute seven out of eight NSVAIGXXS motifs (El Tahir et al, 2000) and two additional similar sequences AAVAVGAGS and SAVTYGAAS (Figure 2B). One of the NSVAIGXXS motifs identified was part of the outer sheet.
Figure 2.

(A) One level of the β-roll in the trimer viewed along the z-axis showing the packing of the oligomeric core by large hydrophobic residues and the packing of the monomeric interior by small hydrophobic residues. The conserved (i−i+3) hydrogen bonds are shown with dashed lines. The colouring of the monomers is the same as in Figure 1C. (B) Alignment of the full β-roll repeats. The NSVAIGXXS repeats are shown in bold and the totally conserved Gly is boxed. The turns T1 and T2 are marked on top of the alignment and the residues making β-strands are in grey boxes. The consensus sequence is marked below the alignment.
The structure-based alignment of the LPBR coils not only revealed the structural basis for the conservation of the NSVAIGXXS pattern but also further sequence conservation within the coil (Figure 2B). The totally conserved Gly is in the i position in the T2 turns. The conformation of T2 does not allow other residues at this position (Figure 1), which is underlined as a clustering in the Ramachandran plot (see Supplementary data, Figure 1). In the equivalent position in T1, Gly is not conserved because the conformation is that of a normal β-strand. Ser residues at i+3 stabilise turns by hydrogen bonding to the (Gly) carbonyl oxygen at i in five (T1) or six (T2) out of nine turns. The small conserved residues form the interior of the monomer, where the larger residues pack into the oligomeric core. The monomeric core is formed by the i+3 residues in T1 and T2, with the middle residues in the outer and inner strands, respectively. In a tight β-roll, there is no space inside the monomer for large residues, and therefore there is a strong preference for small ones (Figure 2), especially Ser in the turns, and Ala in the middle of the strands. Conversely, the intertrimeric core is formed by the remaining inner strand positions 1 and 3 (Figure 2), which prefer having Val (6/9) and Ile (5/9) in these positions, respectively.
The N-terminal residues are not seen (26–31), but the N-terminus of the model (35-TAVQIS-40) forms the N-terminal inner β-strand and is trapped under the loop 41–50. The rest of the proline-rich loop (52-PVRPPVPGAG-61) is disordered, but one possibility is that the chain reverses direction at Y51, forming another IS and then loops around clockwise to connect to G62 (see Supplementary data, Figure 2). An alternative arrangement, which we prefer because it does not break the strand pattern of the LPBR, is that the proline-rich loop loops around in an anticlockwise manner to join G62 (Figure 3). In both cases, the N-terminus of one monomer is trapped under the 41–50 loop of the anticlockwise adjacent one. This clearly increases mechanical stability. The equivalent loop in Y. pseudotuberculosis YadA is about 40 amino acids long and is the larggest difference between the two YadAs (Skurnik and Wolf-Watz, 1989).
Figure 3.
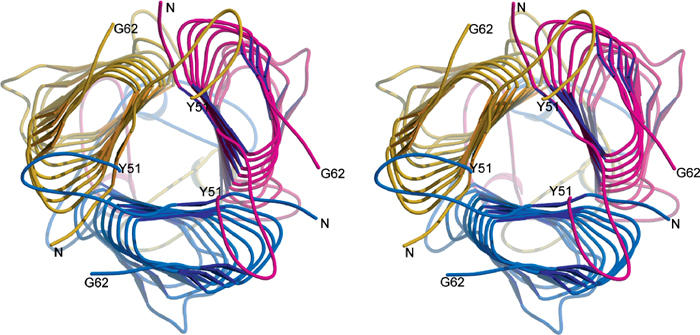
The YadA head domain viewed down the z-axis from the N-terminus. The twisting of the monomers and the extension of the coils in different directions at the top and the bottom of the LPBRs is seen. On the top of the trimer, strands 32–40 are trapped under loops 41–50. The colours indicate the most plausible connectivity of a separate N-terminal loop with the rest of the monomer. The colouring of the monomers is the same as in Figure 1C.
The neck region
The neck region makes the transition from the globular head region to the narrower stalk domain (Figure 4A). Residues 194–200 of one monomer run below the inner sheet of the adjacent monomer in an anticlockwise direction. This is followed by a loop consisting of residues 201–209 under the third monomer and a C-terminal helix, which together resemble a safety pin (Figure 4A). The arrangement presumably serves exactly this purpose: The region from 194 to 209, which extends from monomer A through B to C, actually pins all three monomers together, partly explaining the extreme stability of the YadA trimer. The hydrophobic residues in the neck region continue the hydrophobic core of the trimer: L196 and L199 end the hydrophobic ladder under V185′ and I187′, respectively; V209 is hydrogen bonded to its symmetry-related counterparts and together they stack around the crystallographic three-fold. Finally, L214 starts the leucine zipper arrangement of the stalk domain. Each ‘safety -pin' is tied to the β-roll trimer and to each other by numerous main-chain interactions, and by a multivalent ion network (Figure 4B). Another network is around R194 in the monomer–monomer interface, where R194 ties a small protruding turn 189–194 to the LPBR. Such networks have been found in many thermostable proteins, for example, Sulfolobus acidocaldarius inorganic pyrophosphatase and Pyrococcus furiosus glutamate dehydrogenase (Yip et al, 1995; Leppänen et al, 1999).
Figure 4.
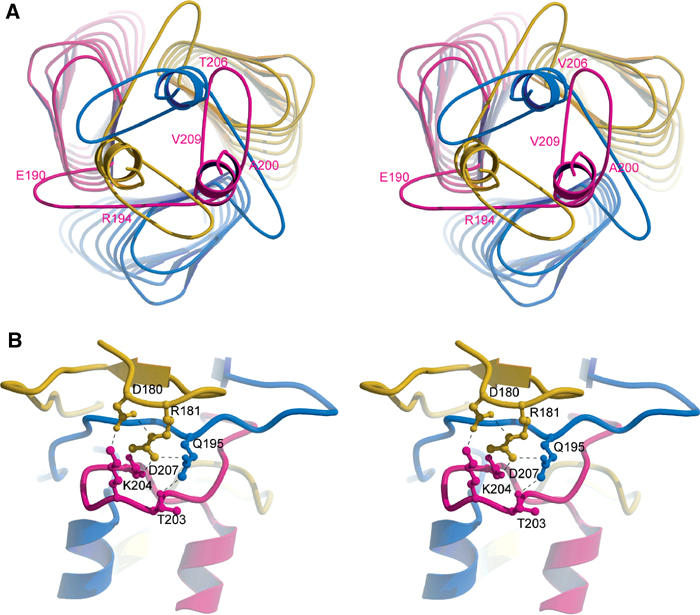
(A) Organisation of the neck region in the C-terminus of the head domain, viewed from the C-terminus along the z-axis. The safety-pin structures as well as the beginnings of the stalk domain helices are shown. Some residues in the neck region are numbered for the magenta monomer for clarity. (B) The neck region (viewed perpendicular to (A) showing one multi-centre ionic network in the safety-pin region. It ties the three ‘safety pins' to the central β-roll assembly. The colouring of the monomers is the same as in Figure 1C.
YadA and collagen binding
Mutagenesis of the surface residues of the YadA LPBR followed by collagen-binding studies was carried out to find the collagen-binding surface (Table II). A set of mutants was designed to make a grid over the collagen-binding area within the 80–190 region defined earlier by deletion mutagenesis (Tamm et al, 1993; Roggenkamp et al, 1996). We chose this approach to find the orientation of the collagen triple helix with respect to YadA and to locate a possible binding site. Moreover, because YadA proteins from different sources are almost 100% identical throughout the collagen-binding domain (Skurnik and Wolf-Watz, 1989), multiple sequence alignment gave no indication about possible binding residues. We primarily mutated charged and hydrophilic surface residues. These were assumed to be most probable ones to interact with the ligand, as earlier data suggested that YadA binds a charged region of collagen (Schulze-Koops et al, 1995). In addition, double mutants were created to introduce large local changes to the surface and thus ensure a measurable effect on binding. Although the deletion of the residues 191–221 abolishes collagen binding (Roggenkamp et al, 2003), no mutations were performed to the neck region, because it appears to be needed for trimeric structure, not directly for the binding.
Table 2.
Apparent dissociation constants for the YadA type I collagen binding
| Mutation | RUmaxa | KD(app) (μM) | % of wt |
|---|---|---|---|
| wt | 3343±160 | 0.28±0.06 | 100 |
| V98D, N99A | 356±37 | 7.34±2.72 | 4 |
| K108A, L110A | 3378±134 | 2.84±0.48 | 11 |
| Q124A, K125A | 1619±64 | 0.98±0.16 | 30 |
| H159A, H162A | 916±27 | 2.49±0.32 | 12 |
| K148A, D150A | 1588±74 | 2.0±0.21 | 15 |
| D180A, E182A | 2209±154 | 25.7±3.7 | 1 |
| E190A, S191A | 3371±143 | 0.59±0.08 | 50 |
| E80A, K83A | 2612±96 | 1.51±0.26 | 20 |
| N166A, Y169A | 4599±731 | 1.77±0.64 | 17 |
| R133A |
1208±17 |
2.9±0.08 |
10 |
| a The RUmax appeared to vary depending on the age of the chip and how efficiently it was cleaned after each run. | |||
All the mutations had small but quantifiable effects on the YadA collagen-binding activity when measured with surface plasmon resonance (SPR) and judged by apparent binding constants KD(app). The larggest effect on binding was detected for the mutant D180A-E182A, whose collagen-binding activity was only about 1% of the native YadA26–241 (wt-YadA; Table II), but this may also reflect the change in the structure as D180 has an intermonomeric contact (Figure 4B). Consistent with this, the D180A-E182A mutant migrated faster than wt-YadA on native PAGE (results not shown). Mutation V98A-N99A reduced binding to only 4% of the wt-YadA binding, but the activity most of the mutants was 10–20% of the wt-YadA. Only mutations Q124A-K125 and E190A-S191A reduced binding activity to a lesser extent (Table II). The results qualitatively agree with the collagen affinity blotting (El Tahir et al, 2000) results; the mutants with the lowest binding activity (D180A-E182 and V98A-N99A) also failed to bind type I collagen when immobilised on a nitrocellullose filter (results not shown).
Docking
Protein–protein docking was used to predict the possible orientation of collagen across the YadA surface. A PPG probe peptide was made as described in Materials and methods and docked on the trimeric YadA. The solutions were filtered using experimental data to select the models consistent with the known collagen-binding data obtained with the mutants. In 27 out of the 30 best solutions, the peptide was placed diagonally on the trimeric YadA (Figure 5), so that the peptide had contacts to most of the mutated residues. However, because the docked probe had only one, quite rigid triple helix, none of the docking solutions was such that the probe could have contacts to all the key residues.
Figure 5.

Most likely orientation of the collagen triple helix on the surface of the trimeric YadA head domain. The mutant residues are coloured according to their effect on type I collagen so that the mutants that abolished the binding totally are coloured red, to <20% in orange and the ones that attenuated the binding in violet. The residues D180 and E182, which also had an effect on the trimeric structure, are in blue. The figure was prepared using PyMOL (DeLano, 2002).
Related structures
The Protein Data Bank (PDB, Berman et al, 2000) was searched using Dali (Holm and Sander, 1993) for structural homologues of YadA. There was one remotely similar structure, a model for a right-handed β-roll structure for an antifreeze protein from Lolium pernenne (Kuiper et al, 2001), but we found no homologues among experimental protein structures. The three existing metalloprotease β-roll substructures (Baumann et al, 1993; Baumann, 1994; Aghajari et al, 2003) were not found by Dali because of differences in topology to the YadA LPBR. The metalloproteases have a β-roll structure as part of their C-terminal β-sandwich domain, but where the YadA β-roll is left-handed, twisted and highly compact, the metalloprotease β-roll is straight, right-handed and larger. The repeating unit in metalloproteases is 18 residues, unlike the 14 residues in YadA.
To study whether other proteins in the sequence databases could have a YadA-like LPBR, we extended the NSVAIGXXS motif to cover the whole β-roll coil (Figure 2B). We used X(3)-[SGA]-[VI]-[AT]-[IVY]-GXX-[AS]-X-[ATV]-X(0,3) as a PROSITE search pattern (Gattiker et al, 2002) in the Swiss-Prot and TrEMBL sequence databases (Boeckmann et al, 2003). The motif was found twice or more in 30 sequences from 30 entries, the maximum number of hits per sequences being nine (Table III). The search identified several proteins, including five YadA variants, already been known to belong to the YadA protein family (YadA-pfam), because they share homologous C-terminal membrane anchors and overall architecture (Hoiczyk et al, 2000). Indeed, a repeat of approximately 14 residues had already identified in these proteins (Hoiczyk et al, 2000). However, more careful analysis of the identified sequences revealed additional YadA-like repeats in these proteins, where almost any small amino acid (Gly, Ala, Ser, Thr) was preferred at the IS interdigitating positions, any hydrophobic residue or Thr in IS position 1 and hydrophobic Thr, Phe or Tyr in IS position 3. Repeating the search with X(3)-[GAST]-[VIMLT]-[GAST]-[VIMLTYF]-GXX-[GAST]-X-[GASTV]-X(0,3) yielded evidence for additional repeats in the already identified proteins (Table III).
Table 3.
Database searching with YadA β-roll -structure motif found 60 sequencesa
| Swiss Prot ACb | Protein | Organism | Size (a.a.) | Motifsc | Extended Motifsd | Arrayse | Necksf |
|---|---|---|---|---|---|---|---|
| Large domains (13–<30 motifs/array) | |||||||
| Q8RQ61 | Putative surface protein | Actinobacillus actinomycetemcomitans (Haemophilus actinomycetemcomitans) | 1264 | 2 | 14 | 2 | 3 |
| Q89IU6 | Blr5538 protein | Bradyrhizobium japonicum | 762 | 7 | 17 | 2 | 3 |
| Q8FCB2* | Putative adhesin | Escherichia coli 06 | 1778 | 4 | 31 | 4 | 13 |
| Q8XDG4* | Putative adhesin (Hypothetical 160.1 kDa protein) | Escherichia coli Q157:H7 | 1588 | 3 | 26 | 4 | 11 |
| Q9F3X5* | MapB protein (Hsf) | Pasteurella multocida | 2712 | 9 | 23 | 2 | 9 |
| Q9F3X6* | MapA protein (Hsf) | Pasteurella multocida | 1299 | 7 | 23 | 2 | 4 |
| Q92KQ7* | Hypothetical protein R00458 | Rhizobium loti (Sinorhizobium meliloti) | 1291 | 5 | 19 | 1 | 7 |
| Q98HJ2* | Hypothetical protein mll0950 | Rhizobium loti (Mesorhizobium loti) | 1953 | 8 | 50 | 7 | 8 |
| Q9FDA0* | Putative outer membrane protein XadA | Xanthomonas oryzae (pv. oryzae) | 1265 | 1 | 34 | 4 | 4 |
| Q8PCQ5* | Outer membrane protein | Xanthomonas campestris (pv. campestris) | 2351 | 6 | 90 | 7 | 7 |
| Q9LAX0* | Hypothetical 127.4 kDa protein | Xanthomonas campestris (pv. pelargonii) | 1328 | 9 | 46 | 2 | 3 |
| Q8PGS0* | Outer membrane protein | Xanthomonas axonopodis (pv. citri) | 2190 | 5 | 80 | 7 | 7 |
| Q8PGR8* | Outer membrane protein | Xanthomonas axonopodis (pv. citri) | 1114 | 3 | 30 | 3 | 3 |
| YadA-like domains (6–11 motifs/array) | |||||||
| Q89J37 | Blr5449 protein | Bradyrhizobium japonicum | 372 | 2 | 3 | 1 | 1 |
| Q9LA53* | Immunoglobulin-binding protein EibE | Escherichia coli | 478 | 1 | 7 | 1 | 1 |
| Q9LA56* | Immunoglobulin-binding protein EibC | Escherichia coli | 504 | 1 | 6 | 1 | 1 |
| Q9MCI8 | Immunoglobulin-binding protein EibE | Bacteriophage P-EibD | 511 | 1 | 6 | 1 | 1 |
| Q8KQM8* | Haemagglutinin | Moraxella catarrhalis | 2314 | 2 | 10 | 2 | 4 |
| Q8KQM9* | Haemagglutinin | Moraxella catarrhalis | 1964 | 4 | 9 | 2 | 3 |
| Q8ZL64* | Putative inner membrane protein | Salmonella typhimurium | 1461 | 2 | 17 | 4 | 11 |
| Q87DF4 | Outer membrane protein XadA | Xylella fastidiosa | 997 | 1 | 19 | 4 | 4 |
| Q9PC04* | Surface protein | Xylella fastidiosa | 1190 | 2 | 21 | 3 | 8 |
| Q9PD63* | Surface exposed outer membrane protein | Xylella fastidiosa | 1004 | 1 | 22 | 4 | 4 |
| Q87D62 | Surface protein | Xylella fastidiosa | 1315 | 3 | 13 | 4 | 9 |
| Q8CL86 | Hypothetical | Yersinia pestis | 736 | 3 | 13 | 3 | 5 |
| Q8ZHJ0* | Putative surface protein | Yersinia pestis | 658 | 2 | 10 | 3 | 5 |
| Q8CKM1* | Hypothetical | Yersinia pestis | 641 | 2 | 10 | 3 | 4 |
| Small arrays of 4–5 motifs | |||||||
| Q8G378 | Outer membrane protein | Brucella suis | 740 | 2 | 6 | 2 | 5 |
| Q8YEK6 | Cell surface protein | Brucella melitensis | 365 | 1 | 3 | 1 | 2 |
| Q8XPL8* | Putative haemagglutinin-related protein | Ralstonia solanacearum (Pseudomonas solanacearum) | 1309 | 2 | 9 | 2 | 12 |
| Q9F2D8* | SapB protein (putative autotransporter) | Salmonella typhi | 1107 | 2 | 10 | 3 | 9 |
| Q8DH28 | Tlr2132 protein | Synechococcus elongatus (Thermosynechcoccus elongatus) | 276 | 4 | 5 | 2 | 2 |
| Q9PD50* | Surface protein | Xylella fastidiosa | 2059 | 4 | 12 | 3 | 14 |
| Q87DE1 | Surface protein | Xylella fastidiosa | 2052 | 1 | 6 | 8 | 16 |
| No neck sequence | |||||||
| Q9X6K5 | Hypothetical 13.0 kDa protein | Klebsiella ocytoca | 153 | — | 6 | (1)g | 0 |
| Q9XUF9 |
C49C3.4 protein |
Caenorhabditis elegans |
1566 |
— |
5 |
(1)g |
0 |
| aThe five YadA entries and 19 completely repeating Gly-rich sequences (for example GTGTGTGTGTGTGT) are excluded. The remaining hits with at least two subsequent motifs are listed. The YadA protein family members are marked with *. | |||||||
| bSwiss-Prot/TrEMBL Accession code. | |||||||
| cNumber of YadA-like motifs. | |||||||
| dNumber of extended YadA-like motifs. | |||||||
| eNumber of arrays (at least three subsequent motifs followed by a neck region). | |||||||
| fNumber of neck regions. | |||||||
| gNo neck region following array of motifs. | |||||||
We therefore searched the protein sequence databases with the extended motif. The search yielded 60 proteins with more than two subsequent motifs (i.e. arrays; Table III). Consistent with identification of these proteins as members of the YadA family, all except two (from Klebsiella oxytoga and Caenorhabditis elegans) also had neck-like sequences (Hoiczyk et al, 2000), usually after the motifs but also elsewhere in the sequence, and sometimes in multiple copies (Table III). We grouped the other sequences into three categories according to the size of the largest YadA-like array: sequences with large (>13 motifs), YadA-like (6–11 motifs) or small (<6 motifs) arrays. In almost every case, the arrays were found in the middle of the sequence and some arrays had insertions, that is, nonrepeating regions interrupting the array. Among the proteins with large YadA-like arrays, almost half of the sequence (44%) of XadA (Table III) was composed of extended YadA-like motifs (34 in total), compared to only one exact YadA-like motif. This shows the importance of the extension of the search pattern.
The most interesting category is the one with YadA-like arrays, as these should form YadA-like domains. The shortest sequences in this category had one array of 3–7 extended motifs, surrounded by a nonrepetitive region, similar to YadA. These proteins include Escherichia coli and bacteriophage P immunoglobulin-binding proteins and Blr5447 protein form Bradyrhizobium japonicum. These proteins are most alike to YadA, as they have a single array of motifs followed by a single neck region. The position of the array in the middle of the sequence and the size of the protein also resemble YadA. The sequences from human pathogens Salmonella typhimurium and Moraxella catarrhalis as well as two surface proteins from the plant pathogen Xylella fastidiosa have the YadA-like array at the beginning of the sequence, then a large nonrepetitive sequence (including several neck regions) and smaller array(s) near the end of the sequence. Finally, Xyella fastidiosa XadA and Q9PD63 (Table III) have four and Y. pestis Q8CL86 has three arrays in a row, interrupted only by a nonrepetitive region after every neck region.
Discussion
The unique features of the YadA head domain are its left-handed parallel β-roll, LPBR, and its trimeric assembly. No other LPBR protein structure has been solved before and, furthermore, only one left-handed β-helical structure is known (Raetz and Roderick, 1995). The YadA β-roll is the tightest β-roll structure, and the first oligomeric one solved so far. In addition to the extremely hydrophobic core of the protein, trimeric YadA is further stabilised by the interactions at both ends of the assembly of the rolls, where the coils tie the oligomer together like a lock nut. At the C-terminus, the neck region ties the monomers together in a clockwise manner, whereas the N-terminal coil runs counterclockwise, when viewed along the z-axis from the N-terminus towards the C-terminus (Figure 3). This leads to a mechanically very stable arrangement. As with a lock nut, the protein cannot easily be unfolded by twisting, which surely must occur for the β-strands to unfold. In addition, the multi-ion networks stabilise the protein (Figure 4B).
The trimer appears to be essential for YadA collagen-binding activity: the role of the previously identified NSVAIGXXS motif (El Tahir et al, 2000) is structural; they form the turns and hydrophobic core of the YadA β-roll and trimer. The V to D and I to E mutations in these motifs prevented collagen binding by breaking apart the essential trimer; three charged residues per mutation would have had to be buried in the centre of the trimer (Figure 2A). Nonetheless, changes in the N-terminal (upper) motifs in the roll allowed residual (0.5–8%) collagen binding, but similar mutants in motifs 6–8 did not bind collagen at all (El Tahir et al, 2000). Presumably, charged mutations in the ‘upper' regions only partially prevented trimerisation, unlike mutations in the ‘lower' region. In agreement with this, deletion of residues 83–104 did not cause total breakdown of the trimer, as the trimer is seen on SDS–PAGE (Tamm et al, 1993). However, this variant eliminates 1.5 turns of the LPBR (Figure 3B). It places the inside strand on the outside and vice versa, in addition to eliminating V98-N99, which are important for collagen binding (Table II). Not surprisingly, this mutant did not bind collagen (Tamm et al, 1993).
The central assembly of LPBRs binds collagen. The rigid collagen fibril is too large to interact with the narrower neck region (190–221), and the very N-terminal part (29–85) is not required for collagen binding (Roggenkamp et al, 1996). Our binding studies using SPR demonstrated that YadA26–241 bound immobilised type I collagen with a KD(app) of 0.3 μM, consistent with earlier collagen-binding studies with YadA expressing cells (Emödy et al, 1989) and experiments with longer YadA fragments (data not shown). Mutational analysis showed that even removing one or two side chains affected the YadA collagen interaction (Table II), and in most cases the effect on KD(app) was about one order of magnitude. Most (12/19) of the mutated residues were charged. The fact that most of the collagen-binding surface of YadA is charged and that mutating these residues reduced the collagen binding is in agreement with an earlier study that YadA would interact with a charged stretch of collagen type I (Schulze-Koops et al, 1995). The most critical residues found in this study, however, included both hydrophobic and hydrophilic residues. Using the experimental data to filter the docking results of the PPG peptide on YadA trimer resulted in a model where the collagen triple helix lies diagonally on the YadA between two loops which protrude from the surface in the C-terminal part of LPBR (Figure 5). Moreover, the peptide lies close to H159 and H162, both of which were shown to be important in the binding of collagen to YadA on the surface of bacteria (Roggenkamp et al, 1995). The peptide makes contact with residues from two monomers including those in the ‘safety pin' (Figure 4B), further explaining the requirement of the trimer for the binding (see above).
If the crossing angle between YadA and collagen is about 30°, as our model suggests, a steric issue emerges. Collagen triple helices assemble to form a 10–300-nm-diameter fibril, which is longer and more rigid than YadA. Consequently, the fibril would collide with the surface of the YadA-expressing bacteria. We therefore suggest that the YadA head groups or their coiled-coil stalks bend upon interaction with collagen, thus avoiding the collision. This would expose an array of collagen-binding surfaces and, consequently, the effective binding constant for collagen on the bacteria would be much tighter than for collagen on a single YadA. Such an arrangement also indicates why the collagen peptide cannot interact with all the residues found to be involved in binding. The less important residues (E80, K83, K108, L110, Q124, K125, N166 and Y169) would interact with adjacent triple helices in the collagen fibril. Finally, this may explain how YadA can be so polyfunctional in such a small molecule; it may be able to bend in different ways exposing different surfaces and so allowing it to bind different ECM molecules.
The YadA LPBR has no structural similarity to other known collagen-binding proteins. Some integrins bind collagen in a metal-dependent manner via the MIDAS site (Emsley et al, 2000), but no metals are needed for YadA collagen binding. The collagen-binding site of the von Willebrand factor domain A, structurally similar to integrin, has been mapped to a shallower groove (Nishida et al, 2003), but again there are no striking similarities to the YadA structure. The same is true for the binding region of Clostridium histolyticum class I collagenase (Wilson et al, 2003), which binds collagen on a tyrosine-rich surface. The catalytic domain of matrix metalloproteinases, significantly larger than the YadA head domain, is a five-stranded β-pleated sheet, and binds triple-helical coiled-coil collagen in a large cleft (Bode et al, 1999). Finally, the S. aureus CBD binds collagen in a groove that fits the collagen triple-helix structure (Symersky et al, 1997), but no such cleft or groove occurs in YadA. However, the YadA surface, formed from layers of parallel β-strands, presents a repeating binding flat surface, which can interact with the similarly flat surface of collagen. This is especially true for YadA-coated bacteria, if the YadA headgroup bends as we suggest.
The discovery of the YadA β-roll complete sequence–structure motif allowed us to search for other similar, β-roll forming sequences, even though β-rolls, like β-helices, are difficult to find by sequence comparison (cf Heffron et al, 1998). We therefore specifically searched for an array of YadA motifs followed by a YadA-like neck region, as the neck region appears to be an integral part of the YadA head domain. Such arrangements were found in a number of bacterial sequences, especially surface proteins and adhesins, from human and animal pathogens, plant pathogens and symbionts, and in cyanobacteria. Although many of the proteins found had already been identified as members of the YadA-pfam based on the similarity of the membrane anchor (Hoiczyk et al, 2000), this is the first time that it is clear that some of the head domains also share similarities with YadA. Residues 650–1206 of the large YadA-pfam member XadA (Table III) had been modelled as a large, right-handed β-helix (Ray et al, 2002). This stretch includes two YadA-like arrays, with a total of 15 extended YadA-like motifs and one nonrepetitive region after the first neck region between the arrays. The almost perfect YadA-like spacing with single neck regions next to arrays strongly suggests that the XadA model is incorrect.
The other class of proteins in YadA-pfam, the E. coli immunoglobulin-binding proteins, not only have a very similar overall architecture to YadA (head, coiled-coil and membrane anchor) but also have a strikingly similar organisation of YadA-like motifs in the N-terminal half of the sequence. Therefore, as immunoglobulin-binding proteins are also known to form extremely stable multimers (Sandt and Hill, 2001), we propose that they actually have YadA-like head domains, which, as in YadA, contribute to their extreme stability. This prediction contradicts earlier reports (Sandt and Hill, 2001).
Finally, we identified some proteins that were not known to share any similarities with the YadA head domain. In human and animal pathogens, this might mean that these proteins could bind ECM molecules, as these functions have been mapped to the YadA head domain. Among the plant pathogens and symbionts, YadA-like head domain structures might be involved in adhesion or in avoiding plant defence mechanisms. YadA-like proteins may thus be a relatively common means by which bacteria evade host responses.
We report here the first atomic structure of the head domain of a member of the lollipop adhesin family. It is a novel left-handed parallel β-roll. The head domain consists not only of the β-roll but also of the very conserved coiled neck region, which seems to stabilise the trimeric assembly of the β-roll structures. This structure may form the core of the binding domains of the other proteins. We also show that the central β-roll part binds triple-helical collagen, describe the collagen-binding surface and identify the surface residues involved in binding. Moreover, we have measured, for the first time, binding constants for the YadA collagen interaction. We have initiated cocrystallisation experiments with collagen model peptide together with more mutational and binding analysis to elucidate the interactions between YadA and ECM molecules in detail.
Materials and methods
Crystallisation of Se-Met-YadA26–241-2M and data collection
Construction of the expression vectors, production, purification and the crystallisation of YadA have been described before (Nummelin et al, 2002). Briefly, the recombinant collagen-binding fragment of YadA, YadA26–241, was expressed in E. coli and purified utilising metal affinity and size exclusion chromatography. The Se-Met-YadA26–241-2M crystallised both in sitting and in hanging drops from 11% polyethylene glycol 8000, 0.2 M sodium acetate, 0.1 M Tris–HCl (pH 6.7). The hexagonal crystals grew to a maximal size of 100 μm within 2 weeks.
For data collection, the crystal was soaked in well solution supplemented with 20% ethylene glycol and the data collection was carried out under in cryogenic conditions. The native data were collected at beam line X11 DESY, EMBL, Hamburg, using a MarCCD detector. The MAD data were collected at beam line BM-14 in ESRF, Grenoble, using a MarCCD detector (Table I). The data were indexed and the native data were rescaled in space group R32 (Table I) with the HKL package (Otwinowski and Minor, 1995).
Solving the Se-Met-YadA26–241-2M structure
The Matthews coefficient (Matthews, 1968) indicated 36% solvent content and one molecule in the asymmetric unit and, in agreement with this, SOLVE (Terwilliger and Berendzen, 1999) found two selenium sites. Amino acids 63–194 could be traced with ArpWarp (Perrakis et al, 1999) and, after one round of slow cool refinement (CNS, Brünger et al, 1998) with a grouped temperature factor refinement, the protein model was further extended to residue 218, and Rfree and Rwork were at this stage 28.8 and 25.9%, respectively. A few more rounds of refinement lowered Rfree and Rwork to 26.5 and 24.2%, respectively, and then water atoms were added after an individual temperature factor refinement in two stages, giving R-factors of 23.9 and 20.1%. In the end, residues 33–50 were added to the N-terminus, resulting in final R-factors of 22.9 and 19.7% after a minimisation refinement followed by individual temperature factor refinement. Refinement and model building were carried out with CNS and O, respectively (Jones et al, 1991; Brünger et al, 1998).
Molecular replacement
To solve the native YadA26–241 structure, the Se-Met-YadA26–241-2M structure without waters was used as a search model for a molecular replacement. Rotation and translation search gave two top solutions, and one of them was correct as judged by an Rfree of 36% after rigid body minimisation. Simulated annealing refinement of the structure lowered the R-factors to 28%, after which the residues 219–221, 32, 51 and 62 together with 100 waters were added to the model in two rounds of refinement, resulting in R-factors of 22.1 and 22.0% using individual temperature factor refinement. Finally, alternate conformations were added to the model and all the water atoms were checked, resulting in final R-factors of 20.4 and 19.5% (Table I). The quality of the model was analysed using PROCHECK (Laskowski et al, 1993) and the hydrogen bonding by HBPLUS (McDonald and Thornton, 1994).
Site-directed mutagenesis
Mutations were performed by inverse polymerase chain reaction (PCR; Byrappa et al, 1995). Plasmid pHN-1 with the yada26–241 gene from Y. enterocolitica O:3 (Nummelin et al, 2002) was used as a template and Pfu DNA polymerase (Promega) for amplification. The PCR products were purified from agarose gel, treated with DpnI to digest the template DNA and purified again from agarose gel, self-ligated and transformed to E. coli strain XL-1 blue (Promega). Transformants were tested for YadA expression and the productive clones were sequenced. Finally, the clones were transformed to E. coli production strain M15(pREP4) and the production and purification were performed as for the wild type (Nummelin et al, 2002). The purified mutant proteins were analysed by native and SDS–PAGE to ensure the native state and full-length protein size.
Collagen-binding studies
SPR using a Biacore 2000 (Biacore AB, Sweden) was used to study the interaction of YadA26–241 and the mutants with type I bovine collagen (Sigma) and to determine the apparent dissociation constants (KD(app)). The coupling of the collagen to a sensor chip CM5 (Biacore AB) was carried out according to the manufacturer's instructions via primary amino groups. First, collagen was dissolved in 0.1 M HCl to a concentration of 1 mg/ml, which was then further diluted with 10 mM Na-acetate (pH 5.5) buffer to a final concentration of 50 μg/ml. After activation, this solution was passed over the activated surface with a flow rate of 5 μl/min, resulting in 3700 response units (RU) of immobilised collagen.
Serial dilutions of each protein (YadA26–241 and the mutants) were passed over the immobilised collagen and the binding was monitored in a running buffer of 10 mM HEPES (pH 7.4), 150 mM NaCl and 0.05% P-20 surfactant versus time. The measurements for the native YadA26–241 and for the mutants Q124A-K125A, K148A-D150A, E190A-S191A, N166A-Y169A and R133A were performed with seven concentrations between 10 and 5000 nM. The rest of the mutants were measured using six or seven higher concentrations (1000–50 000/100 000 nM) as responses were too weak at the lower concentrations. The binding of mutants to collagen was also assayed by collagen affinity blotting using purified protein as described earlier (El Tahir et al, 2000).
Data analysis
The response immediately after injection of 180 s was assumed to correspond to the equilibrium state. The response for each concentration was corrected for bulk errors by subtracting the response for an uncoupled reference surface at that concentration. Then a plot of corrected responses (RUcorr) at a specific protein concentration [P] versus protein concentration [P] for each case was nonlinearly fitted to a one-site binding model, represented by (Matsushita et al, 2001)
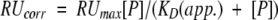 |
1 |
Docking
Protein–protein docking was performed with the soft docking program BIGGER (Palma et al, 2000). As a starting model for collagen probe, we used the structure of (Pro-Hyp-Gly)5Pro-Hyp-Ala-(Pro-Hyp-Gly)4 (PDB code 1CAG), but because BIGGER did not recognise Hyp, these residues were mutated to Pro in O and the modified peptide (PPG)5-PPA-(PPG)4 was used as a docking search probe. The docking target was the refined model of the trimeric YadA26–241 excluding waters. The docking results were then combined with the experimental collagen-binding results by filtering the search results so that the distance from the most critical residues (V98, N99, D180 and E182) was set to 4 Å, the distance from least critical residues (E190, S191, Q124 and K125) to 6 Å and from the rest of the mutated residues to 5 Å. The filtered results were then visually inspected.
Figure preparation
Unless otherwise indicated in the figure legends, all images were composed with MOLSCRIPT 2.0 (Kraulis, 1991) and rendered with Raster3D (Merritt and Bacon, 1997).
PDB accession code
The coordinates and structure factors for YadA26–241 are available in the Protein Data Bank under accession code 1P9H.
Supplementary Material
Supplementary Figure 1
Ramachandran plot. Clusters for conserved Gly residues in the right upper corner (G, plus one in the left-lower corner) and for the turn residues in the left-handed helix area (T) are indicated.
Supplementary Figure 2
An alternative way for connectivity of a separate N-terminal loop with the rest of the monomer. The colouring is the same as in Figure 1C.
Acknowledgments
We thank Dr Yasmin El Tahir for helpful discussions and M.Sc. Tomas Strandin for help with Biacore measurements. We also thank beam line scientists at beam lines X11, EMBL, Hamburg, Germany and BM14, ESRF, Grenoble, France for their assistance. This work was supported by ISB (National Graduate School in Informational and Structural Biology), the Academy of Finland (Nos 16815 and 6301s440 to AG and 45820, 50441 and 42105 to MS) and the Sigrid Juselius Foundation.
References
- Aghajari N, Van Petegem F, Villeret V, Chessa J-P, Gerday C, Haser R, Van Beeumen J (2003) Crystal structures of a psychrophilic metalloprotease reveal new insights into catalysis by cold-adapted psroteases. PROTEINS: Struct Funct Genet 50: 636–647 [DOI] [PubMed] [Google Scholar]
- Balligand G, Laroche Y, Cornelis G (1985) Genetic analysis of virulence plasmid from a serogroup 9 Yersinia enterocolitica strain: role of outer membrane protein P1 in resistance to human serum and autoagglutination. Infect Immun 48: 782–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann U (1994) Crystal structure of the 50 kDa metallo protease from Serratia marcescens. J Mol Biol 242: 244–251 [DOI] [PubMed] [Google Scholar]
- Baumann U, Wu S, Flaherty KM, McKay DB (1993) Three-dimensional structure of the alkaline protease of Pseudomonas aeruginosa: a two-domain protein with a calcium-binding parallel beta roll motif. EMBO J 12: 3357–3364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode W, Fernandez-Catalan C, Tschesche H, Grams F, Nagase H, Maskos K (1999) Structural properties of matrix metalloproteinases. Cell Mol Life Sci 55: 639–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31: 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Byrappa S, Gavin DK, Gupta KC (1995) A highly efficient procedure for site-specific mutagenesis of full-length plasmids using Vent DNA polymerase. Genome Res 5: 404–407 [DOI] [PubMed] [Google Scholar]
- China B, Sory M-P, N'Guyen BT, De Bruyere M, Cornelis GR (1993) Role of the YadA protein in prevention of opsonization of Yersinia enterocolitica by C3b molecules. Infect Immun 61: 3129–3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cover TL, Aber RC (1989) Yersinia enterocolitica. N Engl J Med 321: 16–24 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA, USA: DeLano Scientific [Google Scholar]
- El Tahir Y, Kuusela P, Skurnik M (2000) Functional mapping of the Yersinia enterocolica adhesin YadA. Identification of eight NSVAIG-S motifs in the amino-terminal half of the protein involved in collagen binding. Mol Microbiol 37: 192–206 [DOI] [PubMed] [Google Scholar]
- El Tahir Y, Skurnik M (2001) Mini-review: YadA, the multifaceted Yersinia adhesin. Int J Med Microbiol 291: 209–218 [DOI] [PubMed] [Google Scholar]
- Emödy L, Heesemann J, Wolf-Watz H, Skurnik M, Kapperud G, O'Toole P, Wadström T (1989) Binding to collagen by Yersinia enterocolitica and Yersinia pseudotuberculosis: evidence for yopA-mediated and chromosomally encoded mechanisms. J Bacteriol 171: 6674–6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley J, King SL, Bergelson JM, Liddington RC (1997) Crystal structure of the I domain from integrin α2β1. J Biol Chem 272: 28512–28517 [DOI] [PubMed] [Google Scholar]
- Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC (2000) Structural basis of collagen recognition by integrin α2β1. Cell 101: 47–56 [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1999) Further additions to MolScript version 14, including reading and contouring of electron-density maps. Acta Crystallogr D 55: 938–940 [DOI] [PubMed] [Google Scholar]
- Flügel A, Schulze-Koops H, Heesemann J, Kühn K, Sorokin L, Burkhardt H, von der Mark K, Emmrich F (1994) Interaction of enteropathogenic Yersinia enterocolitica with complex basement membranes and the extracellular matrix proteins collagen type IV, laminin-1 and -2, and nidogen/entactin. J Biol Chem 269: 29732–29738 [PubMed] [Google Scholar]
- Foster TJ, Höök M (1998) Surface protein adhesins of Staphylococcus aureus. Trends Microbiol 6: 484–488 [DOI] [PubMed] [Google Scholar]
- Gattiker A, Gasteiger E, Bairoch A (2002) ScanProsite: a reference implementation of a PROSITE scanning tool. Appl Bioinformatics 1: 107–108 [PubMed] [Google Scholar]
- Han YW, Miller VL (1997) Reevaluation of the virulence phenotype of the inv yadA double mutants of Yersinia pseudotuberculosis. Infect Immun 65: 327–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesemann J, Grüter L (1987) Genetic evidence that the outer membrane protein YOP1 of Yersinia enterocolitica mediates adherence and phagocytosis resistance to human epithelial cells. FEMS Microbiol Lett 40: 37–41 [Google Scholar]
- Heffron S, Moe GR, Sieber V, Mengaud J, Cossart P, Vitali J, Jurnak F (1998) Sequence profile of the parallel β helix in the pectate lyase superfamily. J Struct Biol 122: 223–235 [DOI] [PubMed] [Google Scholar]
- Hohenester E, Maurer P, Hohenadl C, Timpl R, Jasonius JN, Engel J (1996) Structure of a novel extracellular Ca2+-binding module in BM-40. Nat Struct Biol 3: 67–73 [DOI] [PubMed] [Google Scholar]
- Hoiczyk E, Roggenkamp A, Reichenbecher M, Lupas A, Heesemann J (2000) Structure and sequence analysis of Yersinia YadA and Moraxella UspAs reveal a novel class of adhesins. EMBO J 19: 5989–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Huizinga EG, van der Plas RM, Kroon J, Sixma JJ, Gros P (1997) Crystal structure of the A3 domain of human von Willebrand factor: implications for collagen binding. Structure 5: 1147–1156 [DOI] [PubMed] [Google Scholar]
- Isberg RR, Tran Van Nhieu G (1995) The mechanism of phagocytic uptake promoted by invasin–integrin interaction. Trends Cell Biol 5: 120–124 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Juliano RL, Haskill S (1993) Signal transduction from the extracellular matrix. J Cell Biol 120: 577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Kuiper MJ, Davies PL, Walker VK (2001) A theoretical model of a plant antifreeze protein from Lolium perenne. Biophys J 81: 3560–3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lee J-O, Rieu P, Arnaout MA, Liddington R (1995) Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18). Cell 80: 631–638 [DOI] [PubMed] [Google Scholar]
- Leppänen V-M, Nummelin H, Hansen T, Lahti R, Schäfer G, Goldman A (1999) Sulfolobus acidocaldarius inorganic pyrophosphatase: structure, thermostability, and effect of metal ion in an archael pyrophosphatase. Protein Sci 8: 1218–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita O, Koide T, Kobayashi R, Nagata K, Okabe A (2001) Substrate recognition by the collagen-binding domain of Clostridium histolyticum Class I collagenase. J Biol Chem 276: 8761–8770 [DOI] [PubMed] [Google Scholar]
- Matthews BW (1968) Solvent content of protein crystals. J Mol Biol 33: 491–497 [DOI] [PubMed] [Google Scholar]
- McDonald IK, Thornton JM (1994) Satisfying hydrogen bonding potential in proteins. J Mol Biol 238: 777–793 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol 277: 505–524 [DOI] [PubMed] [Google Scholar]
- Miller VL, Falkow S (1988) Evidence for two genetic loci in Yersinia enterocolitica that can promote invasion of epithelial cells. Infect Immun 56: 1242–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N, Sumikawa H, Sakakura M, Shimba N, Takahashi H, Terasawa H, Suzuki E, Shimada I (2003) Collagen-binding mode of vWF-A3 domain determined by a transferred cross-saturation experiment. Nat Struct Biol 10: 53–58 [DOI] [PubMed] [Google Scholar]
- Nummelin H, El Tahir Y, Ollikka P, Skurnik M, Goldman A (2002) Expression, purification and crystallization of a collagen-binding fragment of Yersinia adhesin YadA. Acta Crystallogr D 58: 1042–1044 [DOI] [PubMed] [Google Scholar]
- Otwinowski A, Minor W (1995) DENZO. New Haven, CT, USA: Yale University [Google Scholar]
- Palma PN, Krippahl L, Wampler JE, Moura J (2000) BIGGER: a new (soft) docking algorithm for predicting protein interactions. PROTEINS: Struct Funct Genet 39: 372–384 [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Piltz D, Vocke T, Heesemann J, Brade V (1992) Mechanism of YadA-mediated serum resistance of Yersinia enterocolica serotype O3. Infect Immun 60: 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CRH, Roderick SL (1995) A left-handed parallel β helix is the structure of UDP-N-acetylglucosamine acyltransferase. Science 270: 997–1000 [DOI] [PubMed] [Google Scholar]
- Ray SK, Rajeshwari R, Sharma Y, Sonti RV (2002) A high-molecular-weight outer membrane protein of Xanthomonas oryzae pv. oryzae exhibits similarity to non-fimbrial adhesins of animal pathogenic bacteria and is required for optimum virulence. Mol Microbiol 46: 637–647 [DOI] [PubMed] [Google Scholar]
- Roggenkamp A, Ackermann N, Jacobi CA, Truelzsch K, Hoffmann H, Heesemann J (2003) Molecular analysis of transport and oligomerization of the Yersinia enterocolitica adhesin YadA. J Bacteriol 185: 3735–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggenkamp A, Neuberger H-R, Flügel A, Schmoll T, Heesemann J (1995) Substitution of two histidine residues in YadA protein of Yersinia enterocolitica abrogates collagen binding, cell adherence and mouse virulence. Mol Microbiol 16: 1207–1219 [DOI] [PubMed] [Google Scholar]
- Roggenkamp A, Ruckdeschel K, Leitritz L, Schmitt R, Heesemann J (1996) Deletion of amino acids 29 to 81 in adhesion protein YadA of Yersinia enterocolitica serotype O:8 results in selective abrogation of adherence to neutrophils. Infect Immun 64: 2506–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Gierasch LM, Smith JA (1985) Turns in peptides and proteins. Adv Protein Chem 37: 1–109 [DOI] [PubMed] [Google Scholar]
- Rosqvist R, Skurnik M, Wolf-Watz H (1988) Increased virulence of Yersinia pseudotuberculosis by two independent mutations. Nature 334: 522–525 [DOI] [PubMed] [Google Scholar]
- Sandt CH, Hill CW (2001) Nonimmune binding of human immunoglobulin A (IgA) and IgG Fc by distinct sequence segments of the EibF cell surface protein of Escherichia coli. Infect Immun 69: 7293–7303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Koops H, Burkhardt H, Heesemann J, Kirsch T, Swoboda B, Bull C, Goodman S, Emmrich F (1993) Outer membrane protein YadA of enteropathogenic Yersiniae mediates specific binding to cellular but not plasma fibronectin. Infect Immun 61: 2513–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Koops H, Burkhardt H, Heesemann J, von der Mark K, Emmrich F (1992) Plasmid-encoded outer membrane protein YadA mediates specific binding of enteropathogenic Yersiniae to various types of collagens. Infect Immun 60: 2153–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Koops H, Burkhardt H, Heesemann J, von der Mark K, Emmrich F (1995) Characterization of the binding region for the Yersinia enterocolitica adhesin YadA on types I and II collagen. Arthritis Rheum 38: 1283–1289 [DOI] [PubMed] [Google Scholar]
- Skurnik M, Bölin I, Heikkinen H, Piha S, Wolf-Watz H (1984) Virulence plasmid-associated autoagglutination in Yersinia spp. J Bacteriol 158: 1033–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik M, Wolf-Watz H (1989) Analysis of the yopA gene encoding the Yop1 virulence determinants of Yersinia spp. Mol Microbiol 3: 517–529 [DOI] [PubMed] [Google Scholar]
- Symersky J, Patti JM, Carson M, House-Pompeo K, Teale M, Moore D, Jin L, Schneider A, DeLucas LJ, Höök M, Narayana SVL (1997) Structure of the collagen-binding domain from a Staphylococcus aureus adhesin. Nat Struct Biol 4: 833–838 [DOI] [PubMed] [Google Scholar]
- Tamm A, Tarkkanen A-M, Korhonen TK, Kuusela P, Toivanen P, Skurnik M (1993) Hydrophobic domains affect the collagen-binding specificity and surface polymerization as well as the virulence potential of the YadA protein of Yersinia enterocolitica. Mol Microbiol 10: 995–1011 [DOI] [PubMed] [Google Scholar]
- Tertti R, Skurnik M, Vartio T, Kuusela P (1992) Adhesion protein YadA of Yersinia species mediates binding of bacteria to fibronectin. Infect Immun 60: 3021–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JJ, Matsushita O, Okabe A, Sakon J (2003) A bacterial collagen-binding domain with novel calcium-binding motif controls domain orientation. EMBO J 22: 1743–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KSP, Stillman TJ, Britton KL, Artymiuk PJ, Baker PJ, Sedelnikova SE, Engel PC, Pasquo A, Chiaraluce R, Consalvi V, Scandurra R, Rice DW (1995) The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 3: 1147–1158 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Ramachandran plot. Clusters for conserved Gly residues in the right upper corner (G, plus one in the left-lower corner) and for the turn residues in the left-handed helix area (T) are indicated.
Supplementary Figure 2
An alternative way for connectivity of a separate N-terminal loop with the rest of the monomer. The colouring is the same as in Figure 1C.