

Abstract
The Cdc14 family of phosphatases specifically reverses proline-directed phosphorylation events. In Saccharomyces cerevisiae, Cdc14p promotes Cdk1p inactivation at mitotic exit by reversing Cdk1p-dependent phosphorylations. Cdk1p is a proline-directed kinase whose activity is required in all eukaryotes for the transit into mitosis. At mitotic commitment, Cdk1p participates in its own regulation by activating the mitotic inducing phosphatase, Cdc25p, and inhibiting the opposing kinase, Wee1p. We have investigated the ability of Schizosaccharomyces pombe Clp1p, a Cdc14p homolog, to disrupt this auto-amplification loop. We show here that Clp1p is required to dephosphorylate, destabilize, and inactivate Cdc25p at the end of mitosis. Clp1p promotes recognition of Cdc25p by the anaphase-promoting complex/cyclosome, an E3 ubiquitin ligase. Failure to inactivate and destabilize Cdc25p in late mitosis delays progression through anaphase, interferes with septation initiation network signaling, and additionally advances the commitment to mitotic entry in the next cycle. This may be a widely conserved mechanism whereby Cdc14 proteins contribute to Cdk1p inactivation.
Keywords: Cdc14 phosphatase/Cdc25 phosphatase/mitotic exit/phosphorylation/protein degradation
Introduction
In eukaryotes, the cell division cycle is driven by periodic fluctuations in the activity of cyclin-dependent kinases (Cdks) (Ohi and Gould, 1999; Nigg, 2001). Cdk activity depends on its association with a cyclin subunit, and further regulation of the complex occurs via phosphorylation (Berry and Gould, 1996). In particular, phosphorylation on Thr-14 and Tyr-15 in the ATP binding domain by Myt1 and Wee1 kinases, respectively, interferes with Cdk's ability to transfer phosphate to the target substrate (Morgan, 1997). Reversal of these phosphorylation events acts as a rate-limiting step in the activation of Cdk and is achieved by the Cdc25 family of phosphatases (Nilsson and Hoffmann, 2000).
During mitotic commitment, the activity of Cdc25p increases while that of Wee1p is downregulated (Coleman and Dunphy, 1994). Both these events are thought to be mediated at least in part through Cdk1p (cyclin B/Cdc2) phosphorylation (Izumi and Maller, 1993), and hence part of a positive feedback loop that provides a rapid switch for mitotic entry. Schizosaccharomyces pombe Cdc25p similarly becomes hyperphosphorylated and hyperactive at the G2/M transition, and elimination of this phosphorylation reduces its phosphatase activity to basal levels (Kovelman and Russell, 1996). In contrast, mitotic phosphorylation of mammalian Cdc25A, an isoform of Cdc25p, by Cdk1p protects the phosphatase from ubiquitin-mediated proteolysis during mitosis, but does not increase its specific activity (Mailand et al, 2002b). At exit from mitosis, dephosphorylation leads to its recognition and ubiquitination by the E3 ubiquitin ligase, the anaphase-promoting complex/cyclosome (APC/C). Cdc25C is similarly dephosphorylated at mitotic exit, but this dephosphorylation does not lead to its instability (Mailand et al, 2002b). SpCdc25p has been predicted to be destabilized during late mitosis (Ducommun et al, 1990; Moreno et al, 1990), but it has not been shown whether its destabilization is linked to its phosphorylation state. While PP2A phosphatases have been predicted to reverse the activating phosphorylations on Cdc25C (Clarke et al, 1993), it is not known whether PP2A accounts for all phosphatase activity counteracting activating phosphorylations of Cdc25 homologs during the exit from mitosis, nor is it known whether disruption of this Cdk1p auto-amplification loop plays any role in mitotic Cdk1p inactivation.
In Saccharomyces cerevisiae and S. pombe, analogous signal transduction pathways have evolved to control the timing of mitotic kinase inactivation and the timing of septation, respectively (Bardin and Amon, 2001; McCollum and Gould, 2001). Although initially thought to play significantly different roles, these two pathways seem to converge on the negative regulation of mitotic Cdks and the positive regulation of cytokinesis. In budding yeast, the mitotic exit network (MEN) plays essential roles in mitotic Cdk1p inactivation and nonessential roles in cytokinesis. In S. pombe, the septation initiation network (SIN) functions in late mitosis to promote actomyosin ring constriction and septum deposition and is not essential for Cdk1p inactivation (Le Goff et al, 1999). Cdk1p activity both directly and indirectly negatively regulates both these pathways in the yeasts. While considerable information is known about MEN-mediated Cdk1p inhibition, less is known about how, once initiated, SIN can inhibit any mitotic Cdk1p activity that remains in late mitosis.
The highly conserved Cdc14 family of phosphatases plays an important role in the negative regulation of Cdk1p by specifically reversing Cdk-dependent phosphorylation events (Visintin et al, 1998; Kaiser et al, 2002; Gray et al, 2003). In budding yeast, the MEN promotes full activation of Cdc14p, which in turn inactivates mitotic Cdk1p through activation of the APC/C-Cdh1 as well as through stabilization of a Cdk1p inhibitor, Sic1p (Visintin et al, 1998; Jaspersen et al, 1999). The S. pombe Cdc14p homolog, Clp1p (also named Flp1p), is not essential for mitotic Cdk1p inactivation at mitotic exit as in budding yeast. However, it does play nonessential roles in both cytokinesis and Cdk1p inhibition as clp1Δ cells display cytokinesis defects and advance prematurely into mitosis, and cells overproducing Clp1p delay Cdk1p activation (Cueille et al, 2001; Trautmann et al, 2001). This Cdk1p inhibition occurs via a mechanism different from the one used in S. cerevisiae as Clp1p is not required for dephosphorylation of the Cdh1p homolog, Ste9p, or for stabilization of the Cdk1p inhibitor, Rum1p (Cueille et al, 2001). Rather, Clp1p negatively regulates Cdk1p at the G2/M transition principally by promoting the inhibited, Tyr-15 phosphorylated form of Cdk1p (Trautmann et al, 2001).
The negative regulation of Cdk represents a conserved feature of Cdc14 family members (Gruneberg et al, 2002; Kaiser et al, 2002; Mailand et al, 2002a). The mechanism of action, however, may differ and be tailored to the unique challenges faced by different organisms. In this report, we have investigated the ability of S. pombe Clp1p to regulate Cdk1p activity negatively. Our results indicate that Clp1p functions in part to antagonize Cdc25p activation. This aspect of Cdc25p function may represent a conserved mechanism by which Cdc14 phosphatases reset Cdk1p activity to low levels, an event necessary for a coordinated mitotic exit and entry into the subsequent cycle.
Results
Clp1p inactivates Cdc25p at mitotic exit
We and others have previously shown that the S. pombe Cdc14p homolog, Clp1p phosphatase, regulates the G2/M transition by promoting the inhibited, Tyr-15 phosphorylated form of Cdk1p, and that the Wee1p inhibitory kinase is important for this effect (Cueille et al, 2001; Trautmann et al, 2001). To address whether or not Clp1p also affects Cdk1p Tyr-15 phosphorylation by influencing the function of the counterbalancing phosphatase, Cdc25p, we created two populations of synchronous cells by centrifugal elutriation both expressing endogenously tagged Cdc25p-MYC, either containing the clp1+ gene or a deletion of it. Cdc25p levels were elevated in the absence of clp1+ at all cell cycle stages examined (Figure 1A).
Figure 1.
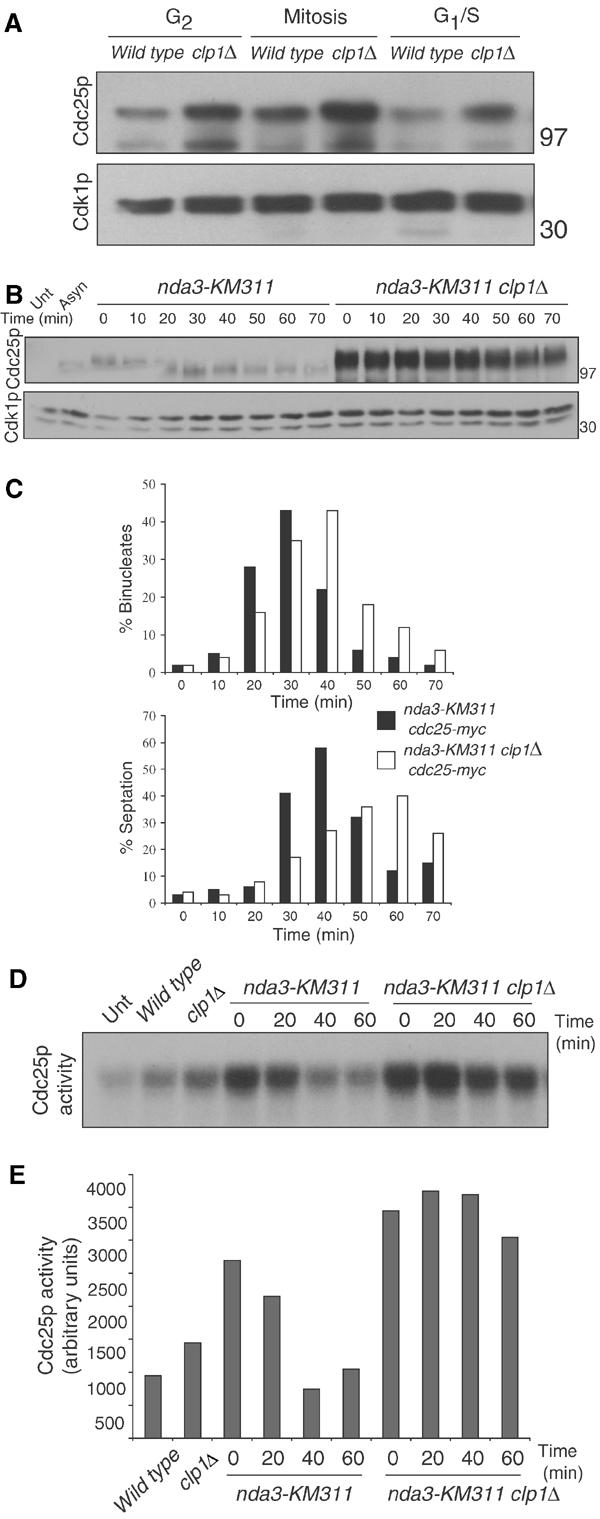
clp1+ regulation of Cdc25p. (A) cdc25-myc (KGY 3377) or cdc25-myc clp1Δ (KGY 3499) were grown to mid-log phase and synchronized by centrifugal elutriation. Samples were taken after synchronization and processed for protein (A) and cell cycle stage by DAPI staining and cell measurement analysis as follows: G2 cells were uninucleate and 8–12 μm in length, mitotic cells were uninucleate or binucleate cells with condensed chromatin, and G1/S cells contained two interphase nuclei and a septum (data not shown). Cdc25p-myc and Cdk1p (serves as loading control in each of our blots) were detected by immunoblotting with 9E10 and anti-PSTAIR antibodies, respectively. Numbers adjacent to the blots indicate the position of molecular weight standards on this and all subsequent blots. (B) nda3-KM311 cdc25-myc (KGY 3916) and nda3-KM311 cdc25-myc clp1Δ (KGY 4213) were grown to mid-log phase and synchronized by cold shift to 18°C for 7 h. Cells were then released to the permissive temperature (32°C), and samples were collected at the indicated time points. Extracts were prepared from these and processed for immunoblot analysis. (C) Completion of mitotic exit was monitored by determining binucleate formation (top panel) and septation index (lower panel). For the Cdc25p immunoblot, samples were immunoprecipitated with 9E10 antibody and immunoblotted with anti-Cdc25p antibodies. (D) Cells were synchronized as in (B) and samples were collected at the indicated time points. Extracts were prepared and immunoprecipitated with 9E10 antibody. Samples were processed for Cdc25p activity and quantified and normalized to basal levels of activity (Unt) in (E) indirectly via activation of Cdk1p's histone H1 activity as described in Materials and methods. In this experiment, Cdc25p activity was a saturable reaction (data not shown).
We next examined whether Cdc25p, a highly phosphorylated protein during mitosis (Moreno et al, 1990; Kovelman and Russell, 1996), was a target for dephosphorylation by Clp1p at mitotic exit. We utilized a reversible, cold-sensitive β-tubulin mutant, nda3-KM311, to arrest cells in an early mitotic state either in the absence or presence of clp1+. These cells were then released to the permissive temperature and monitored for Cdc25p levels and phosphorylation state. As the control cells exited mitosis and septated (Figure 1 C), Cdc25p became hypophosphorylated and slightly destabilized by the end of the time course. In the absence of clp1+, Cdc25p remained at high levels and in the hyperphosphorylated state (Figure 1B).
Clp1p may target Cdc25p at mitotic exit for dephosphorylation, and hence inactivation, to help downregulate Cdk1p activity. To test whether the hyperphosphorylated Cdc25p observed in the absence of clp1+ at mitotic exit is still active, we looked at Cdc25p activity from an independent but representative nda3-KM311 block/release experiment as before. In clp1Δ cells, Cdc25p activity was elevated and persisted for the duration of the time course, whereas wild-type cells inactivated Cdc25p within 40 min after release (Figure 1D and E).
Cdc25p localizes within the nucleus where it presumably encounters its substrate, Cdk1p, from the late stages of interphase until the late stages of anaphase (Lopez-Girona et al, 1999). To determine whether the lack of Clp1p led to increased amounts of nuclear Cdc25p, the cdc25+ endogenous locus was tagged at its C-terminus with a gfp cassette (Bahler et al, 1998) in the presence or absence of clp1+. clp1Δ cells showed a higher percentage of binucleates with nuclear Cdc25p-GFP staining (Figure 2A, arrows). To quantitate this, we created a Cdc25p-GFP strain, which also contained endogenously gfp-tagged sid4+. Sid4p is a constitutive spindle pole body component (a subcellular localization that we have not observed for Cdc25p-GFP), and served as a marker for mitotic progression (Tomlin et al, 2002). In clp1+ cells, Cdc25p-GFP nuclear staining was observed in only 17% of late anaphase cells, whereas in the absence of clp1+ this percentage increased to 67% (Figure 2B). At this time, we cannot distinguish whether the increased Cdc25p levels observed in the absence of clp1+ enhanced our ability to detect late anaphase nuclear fluorescence, as opposed to Clp1p specifically regulating rates of Cdc25p nuclear import/export (see Discussion).
Figure 2.
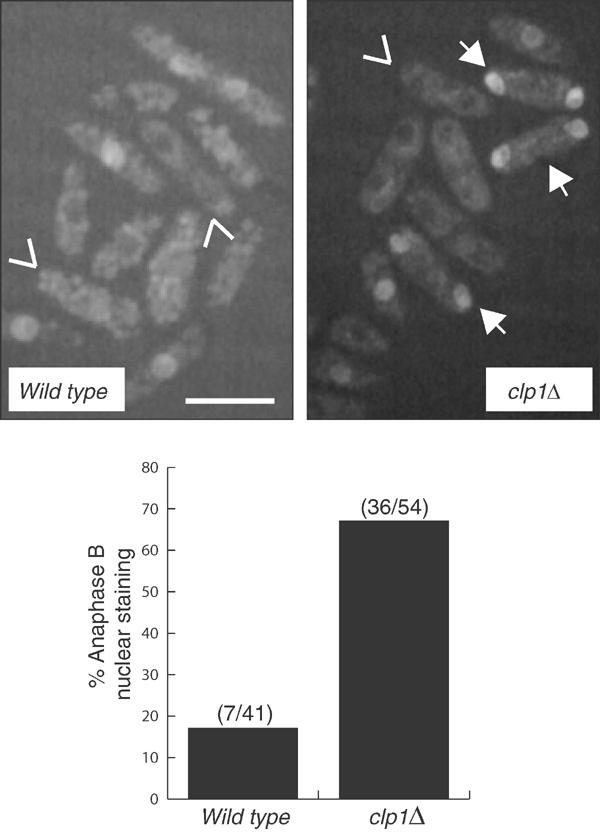
Cdc25p remains nuclear in clp1Δ anaphase B cells. (A) Compression of image stacks of cdc25-GFP (KGY 4337) and cdc25-GFP clp1Δ (KGY 4341) asynchronous cells grown at 25°C in YE medium. Tail-less arrows (∨) indicate binucleate cells without Cdc25p-GFP nuclear fluorescence and arrows (↓) indicate binucleate cells with nuclear Cdc25p-GFP. Scale bar indicates 10 μm. (B) Percentage of anaphase B cells with nuclear Cdc25p-GFP from cdc25-GFP sid4-GFP (KGY 877) and cdc25-GFP sid4-GFP clp1Δ (KGY 148) asynchronous cells grown at 25°C in YE medium. Anaphase B cells were those with two well-separated spindle pole bodies (Sid4p-GFP) and no septum.
Clp1p promotes the destabilization and ubiquitination of Cdc25p
Cdc25p homologs in higher eukaryotes are degraded via ubiquitin-mediated proteolysis, often in response to DNA damage checkpoints (Donzelli and Draetta, 2003). To address whether S. pombe Cdc25p is destabilized during the G1 phase and whether or not this was clp1+ dependent, we expressed the catalytically inactive Cdc25p-C480S from the inducible nmt promoter in cells arrested in G1 with a cdc10-V50 allele. Thiamine (repressing conditions) was then added to the cultures along with cyclohexamide to prevent protein translation, and Cdc25p levels were monitored via immunoblotting. Cdc25p was nearly undetectable (see Asterisk in Long Exposure) in cdc10-V50 arrested cells (Figure 3A). However, in the absence of clp1+, Cdc25p was stabilized. FACS analysis confirmed that these cells remained in G1 at the end of the time course (Figure 3B).
Figure 3.
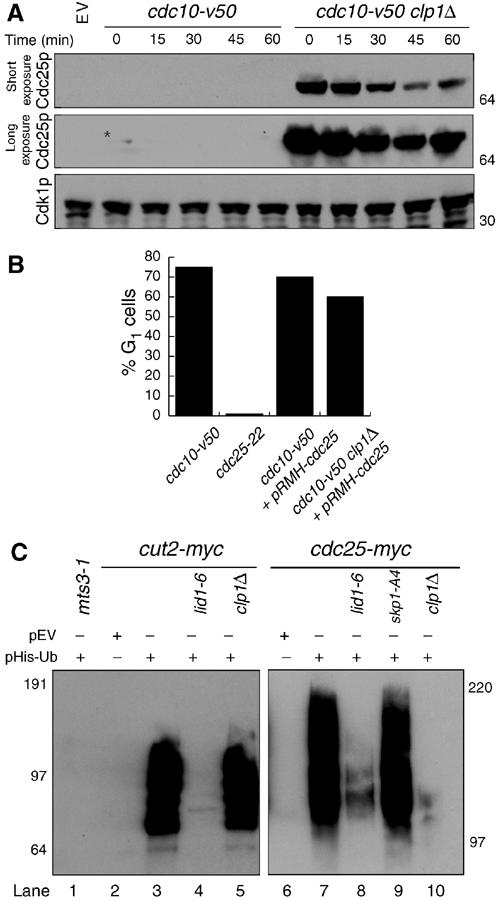
Cdc25p destabilization and ubiquitination requires Clp1p and APC/C. (A) cdc10-V50 (KGY 1744) and cdc10-V50 clp1Δ (KGY 2752) cultures expressing pREP41myc-cdc25C480S were grown in the absence of thiamine for 20 h at the permissive temperature (25°C), and then shifted to the nonpermissive temperature (36°C) for 3.5 h, at which point excess thiamine (4 μM) and cyclohexamide (100 μg/ml) were added to the cultures. Samples were taken at the indicated time points. Extracts were prepared and immunoblotted with 9E10 and anti-PSTAIR to detect Cdc25p and Cdk1p, respectively. (B) Percentage of G1 cells from (A) as determined by Sytox Green (Molecular Probes) staining and flow cytometry. (C) In vivo ubiquitination assays. mts3-1 (KGY 574) (lane 1), mts3-1 cut2-myc (KGY 1923) (lane 3), mts3-1 cut2-myc lid1-6 (KGY 1948) (lane 4), mts3-1 cut2-myc clp1Δ (KGY 878) (lane 5), mts3-1 cdc25-myc (KGY 4366) (lane 7), mts3-1 lid1-6 cdc25-myc (KGY 100) (lane 8), mts3-1 skp1-A4 cdc25-myc (KGY 102) (lane 9), and mts3-1 clp1Δ cdc25-myc (KGY 110) (lane 10) strains transformed with pREP1-His6-ubiquitin, and mts3-1 cut2-myc (KGY 1923) (lane 2) and mts3-1 cdc25-myc (KGY 4366) (lane 6) transformed with empty vector were grown at 25°C for 22 h in the absence of thiamine to induce His6-ubiquitin expression and then shifted to 36°C for an additional 4 h. Samples were collected and extracts were prepared. Ubiquitin conjugates were purified on Ni-NTA beads, separated on SDS–PAGE, and immunoblotted with 9E10 antibodies to detect Cut2p and Cdc25p.
Human Cdc25A and Cdc25C have recently been reported to be APC/C substrates, and in the case of Cdc25A, the SCF-ubiquitin-dependent pathways as well (Donzelli and Draetta, 2003). To examine whether destabilization of Cdc25p was due to ubiquitination, and which ubiquitin ligase(s) may be responsible, we employed an in vivo ubiquitination assay. As previously reported (Nefsky and Beach, 1996), Cdc25p is ubiquitinated in vivo (Figure 3C, lane 7). The use of temperature-sensitive mutations in lid1-6, an APC/C mutant (Berry et al, 1999), and skp1-A4, a mutation that renders the SCF defective (Yamano et al, 2000), revealed that APC/C activity (Figure 3C, lane 8) was required for the in vivo ubiquitination of Cdc25p, while the SCF was dispensable (Figure 3C, lane 9). Interestingly, Clp1p was also required for Cdc25p ubiquitination (Figure 3C, lane 10). This is not due to a general APC/C defect in clp1Δ cells as ubiquitination of the APC/C substrate Cut2p was unaffected (Figure 3C, lane 5).
Clp1p reverses Cdk1p-dependent phosphorylation events on Cdc25p
Knowing that Cdc14p homologs specifically remove phosphorylation events placed by proline-directed kinases (Visintin et al, 1998; Kaiser et al, 2002; Gray et al, 2003), coupled with evidence from Xenopus and human cells demonstrating that Cdk1p phosphorylates Cdc25p homologs both in vitro and in vivo (Izumi and Maller, 1993), we tested whether recombinant S. pombe Cdk1p could phosphorylate Cdc25p in vitro. Recombinant Cdk1p complex, but not the kinase inactive complex, phosphorylated recombinant MBP-Cdc25p, but not MBP alone or a mutant of Cdc25p in which all 15 of the putative S/T-P sites had been mutated to nonphosphorylatable alanine residues (Figure 4A).
Figure 4.
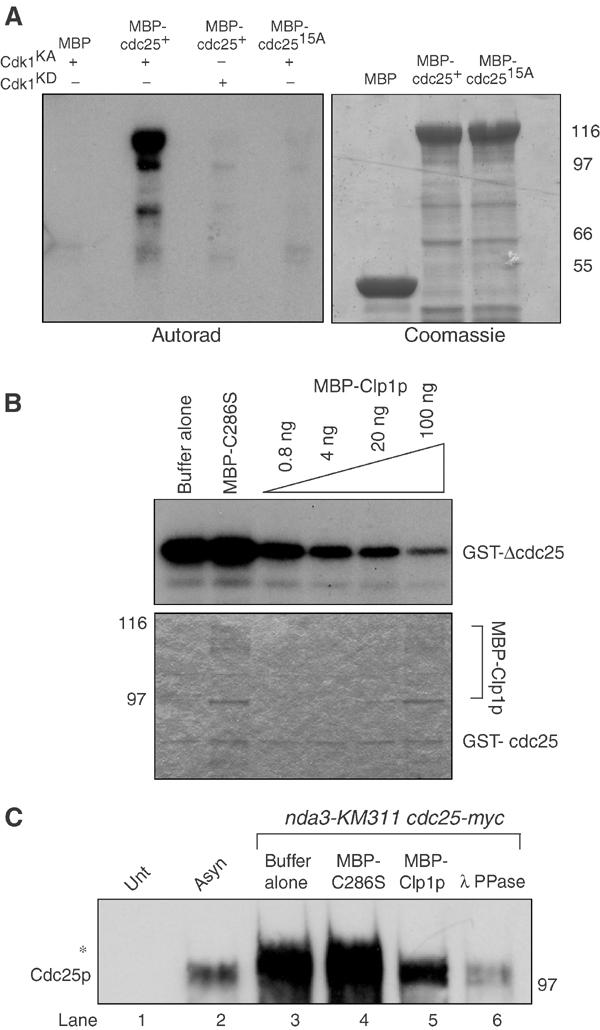
Clp1p reverses Cdk1p-dependent phosphorylation of Cdc25p. (A) Approximately 1–2 μg of MBP, MBP-Cdc25p, or MBP-Cdc25p-15A was phosphorylated in vitro with baculoviral produced and purified recombinant active (KA) or kinase dead (KD) Cdk1p complex. Reactions were separated by SDS–PAGE and analyzed by Coomassie blue staining and autoradiography. (B) Approximately 100 ng of GST-ΔCdc25p containing amino acids 1–197 was phosphorylated by recombinant Cdk1p complex in vitro as in (A), and subsequently incubated with the indicated amounts of MBP-C286S or MBP-Clp1p. Reactions were separated by SDS–PAGE, and analyzed by Coomassie blue staining (lower panel) and autoradiography (upper panel). (C) Cell pellets from nda3KM311 cdc25-myc (KGY 3916) arrested at the restrictive temperature (18°C), and wild type (KGY 246) (Unt) and cdc25-myc (KGY 3377) (Asyn) grown to mid-log phase were collected. Extracts were prepared and immunoprecipitated with 9E10 antibodies and subsequently incubated with phosphatase buffer alone (lanes 1, 2, and 3), 100 ng MBP-C286S (lane 4), 100 ng MBP-Clp1p (lane 5), or 0.5 μg Lambda phosphatase (lane 6). Reactions were separated by SDS–PAGE and immunoblotted with anti-Cdc25p antibodies. The asterisk (*) indicates the position of the hyperphosphorylated Cdc25p.
We next examined whether or not recombinant MBP-Clp1p could dephosphorylate recombinant GST-Cdc25p 1-197, which was phosphorylated in vitro by active Cdk1p complex. MBP-Clp1p, but not a phosphatase dead version in which the catalytic Cys-286 is replaced by serine (MBP-C286S) (Denu et al, 1996), removed radioactive phosphate from Cdc25p in a dose-dependent fashion (Figure 4B). Furthermore, both MBP-Clp1p and λ phosphatase, but not MBP-C286S, could dephosphorylate Cdc25p immunoprecipitated from mitotically arrested S. pombe cells (Figure 4C). Approximately five times the amount of λ PPase was added (∼500 ng) to achieve similar levels of dephosphorylation.
Clp1p and Cdc25p interact in vivo
To determine whether we could detect an interaction between Clp1p and Cdc25p in vivo, we employed a reciprocal coimmunoprecipitation assay. In this experiment, catalytically inactive Clp1p (C286S) was expressed because it has been predicted to bind with higher affinity to its substrate (Tonks and Neel, 1996). Cdc25p-MYC specifically associated with a minor amount of Clp1p-HA when expressed from a multicopy plasmid (Figure 5A). We obtained similar results when wild-type Clp1p was expressed (data not shown). The low level of interaction between Cdc25p and Clp1p likely represents both the transient nature of the interaction as well as the possibility that many proteins may compete for Clp1p binding in vivo.
Figure 5.
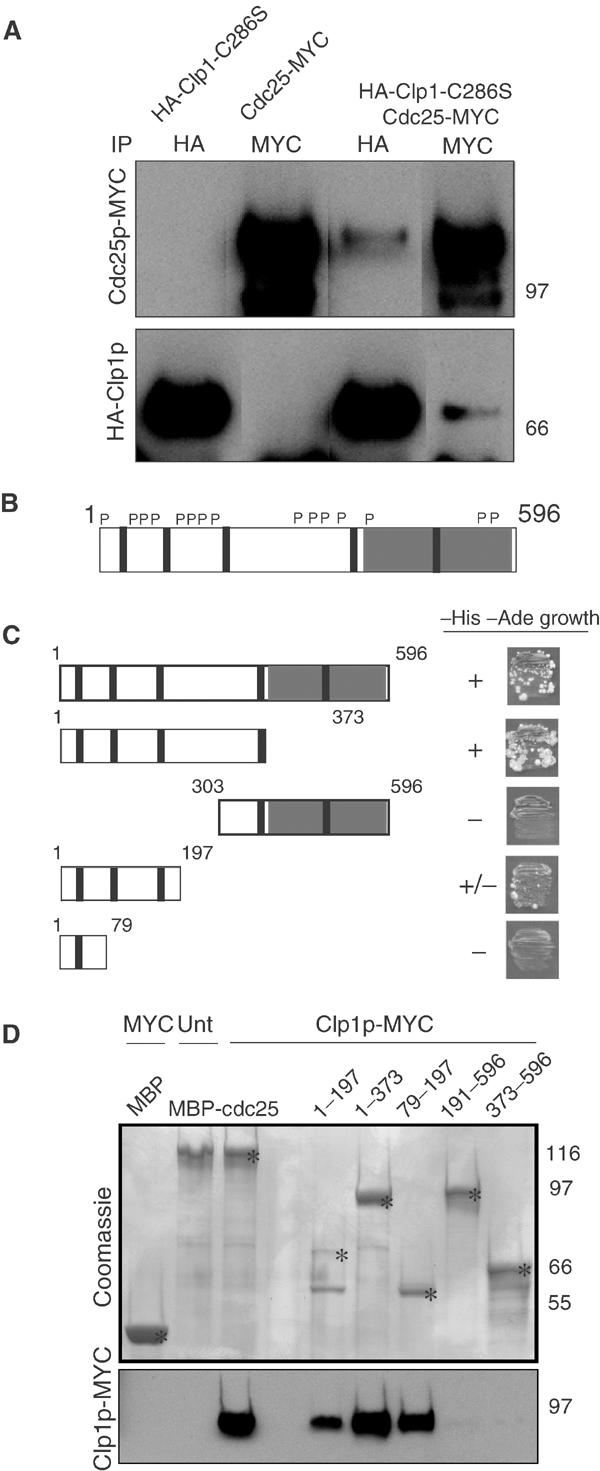
Clp1p interacts with the N-terminus of Cdc25p. (A) wild type (KGY 246) or cdc25-myc (KGY 3377) cells were transformed with empty vector or pREP41- HA-clp1C286S, and expression was induced by growth in media lacking thiamine for 18 h at 32°C. Samples were collected and extracts were prepared. Native lysates were split, immunoprecipitated with 9E10 and 12CA5 antibodies, and separated by SDS–PAGE. Membranes were probed with 9E10 and 12CA5 antibodies to detect MYC- and HA-tagged proteins, respectively. (B) Schematic representation of Cdc25p depicting carboxy terminal catalytic domain (gray box), putative Cdk1p phosphorylation sites (P), and five putative destruction boxes (∣). (C) Strain PJ206 (KGY 1296) was cotransformed with bait plasmid pGBT9:clp1 and the indicated cdc25 fragments in the prey plasmid, pGAD424, and then screened for ability (±) to support growth on –His –Ade (double selection) plates. (D) Extracts were prepared from either wild type (KGY 246) (Unt) or clp1-MYC (KGY 2882) (Clp1p-MYC) strains grown in YE media at 32°C, and incubated in the presence of the indicated MBP-Cdc25p fragments bound to amylose beads. Beads were washed extensively and samples were separated by SDS–PAGE. Proteins were either detected by Coomassie blue staining or transferred to PVDF and subsequently probed with 9E10 antibodies to detect Clp1p. Asterisks (*) indicate the position of the predicted size of the fusion protein on Coomassie-stained gel.
To identify which domains of Cdc25p (Figure 5B) might be required for interaction with Clp1p, we utilized the yeast two-hybrid assay. Interaction of Clp1p and Cdc25p depended on the N-terminal 200 amino acids of Cdc25p (Figure 5C). To confirm this, Cdc25p fragments produced as MBP fusions were tested for their ability to bind Clp1p-MYC in cell lysates. All MBP-Cdc25p fragments containing the N-terminus but not MBP alone were able to interact with Clp1p-MYC, confirming the two-hybrid data (Figure 5D).
clp1Δ cells delay mitotic Cdk1p inactivation and advance G2 due to stabilized Cdc25p
Having established that in the absence of clp1+ Cdc25p is upregulated during many stages of the cell cycle, we wanted to know whether this resulted in higher Cdk1p activity at inappropriate times. To this end, we examined synchronous cell populations either in the presence or absence of clp1+ using a cdc25-22 arrest/release protocol. At 90 min after the release of the wild-type population, the completion of cell division had nearly been reached (Figure 6A). However, in the absence of clp1+ at the 90 min time point, the septation peak had not been reached and did not occur for an additional 20 min (Figure 6A and data not shown). This could result from a septation defect or a delay at any point before this. To pinpoint the delay, we examined the microtubule cytoskeleton of the cells and quantitated the percentages of metaphase and anaphase B spindles (Figure 6A and data not shown). In the presence of clp1+, anaphase began at 30 min and spindles disassembled by 60 min as septation began (Figure 6A). This was in marked contrast to the timing of anaphase in the absence of clp1+ where spindle elongation began with a 10 min delay, and elongated spindles persisted until the end of the time course (Figure 6A). As spindle disassembly is inhibited by elevated Cdk1p activity (Wheatley et al, 1997), we examined Cdk1p activity through immunoprecipitation kinase assays. clp1Δ cells delayed Cdk1p inactivation for 20–30 min compared to control cells (Figure 6B).
Figure 6.
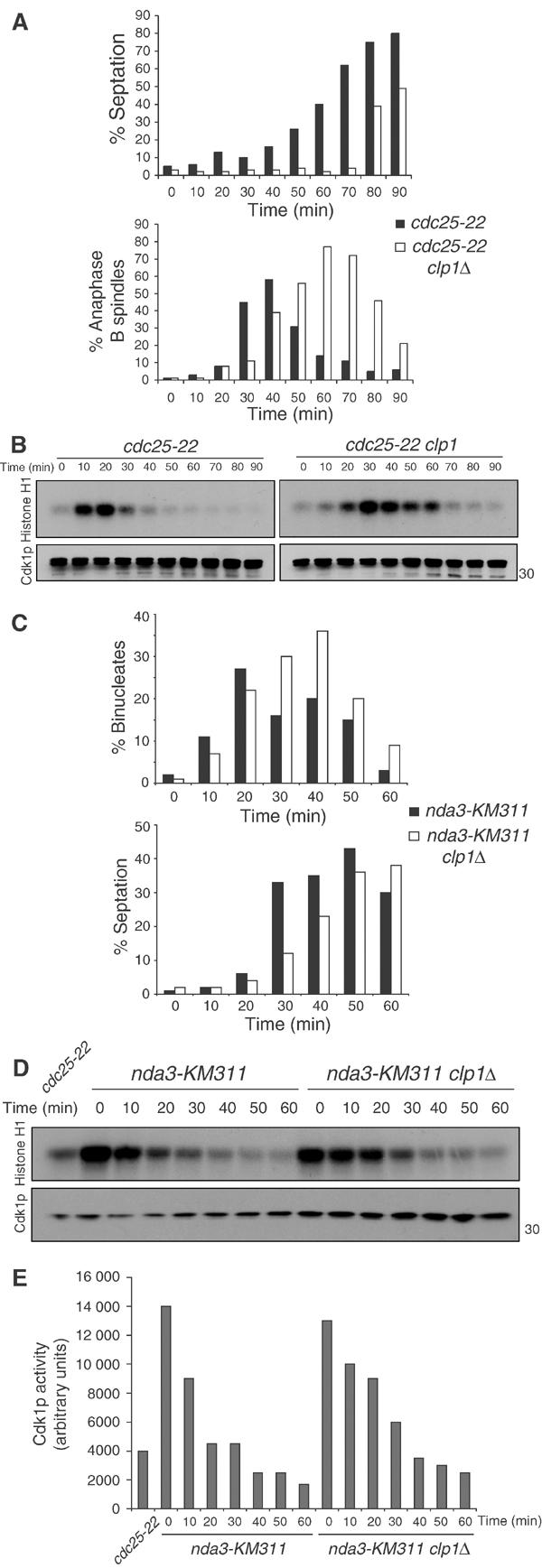
clp1Δ cells delay Cdk1p inactivation at the end of mitosis. cdc25-22 (KGY 851) and cdc25-22 clp1Δ (KGY 3380) cells were arrested in G2 by incubation at 36°C for 4 h. Cultures were then released to 25°C, and samples were taken at the indicated time points and fixed with ethanol or frozen. (A) Septation index was calculated using a light microscope (top panel). Ethanol-fixed samples were subjected to indirect immunofluorescence with anti-TAT1 antibodies (bottom panel). The presence of elongated spindles and separated DNA masses was assessed in at least 200 cells. (B) Cell pellets were lysed under native conditions and immunoprecipitated for Cdk1p with 4711 antibody. Samples were processed for histone H1 kinase activity and immunoblotted for Cdk1p levels with anti-PSTAIR. (C) nda3-KM311 (KGY 3612) and nda3-KM311 clp1Δ (KGY 3783) were arrested in prometaphase by incubation at 18°C for 6.5 h. Cells were released at 32°C, and ethanol-fixed samples and cell pellets were collected at the indicated time points. Septation index was determined as in (A) (C, lower panel) and fixed cells were stained with DAPI to visualize nuclei. The presence of separated DNA masses and no septum was assessed in at least 200 cells (C, top panel). (D) Cell pellets were analyzed as in (B) and Cdk1p activity was quantified (E).
A second method of synchronization was used to assess anaphase progression in clp1Δ cells to eliminate the possibility that the observed delay was specific to the cdc25-22 allele. nda3-KM311 cells were arrested in early mitosis in the presence or absence of clp1+, and then released to the permissive temperature to allow anaphase progression. clp1Δ cells delayed septation by 10 min (Figure 6C), and mitotic Cdk1p activity was not returned to interphase levels until 40 min after release (Figure 6D and E). This is in contrast to wild-type cells, which inactivated Cdk1p 20 min after release (Figure 6D and E). The more pronounced exit defect observed with the cdc25-22 arrest/release protocol likely represents the better synchrony achieved with this protocol.
To examine whether the mitotic exit delay was at least in part due to hyperactive Cdc25p in clp1Δ cells, we again utilized the cdc25-22 arrest/release protocol either in the presence or absence of clp1+. These cells were released to the permissive temperature to allow mitotic entry. As soon as cells entered mitosis, as judged by the formation of mitotic spindles and an elevation of Cdk1p kinase activity (Figure 7B and data not shown), the cultures were divided in half. One half of the culture was shifted to the restrictive temperature to once again inactivate Cdc25-22p, while the other half was left to progress through mitosis at the permissive temperature. As before, when kept at the permissive temperature, clp1Δ cells exited mitosis with a 20–30 min delay as judged by spindle disassembly and mitotic Cdk1p inactivation (Figures 7A and B). However, when Cdc25-22p was inactivated subsequent to Cdk1p activation, clp1Δ cells disassembled mitotic spindles and inactivated Cdk1p with only a 10 min delay (Figures 7A and B). This slight delay could result from slow inactivation of Cdc25-22p, or it could suggest that Clp1p functions through additional mechanisms to promote Cdk1p inactivation. These data argue that the mitotic exit defect of clp1Δ cells is due in part to the persistence of Cdc25p activity.
Figure 7.
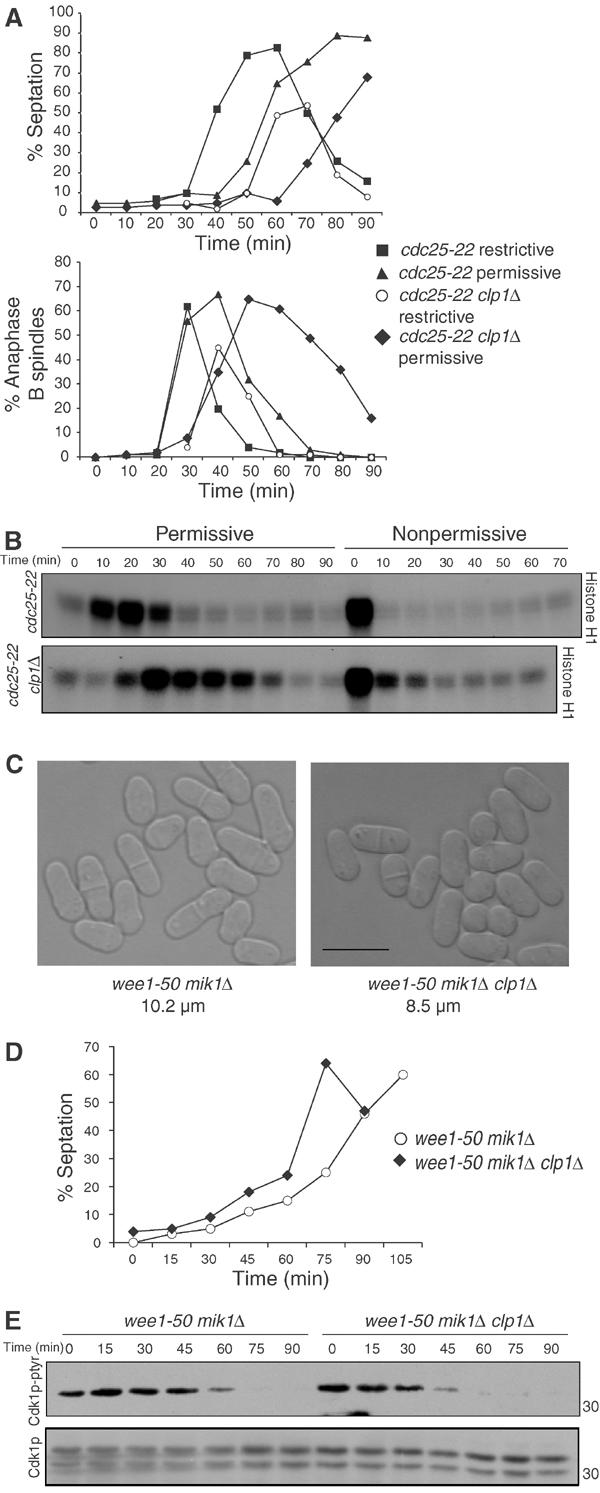
Exit delay and G2 advancement depend in large part on Cdc25p. (A) cdc25-22 (KGY 851) and cdc25-22 clp1Δ (KGY 3380) were arrested in G2 by incubation at 36°C for 4 h and released to 25°C to undergo a synchronous mitosis. The timing of mitotic entry was judged by histone H1 kinase assays as well as microtubule staining for metaphase spindles in several previous experiments and confirmed in this one. After cells entered mitosis (20 min for wild type and 30 min for clp1Δ), the cultures were spilt in half, and incubated either at 25°C (permissive) or 36°C (nonpermissive). Ethanol-fixed samples and cell pellets were taken at the indicated time points. (A) Septation was determined by light microscopy (top panel). Ethanol-fixed samples were subjected to indirect immunofluorescence with anti-TAT1 antibodies (bottom panel). The presence of elongated spindles and separated DNA masses was assessed in at least 200 cells. (B) Cell pellets were lysed under native conditions and immunoprecipitated for Cdk1p with anti-4711. Immunecomplexes were processed for histone H1 kinase activity and immunoblotted for Cdk1p with anti-PSTAIR antibodies. (C) Differential interference contrast images of wee1-50 mik1Δ (KGY 612) and wee1-50 mik1Δ clp1Δ (KGY 98) grown at 25°C in YE medium. Cell lengths at septation were scored from at least 20 cells using Openlab software (Improvision). The scale bar indicates 10 μm. (D, E) wee1-50 mik1Δ (KGY 612) and wee1-50 mik1Δ clp1Δ (KGY 98) cells grown at 25°C were synchronized by centrifugal elutriation. Cells were released to the restrictive temperature (36°C), and samples taken at the indicated time points were processed for % septation (D) and for protein. (E) Lysates were prepared under native conditions and separated on SDS–PAGE. Membranes were immunoblotted for Tyr-15 phosphorylated Cdk1p and total Cdk1p with phospho-Tyr-15 Cdk1p and PSTAIR antibodies, respectively.
We and others have shown previously that clp1Δ cells enter mitosis at a premature cell size (Cueille et al, 2001; Trautmann et al, 2001). This could result from inhibition of Wee1p, activation of Cdc25p, or a combination of the two. As we have demonstrated defective downregulation of Cdc25p levels in clp1Δ cells, we wanted to determine whether elevated Cdc25p activity was contributing to the G2 advancement of clp1Δ cells. wee1-50 mik1Δ cells lose Tyr-15 kinase activity upon shift to the restrictive temperature, and therefore the rate at which these cells enter mitosis is entirely dependent upon the amount of Cdc25p activity (Rhind et al, 1997). We examined the rates of mitotic entry in wee1-50 mik1Δ cells in the presence or absence of clp1+. A synchronous population of G2 cells was obtained by centrifugal elutriation and immediately shifted to the restrictive temperature to inactivate Wee1p kinase activity. clp1Δ cells dephosphorylated Tyr-15 of Cdk1p 15 min earlier than wild-type cells (Figure 7E). They also reached the peak of septation 15–30 min earlier than control cells (Figure 7D). Even at the permissive temperature, the triple mutant (wee1-50 mik1Δ clp1Δ) divides at a smaller cell size (8.5 μm as opposed to 10.2 μm) than the double mutant (Figure 7C). Together, these data suggest that loss of clp1+ function acts additively with loss of wee1+ function, and suggests that increased Cdc25p activity during G2 advances mitotic commitment in the absence of clp1+.
Discussion
In this report, we have investigated the mechanism by which S. pombe Clp1p, a Cdc14 family phosphatase, inhibits Cdk1p activity. Unlike S. cerevisiae, Cdc14p, which dephosphorylates and activates the APC/C activator Cdh1p and the Cdk1p inhibitor, Sic1p, to inhibit Cdk1p (Morgan, 1999), Clp1p is not required for dephosphorylation of the homologous S. pombe proteins (Cueille et al, 2001), suggesting an alternative mechanism by which Clp1p mediates Cdk1p inhibition. Indeed, our findings suggest that Clp1p contributes to Cdk1p inhibition at least in part by dephosphorylating, destabilizing, and inactivating Cdc25p, thereby disrupting the Cdk1p positive feedback loop. The increased stability and activity of Cdc25p in clp1Δ cells may help explain why clp1Δ mutants advance prematurely into mitosis, delay mitotic Cdk1p inactivation, and inefficiently activate the SIN (Figure 8).
Figure 8.
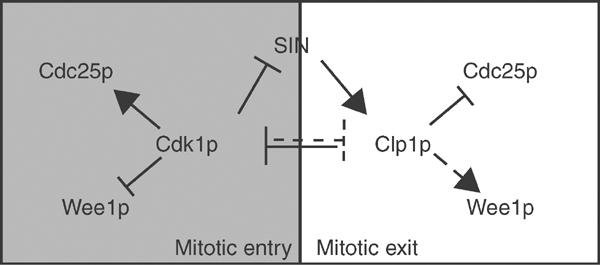
Positive and negative regulation of Cdk1p during mitosis. During mitosis, Cdk1p stimulates its own activity by activating its positive regulator (Cdc25p) and inactivating its negative regulators (Wee1p, SIN, Clp1p). As Cdk1p activity is sufficiently downregulated at mitotic exit, SIN and Clp1p function together to combat Cdk1p activation by reversing the auto-amplification loop and promoting cytokinesis.
Clp1p dephosphorylates and inactivates Cdc25p in late mitosis. One question that remains is what kinase(s) is responsible for mitotic Cdc25p activation. As Cdc14p homologs dephosphorylate residues immediately upstream of prolines (Visintin et al, 1998; Kaiser et al, 2002; Gray et al, 2003) and Cdk1p is a proline-directed kinase (Holmes and Solomon, 1996), it is tempting to speculate that Cdk1p plays an important role in Cdc25p activation. Consistent with this possibility, Cdc25p is an excellent substrate for Cdk1p in vitro. Additionally, elimination of Cdk1p sites in Xenopus Cdc25C abolishes its mitotic hyperphosphorylation, and in vitro Cdk1/cyclin B phosphorylation of human Cdc25C stimulates its phosphatase activity (Hoffmann et al, 1993; Izumi and Maller, 1993) Complicating this scenario, however, is evidence that the Polo kinase, Plk1, phosphorylates Cdc25C and is capable of stimulating its activity (Kumagai and Dunphy, 1996). Cdc25p phosphorylation and activation may occur in two steps, requiring both Cdk1p and Plk activities. Consistent with this, the polo box domain, contained in all Plks, binds residues in Cdc25C that have been previously phosphorylated by Cdk1p (Elia et al, 2003). Cdc14p homologs may dephosphorylate only Cdk1p sites of phosphorylation, but this may additionally disrupt Polo binding and prevent further phosphorylation of Cdc25p. Identification of both Polo and Cdk1p phosphorylation sites in Cdc25p will be needed to clarify how the feedback loop operates in S. pombe.
In addition to a role in dephosphorylation, Clp1p is required for Cdc25p ubiquitination via the APC/C. Cdc25p is highly stabilized in clp1Δ cells compared to wild-type cells, particularly in late mitosis/G1. In mammals, Cdc25A and Cdc25C are both degraded via the APC/C in times of cell stress or insult (Donzelli and Draetta, 2003). Additionally, Cdc25A is protected from APC/C recognition by Cdk1p phosphorylation during mitosis (Mailand et al, 2002b). Dephosphorylation at mitotic exit leads to its degradation. A similar mechanism may operate in S. pombe. While APC/C-Cdh1p recognizes the KEN boxes in Cdc25A and Cdc25C (Chen et al, 2002; Donzelli et al, 2002), S. pombe Cdc25p recognition appears different as Cdc25p lacks a KEN box but instead possesses five potential D boxes. In the future, it will be interesting to assess the role of these motifs in Cdc25p stability and to determine whether phosphorylation protects Cdc25p from ubiquitination.
Although our data implicate the APC/C in S. pombe Cdc25p degradation as occurs in higher eukaryotes (Donzelli and Draetta, 2003), the HECT domain containing ubiquitin ligase, Pub1p, has also been shown to be involved in Cdc25p destabilization (Nefsky and Beach, 1996). pub1Δ mutants accumulate Cdc25p to high levels and, interestingly, display a semi-wee phenotype (Nefsky and Beach, 1996), similar to that seen in clp1Δ mutants. Indeed, we were still able to detect low levels of Cdc25p ubiquitination in the absence of both APC/C and Clp1p function (Figure 3C), which may be attributable to Pub1p activity (Nefsky and Beach, 1996). Thus, the APC/C may function in concert with Pub1p to regulate Cdc25p stability in S. pombe.
Our results indicate that Clp1p may additionally affect Cdc25p nuclear exclusion. Cdc25p homologs are regulated via their subcellular localization, being cytoplasmic during the majority of interphase and nuclear in mitosis. Regulated nuclear export of Cdc25p and interaction with 14-3-3 proteins during interphase and in times of DNA damage checkpoint activation prevent its nuclear accumulation and premature Cdk1p activation (Takizawa and Morgan, 2000). In mammals, Plk1 phosphorylates and inactivates the Cdc25C nuclear export sequence (NES) to allow Cdc25C nuclear retention and mitotic entry (Toyoshima-Morimoto et al, 2002). How the balance is swayed to allow S. pombe Cdc25p to accumulate in the nucleus and then be removed in late mitosis is unknown, but it too may result from post-translational modifications and interactions with 14-3-3 proteins. In S. pombe, 14-3-3 mutants fail to remove Cdc25p properly from the nucleus during late mitosis as in clp1Δ mutants (Lopez-Girona et al, 1999). It seems plausible that 14-3-3 proteins could fail to interact with mitotic, hyperphosphorylated Cdc25p, and dephosphorylation during mitotic exit by Clp1p might allow for 14-3-3 recognition and nuclear exclusion of Cdc25p.
One question arising from our work is how Clp1p disruption of the Cdk1p feedback loop is restricted temporally to allow the burst of Cdc25p activity required for the G2/M transition. Although released from its nucleolar inhibition early in mitosis, Clp1p is highly phosphorylated at this stage (Cueille et al, 2001), and this phosphorylation may inhibit its phosphatase activity. Clp1p serves as an excellent substrate in vitro for Cdk1p (our unpublished results), and this may couple Clp1p activation to Cdk1p inactivation. The regulation of Cdc14 family members by phosphorylation may be a conserved mechanism as Cdc14A is also hyperphosphorylated in mitosis (Kaiser et al, 2002).
Our results suggest that Clp1p functions in late mitosis to combat Cdk1p activity by promoting its Tyr-15 phosphorylation. As full Cdc13p destruction is never witnessed in late mitosis of wild-type S. pombe cells (Chang et al, 2001), and the Cdk1p inhibitor Rum1p plays no obvious roles in Cdk1p inactivation (Blanco et al, 2000), S. pombe cells may utilize Tyr-15 phosphorylation of Cdk1p as a mechanism to further inactivate Cdk1p. Evidence from S. cerevisiae points to the fact that in times of prolonged spindle checkpoint activation, cells can adapt and inactivate Cdk1p through inhibitory phosphorylation, as occurs in cdc55Δ mutants (Minshull et al, 1996). Bypass of this arrest does not affect the cyclin B, Clb2p, proteolysis, but instead relies on Tyr-19 phosphorylation to inhibit Cdk1p activity (Minshull et al, 1996).
Failure to inactivate and degrade Cdc25p at mitotic exit in clp1Δ cells results in a failure to inactivate properly Cdk1p in late mitosis and advances the G2/M transition. As Cdk1p activity acts antagonistically to SIN function (Guertin et al, 2000; Chang et al, 2001), we propose that Clp1p functions to maintain Cdk1p activity below a certain threshold to allow SIN to stay active and properly execute cytokinesis (Figure 8). Elevated Cdk1p activity in late mitosis may explain why a small percentage of clp1Δ cells fail at cytokinesis (Cueille et al, 2001; Trautmann et al, 2001). Genetic and biochemical evidence additionally suggests that Wee1p is an essential effector of Clp1p-mediated Cdk1p inhibition (Cueille et al, 2001; Trautmann et al, 2001). Clp1p might similarly dephosphorylate and activate Wee1p at mitotic exit and/or promote its stabilization, as Wee1p is known to be destabilized upon mitotic entry in S. pombe (Aligue et al, 1997). In the future, it will be interesting to determine whether Clp1p targets Wee1p.
Studies into the functions of Cdc14 homologs from higher eukaryotes have only recently been described (Trautmann and McCollum, 2002). From these studies however, it is clear that they function to antagonize Cdk-dependent phosphorylation events during the cell cycle (Kaiser et al, 2002). As Cdc25A and Cdc25C are Cdk1p substrates, they are also excellent candidate substrates for one or both of the Cdc14 mammalian homologs. Additionally, because Cdc14A activates the APC/C-Cdh1 in vitro (Bembenek and Yu, 2001), Cdc14A may regulate Cdc25A stability via two mechanisms. It will be interesting to determine whether Cdc25p inactivation is a common mechanism utilized by Cdc14 homologs to combat Cdk1p activity in late mitosis.
Materials and methods
Strains, media, and methods
The S. pombe strains used in this study (Table 1) were grown in YE or minimal medium with the appropriate supplements as previously described (Moreno et al, 1991). Strains were constructed by tetrad dissections or random spore analysis. DNA transformations were carried out by lithium acetate transformation (Keeney and Boeke, 1994). Induction of the nmt promoter (Maundrell, 1993) was achieved by growing cells in thiamine (repressing conditions) and then washing cells three times in medium lacking thiamine (inducing conditions).
Table 1.
Strains used in this study
| Strain | Genotype | Source/Reference |
|---|---|---|
| KGY 98 | h90 wee1-50 mik1::ura4+ clp1::ura4+ uar4-D18 ade6-M21X leu1-32 | This study |
| KGY 100 | h− mts3-1 cdc25-myc::ura4+ lid1-6 ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 102 | h− mts3-1 cdc25-myc::ura4+ skp1-A4 ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 110 | h− mts3-1 cdc25-myc::ura4+ clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 148 | h− cdc25-GFP:kanR sid4-GFP:kanR clp1::ura4+ ura4-D18 ade6-M210 leu1-32 | This study |
| KGY 246 | h− ade6-M210 ura4-D18 leu1-32 | Lab stock |
| KGY 574 | h− mts3-1 leu1-32 | Lab stock |
| KGY 612 | h− wee1-50 mik1::ura4+ uar4D18 ade6M21X leu1-32 | Lab stock |
| KGY 851 | h− cdc25-22 ura4-D18 ade6-M21X leu1-32 | Lab stock |
| KGY 877 | h+ cdc25-GFP:kanR sid4-GFP:kanR ura4-D18 ade6-M210 leu1-32 | This study |
| KGY 878 | h− mts3-1 cut2-myc:kanR clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 1744 | h− cdc10-v50 ura4-D18 leu1-32 | Lab stock |
| KGY 1923 | h− mts3-1 cut2-myc:kanR leu1-32 | Berry et al (1999) |
| KGY 1948 | h− mts3-1 cut2-myc:kanR lid1-6 ade6-M21X ura4-D18 leu1-32 | Berry et al (1999) |
| KGY 2752 | h+ cdc10-v50 clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 2882 | h− clp1-myc:kanR ura4-D18 ade6-M21X leu1-32 | Trautmann et al (2001) |
| KGY 3377 | h− cdc25-myc::ura4+ ura4-D18 leu1-32 | Lopez-Girona et al (1999) |
| KGY 3380 | h− cdc25-22 clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 3381 | h− clp1::ura4+ ade6-M210 ura4-D18 leu1-32 | Trautmann et al (2001) |
| KGY 3377 | h− cdc25-myc::ura4+ ura4-D18 leu1-32 | Lopez-Girona et al (1999) |
| KGY 3499 | h+ cdc25-myc::ura4+ clp1::ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 3612 | h− nda3-KM311 ura4-D18 leu1-32 | Lab stock |
| KGY 3783 | h+ nda3-KM311 clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 3916 | h+ nda3-KM311 cdc25-myc::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 4213 | h+ nda3-KM311 cdc25-myc::ura4+ clp1::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
| KGY 4337 | h− cdc25-GFP:kanR ura4-D18 ade6-M210 leu1-32 | This study |
| KGY 4341 | h− cdc25-GFP:kanR clp1::ura4+ ura4-D18 ade6-M210 leu1-32 | This study |
| KGY 4366 | h+ mts3-1 cdc25-myc::ura4+ ura4-D18 ade6-M21X leu1-32 | This study |
The cdc25 ORF was tagged at its 3′ end with the EGFP-KanR cassette as previously described (Bahler et al, 1998). KanR transformants were screened by whole-cell PCR and then visualized using fluorescent microscopy to assure proper integration and expression of the fusion protein.
Molecular biology techniques
All plasmid constructions were performed by standard molecular biology techniques. All DNA oligonucleotides were synthesized by Integrated DNA Technologies, Inc. (Iowa). Automated sequencing confirmed the presence of mutations and the fidelity of the sequence. All sequences were PCR amplified with Taq-Plus Precision (Stratagene) according to the manufacturer's protocol. Site-directed mutagenesis was carried out with Chameleon (Stratagene) or Quickchange (Stratagene) according to the manufacturer's protocols. The cdc25+ open reading frame was amplified from a genomic clone with oligonucleotides containing Nde1 and Sma1 at the 5′ and 3′ ends, respectively. The PCR product was cut with Nde1 and Sma1 and cloned into the pREP1 vector to yield pKG49. All other constructs were subcloned from this construct by restriction digests or through PCR amplification.
Cytology and microscopy
Cdc25p-GFP cells were grown in YE medium at 25°C and visualized live using an Ultraview LCI confocal microscope equipped with a 488 nm Ar ion laser (Perkin-Elmer). Z-series optical sections were taken at 0.5 μm spacing, and images were captured using Ultraview LCI software (version 5.2, Perkin-Elmer) and processed using Velocity software (version 1.4.2; Improvision). To visualize DNA and microtubules, cells fixed with ethanol were stained with DAPI (4,6-diamidino-2-phenylindole) and anti-TAT1 (tubulin) antibodies, respectively, as described (Chang et al, 2001). FACS analysis was performed as described (Breeding et al, 1998).
Immunoprecipitations and immunoblots
Whole-cell lysates were prepared in NP-40 buffer as described either in native or denaturing conditions (Gould et al, 1991). Native lysates were subjected to immunoprecipitation with anti-Cdc2 (4711), anti-HA, or anti-MYC antibodies. Immunoblot analysis was performed as described (Tomlin et al, 2002). Cdc25 activity assays were performed as described (Kovelman and Russell, 1996), except that lysates were immunoprecipitated with 9E10 antibodies. Immunoprecipitates containing Cdc25p were washed as described in Buffer C (Kovelman and Russell, 1996) supplemented with 0.1% Triton X-100, and incubated with inactive Cdk1p obtained from cdc25-22 (G2) lysates. Cdk1p was then reisolated and histone H1 kinase assays were performed as described (Gould et al, 1991). Histone H1 assays were quantified by Cherkov scintillation counting. For the phosphatase assay, native lysates were prepared as before, and immunoprecipitated with 9E10 antibodies. Beads were washed extensively in NP-40 buffer, and then twice in phosphatase assay buffer (50 mM imidazole (pH 6.9), 1 mM EDTA, 1 mM DTT). Beads were then incubated at 30°C for 45 min in phosphatase assay buffer containing recombinant phosphatases. Reactions were separated by SDS–PAGE and visualized through immunoblot analysis as described (Tomlin et al, 2002). In vivo ubiquitination assays were performed as described (Benito et al, 1998; Berry et al, 1999).
In vitro kinase and phosphatase assays
All recombinant bacterially produced proteins were purified on either glutathione beads (GST) or amylose beads (MBP) as described (Tomlin et al, 2002). Approximately 100 ng of recombinant Cdk1p kinase complex, purified from baculovirus infected insect cells as described (Yoon et al, 2002), was used to phosphorylate approximately 1 μg of bacterially produced MBP-Cdc25p in HB15 buffer supplemented with 10 μM cold ATP and 5 μCi γ P32 ATP. Reactions were incubated at 30°C for 30 min and terminated by the addition of sample buffer. Samples were boiled and separated by SDS–PAGE. Coomassie staining or autoradiography was used for the detection of proteins. For phosphatase assays, bacterially produced, soluble GST-Cdc25p 1-197 was phosphorylated by kinase active Cdk1p complex as before and recovered on glutathione beads. Beads were washed extensively in TB1 buffer (50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 1 mM EDTA, 1 mM DTT) and eluted in TB1 buffer containing 20 mM glutathione. Eluted phosphorylated proteins were incubated in the presence of recombinant phosphatase at 30°C for 45 min in phosphatase assay buffer. Reactions were terminated by the addition of sample buffer and subsequent boiling. Reactions were separated by SDS–PAGE and proteins were visualized by Coomassie staining or autoradiography. Histone H1 kinase assays of Cdk1p immunoprecipitates were performed as described (Gould et al, 1991).
Acknowledgments
We thank P Russell, K Gull, and D McCollum for providing strains and antibodies, R Fisher for providing recombinant cdc2/cdc13 baculoviruses and advice, A Feoktistova for excellent technical assistance, R Carnahan for help with microscopy, S Venkatram and D McCollum for helpful discussions, and all members of the Gould lab for advice. BAW was supported by a Multidisciplinary Basic Research Training Grant in Cancer, T32CA09592. This work was supported by the Howard Hughes Medical Institute, of which KLG is an Investigator.
References
- Aligue R, Wu L, Russell P (1997) Regulation of Schizosaccharomyces pombe Wee1 tyrosine kinase. J Biol Chem 272: 13320–13325 [DOI] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14: 943–951 [DOI] [PubMed] [Google Scholar]
- Bardin AJ, Amon A (2001) Men and sin: what's the difference? Nat Rev Mol Cell Biol 2: 815–826 [DOI] [PubMed] [Google Scholar]
- Bembenek J, Yu H (2001) Regulation of the anaphase-promoting complex by the dual specificity phosphatase human Cdc14a. J Biol Chem 276: 48237–48242 [DOI] [PubMed] [Google Scholar]
- Benito J, Martin-Castellanos C, Moreno S (1998) Regulation of the G1 phase of the cell cycle by periodic stabilization and degradation of the p25rum1 CDK inhibitor. EMBO J 17: 482–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry LD, Feoktistova A, Wright MD, Gould KL (1999) The Schizosaccharomyces pombe dim1(+) gene interacts with the anaphase-promoting complex or cyclosome (APC/C) component lid1(+) and is required for APC/C function. Mol Cell Biol 19: 2535–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry LD, Gould KL (1996) Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog Cell Cycle Res 2: 99–105 [DOI] [PubMed] [Google Scholar]
- Blanco MA, Sanchez-Diaz A, de Prada JM, Moreno S (2000) APC(ste9/srw1) promotes degradation of mitotic cyclins in G(1) and is inhibited by cdc2 phosphorylation. EMBO J 19: 3945–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeding CS, Hudson J, Balasubramanian MK, Hemmingsen SM, Young PG, Gould KL (1998) The cdr2(+) gene encodes a regulator of G2/M progression and cytokinesis in Schizosaccharomyces pombe. Mol Biol Cell 9: 3399–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Morrell JL, Feoktistova A, Gould KL (2001) Study of cyclin proteolysis in anaphase-promoting complex (APC) mutant cells reveals the requirement for APC function in the final steps of the fission yeast septation initiation network. Mol Cell Biol 21: 6681–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Z, Bower J, Lu Y, Leonard SS, Ding M, Castranova V, Piwnica-Worms H, Shi X (2002) Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin-proteasome pathway. Proc Natl Acad Sci USA 99: 1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR, Hoffmann I, Draetta G, Karsenti E (1993) Dephosphorylation of cdc25-C by a type-2A protein phosphatase: specific regulation during the cell cycle in Xenopus egg extracts. Mol Biol Cell 4: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TR, Dunphy WG (1994) Cdc2 regulatory factors. Curr Opin Cell Biol 6: 877–882 [DOI] [PubMed] [Google Scholar]
- Cueille N, Salimova E, Esteban V, Blanco M, Moreno S, Bueno A, Simanis V (2001) Flp1, a fission yeast orthologue of the S. cerevisiae CDC14 gene, is not required for cyclin degradation or rum1p stabilisation at the end of mitosis. J Cell Sci 114: 2649–2664 [DOI] [PubMed] [Google Scholar]
- Denu JM, Stuckey JA, Saper MA, Dixon JE (1996) Form and function in protein dephosphorylation. Cell 87: 361–364 [DOI] [PubMed] [Google Scholar]
- Donzelli M, Draetta GF (2003) Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep 4: 671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF (2002) Dual mode of degradation of Cdc25 A phosphatase. EMBO J 21: 4875–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducommun B, Draetta G, Young P, Beach D (1990) Fission yeast cdc25 is a cell-cycle regulated protein. Biochem Biophys Res Commun 167: 301–309 [DOI] [PubMed] [Google Scholar]
- Elia AE, Cantley LC, Yaffe MB (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299: 1228–1231 [DOI] [PubMed] [Google Scholar]
- Gould KL, Moreno S, Owen DJ, Sazer S, Nurse P (1991) Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J 10: 3297–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CH, Good VM, Tonks NK, Barford D (2003) The structure of the cell cycle protein Cdc14 reveals a proline-directed protein phosphatase. EMBO J 22: 3524–3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruneberg U, Glotzer M, Gartner A, Nigg EA (2002) The CeCDC-14 phosphatase is required for cytokinesis in the Caenorhabditis elegans embryo. J Cell Biol 158: 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Chang L, Irshad F, Gould KL, McCollum D (2000) The role of the sid1p kinase and cdc14p in regulating the onset of cytokinesis in fission yeast. EMBO J 19: 1803–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G (1993) Phosphorylation and activation of human cdc25-C by cdc2—cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J 12: 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JK, Solomon MJ (1996) A predictive scale for evaluating cyclin-dependent kinase substrates. A comparison of p34cdc2 and p33cdk2. J Biol Chem 271: 25240–25246 [DOI] [PubMed] [Google Scholar]
- Izumi T, Maller JL (1993) Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell 4: 1337–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO (1999) Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol 9: 227–236 [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Zimmerman ZA, Charbonneau H, Jackson PK (2002) Disruption of centrosome structure, chromosome segregation, and cytokinesis by misexpression of human Cdc14A phosphatase. Mol Biol Cell 13: 2289–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney JB, Boeke JD (1994) Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics 136: 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovelman R, Russell P (1996) Stockpiling of Cdc25 during a DNA replication checkpoint arrest in Schizosaccharomyces pombe. Mol Cell Biol 16: 86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (1996) Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science 273: 1377–1380 [DOI] [PubMed] [Google Scholar]
- Le Goff X, Utzig S, Simanis V (1999) Controlling septation in fission yeast: finding the middle, and timing it right. Curr Genet 35: 571–584 [DOI] [PubMed] [Google Scholar]
- Lopez-Girona A, Furnari B, Mondesert O, Russell P (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397: 172–175 [DOI] [PubMed] [Google Scholar]
- Mailand N, Lukas C, Kaiser BK, Jackson PK, Bartek J, Lukas J (2002a) Deregulated human Cdc14A phosphatase disrupts centrosome separation and chromosome segregation. Nat Cell Biol 4: 317–322 [DOI] [PubMed] [Google Scholar]
- Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J (2002b) Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21: 5911–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maundrell K (1993) Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123: 127–130 [DOI] [PubMed] [Google Scholar]
- McCollum D, Gould KL (2001) Timing is everything: regulation of mitotic exit and cytokinesis by the MEN and SIN. Trends Cell Biol 11: 89–95 [DOI] [PubMed] [Google Scholar]
- Minshull J, Straight A, Rudner AD, Dernburg AF, Belmont A, Murray AW (1996) Protein phosphatase 2A regulates MPF activity and sister chromatid cohesion in budding yeast. Curr Biol 6: 1609–1620 [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- Moreno S, Nurse P, Russell P (1990) Regulation of mitosis by cyclic accumulation of p80cdc25 mitotic inducer in fission yeast. Nature 344: 549–552 [DOI] [PubMed] [Google Scholar]
- Morgan DO (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13: 261–291 [DOI] [PubMed] [Google Scholar]
- Morgan DO (1999) Regulation of the APC and the exit from mitosis. Nat Cell Biol 1: E47–E53 [DOI] [PubMed] [Google Scholar]
- Nefsky B, Beach D (1996) Pub1 acts as an E6-AP-like protein ubiquitiin ligase in the degradation of cdc25. EMBO J 15: 1301–1312 [PMC free article] [PubMed] [Google Scholar]
- Nigg EA (2001) Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2: 21–32 [DOI] [PubMed] [Google Scholar]
- Nilsson I, Hoffmann I (2000) Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res 4: 107–114 [DOI] [PubMed] [Google Scholar]
- Ohi R, Gould KL (1999) Regulating the onset of mitosis. Curr Opin Cell Biol 11: 267–273 [DOI] [PubMed] [Google Scholar]
- Rhind N, Furnari B, Russell P (1997) Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev 11: 504–511 [DOI] [PubMed] [Google Scholar]
- Takizawa CG, Morgan DO (2000) Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr Opin Cell Biol 12: 658–665 [DOI] [PubMed] [Google Scholar]
- Tomlin GC, Morrell JL, Gould KL (2002) The spindle pole body protein Cdc11p links Sid4p to the fission yeast septation initiation network. Mol Biol Cell 13: 1203–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonks NK, Neel BG (1996) From form to function: signaling by protein tyrosine phosphatases. Cell 87: 365–368 [DOI] [PubMed] [Google Scholar]
- Toyoshima-Morimoto F, Taniguchi E, Nishida E (2002) Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 3: 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann S, McCollum D (2002) Cell cycle: new functions for Cdc14 family phosphatases. Curr Biol 12: R733–R735 [DOI] [PubMed] [Google Scholar]
- Trautmann S, Wolfe BA, Jorgensen P, Tyers M, Gould KL, McCollum D (2001) Fission yeast Clp1p phosphatase regulates G2/M transition and coordination of cytokinesis with cell cycle progression. Curr Biol 11: 931–940 [DOI] [PubMed] [Google Scholar]
- Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell 2: 709–718 [DOI] [PubMed] [Google Scholar]
- Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, Wang Y (1997) CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J Cell Biol 138: 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano H, Kitamura K, Kominami K, Lehmann A, Katayama S, Hunt T, Toda T (2000) The spike of S phase cyclin Cig2 expression at the G1-S border in fission yeast requires both APC and SCF ubiquitin ligases. Mol Cell 6: 1377–1387 [DOI] [PubMed] [Google Scholar]
- Yoon HJ, Feoktistova A, Wolfe BA, Jennings JL, Link AJ, Gould KL (2002) Proteomics analysis identifies new components of the fission and budding yeast anaphase-promoting complexes. Curr Biol 12: 2048–2054 [DOI] [PubMed] [Google Scholar]