

Abstract
The G2 DNA damage checkpoint delays mitotic entry via the upregulation of Wee1 kinase and the downregulation of Cdc25 phosphatase by Chk1 kinase, and resultant inhibitory phosphorylation of Cdc2. While checkpoint activation is well understood, little is known about how the checkpoint is switched off to allow cell cycle re-entry. To identify proteins required for checkpoint release, we screened for genes in Schizosaccharomyces pombe that, when overexpressed, result in precocious mitotic entry in the presence of DNA damage. We show that overexpression of the type I protein phosphatase Dis2 sensitises S. pombe cells to DNA damage, causing aberrant mitoses. Dis2 abrogates Chk1 phosphorylation and activation in vivo, and dephosphorylates Chk1 and a phospho-S345 Chk1 peptide in vitro. dis2Δ cells have a prolonged chk1-dependent arrest and a compromised ability to downregulate Chk1 activity for checkpoint release. These effects are specific for the DNA damage checkpoint, because Dis2 has no effect on the chk1-independent response to stalled replication forks. We propose that inactivation of Chk1 by Dis2 allows mitotic entry following repair of DNA damage in the G2-phase.
Keywords: checkpoint, Chk1, Dis2, DNA damage, PP1
Introduction
Signalling pathways known as checkpoints monitor the cell cycle and prevent the initiation of downstream events when defects are detected. Checkpoints therefore underlie the fidelity by which cells replicate with stable genetic inheritance.
The G2/M transition of the cell cycle is controlled by the cyclin-dependent kinase Cdc2 in complex with cyclin B. Cdc2 is maintained in an inactive state by phosphorylation on Tyr-15 (Y15) by Wee1-family kinases. When conditions are appropriate for mitotic entry, Y15 is dephosphorylated by Cdc25 phosphatases, leading to Cdc2 activation, phosphorylation of key substrates and initiation of mitosis (Dunphy, 1994).
The G2 DNA damage checkpoint consists of a phosphorylation cascade of well-conserved serine/threonine (S/T) protein kinases, which prevents entry into mitosis by delaying Cdc2 Y15 dephosphorylation until completion of DNA repair (O'Connell et al, 2000). In Schizosaccharomyces pombe, the ATR kinase homologue Rad3 phosphorylates the checkpoint effector kinase Chk1 on S345, and this is essential for induction of Chk1 kinase activity (Lopez-Girona et al, 2001; Capasso et al, 2002). Activated Chk1 functions to maintain Cdc2 Y15 phosphorylation by both upregulating Wee1 and downregulating Cdc25 via phosphorylation, with either event being sufficient to elicit an arrest in S. pombe (Raleigh and O'Connell, 2000).
Much information has been gained regarding the signalling pathways involved in establishing a G2 DNA damage checkpoint arrest, but the means by which checkpoint signalling is overcome to allow cell cycle re-entry are poorly understood. In Saccharomyces cerevisiae, the type IIC S/T phosphatases Ptc2 and Ptc3 are required for recovery from a checkpoint induced by DNA double-strand breaks, by dephosphorylating and inactivating the checkpoint effector kinase Rad53 (Leroy et al, 2003). However, the DNA damage checkpoint in S. cerevisiae differs markedly from those of S. pombe and mammalian cells, arresting mitosis by inhibition of the anaphase inhibitor Pds1, rather than by Cdc2 inhibition (Cohen-Fix and Koshland, 1997). Moreover, S. pombe-type IIC phosphatases have been implicated in stress signalling and osmoregulation (Shiozaki and Russell, 1995), but not DNA damage checkpoint control. Type 1 S/T phosphatase (PP1) is implicated in the M-phase entry in Xenopus oocytes, and was recently shown to dephosphorylate Cdc25 at the S287 phosphorylation site essential for DNA damage checkpoint arrest, suggesting a possible role for PP1 in antagonising the DNA damage checkpoint (Margolis et al, 2003). However, in S. pombe, deletion of either of the two PP1 homologues dis2 or sds21 has no discernible effect on cell cycle dynamics, and overexpression of Sds21 causes a cell cycle delay, whereas Dis2 overexpression has no obvious cell cycle effect (Ohkura et al, 1989; Yamano et al, 1994). Deletion of both genes is lethal, but a role for dis2 and sds21 in mitosis was indicated by aberrant chromosome segregation in dis2-11, a cold-sensitive phosphatase-dead mutant that also behaves as a semi-dominant inhibitor of Sds21 (Ohkura et al, 1988, 1989).
We hypothesised that overexpression of proteins involved in release from the G2 DNA damage checkpoint may over-ride the checkpoint, leading to precocious entry into mitosis in the presence of DNA damage. To this end, we screened for cDNAs that, when overexpressed in the presence of the DNA-damaging agent methyl methanesulphonate (MMS), caused mitotic catastrophe. We obtained three isolates of the S. pombe PP1 homologue dis2 together with an isolate of cdc25. We show here that Dis2 is required for the dephosphorylation of Chk1 kinase and normal recovery from G2 DNA damage checkpoint arrest. This is the first identified checkpoint release pathway in S. pombe, and given the high degree of conservation between the G2 checkpoint in S. pombe and higher eukaryotes, we propose that this will be a universal mechanism by which this checkpoint acting through Chk1-mediated inhibition of Cdc2 is terminated.
Results
Dis2 overexpression causes precocious entry into mitosis in the presence of DNA damage
In the presence of DNA damage, the G2-phase S. pombe cells arrest the nuclear cell cycle to allow repair, but continue to grow, resulting in cellular elongation. If the G2 checkpoint is abrogated due to loss-of-function mutations in checkpoint genes, cells enter mitosis in the presence of un- or partially repaired DNA damage. This results in mitotic catastrophe or a ‘cut' phenotype, characterised by unequal division of DNA between daughter cells, and a frequent bisection of the nucleus by the division septum (O'Connell et al, 2000). A loss-of-function mutation in a gene required for checkpoint release, on the other hand, would be predicted to result in the opposing phenotype, that is, a prolonged cell cycle arrest. Gain-of-function defects in checkpoint release genes should mimic classical checkpoint mutants, due to an ability to over-ride checkpoint arrest.
To isolate genes involved in release of the G2 DNA damage checkpoint, wild-type S. pombe cells were transformed with an nmt1 promoter-driven S. pombe cDNA library. Approximately 1.5 × 105 transformant colonies were replica-plated to minimal medium lacking thiamine, to induce cDNA overexpression, with or without the DNA-damaging drug MMS. MMS is lethal to checkpoint mutants, but has little effect on wild-type cells at the concentrations used. After 2 days at 30°C, those colonies selectively dying on MMS plates by mitotic catastrophe were retained, and plasmids were recovered in Escherichia coli. Using this strategy, one isolate of cdc25 and three isolates of dis2, one of two PP1s in S. pombe, were obtained.
To examine the role of dis2 in the DNA damage checkpoint, we analysed the effect of Dis2 overexpression on sensitivity to ionising radiation (IR)-mimetic and ultraviolet (UV) radiation-mimetic DNA-damaging drugs, MMS and 4-nitroquinoline-N-oxide (4-NQO), respectively. Wild-type cells overexpressing Dis2, together with vector only, rad3-136 and chk1Δ controls, were serially diluted and spotted onto plates lacking thiamine±MMS or 4-NQO. Cells overexpressing Dis2 were found to be hypersensitive to both MMS and 4-NQO compared with vector control cells (Figure 1A). As expected, rad3-136 and chk1Δ strains were hypersensitive to both DNA-damaging drugs. As previously reported, Dis2-overexpressing cells appeared essentially wild type in the absence of DNA damage (Figure 1B) (Yamano et al, 1994). However, while vector control cells elongated in the presence of MMS, due to a DNA damage checkpoint, a significant proportion of Dis2-overexpressing cells underwent mitotic catastrophe, although they were slightly elongated, indicating a transient interphase delay prior to mitotic entry. The sensitivity of Dis2-overexpressing cells to a rapidly delivered dose of UV-C or IR was also compared with that of control cells and checkpoint mutants by a survival assay. Dis2 overexpression did not significantly affect colony formation after UV-C radiation, and only mildly sensitised cells to IR (Figure 1C). These results suggest that Dis2 overexpression produces a semidominant checkpoint defect, causing cells to undergo catastrophic mitosis. This effect was readily observed when cells were chronically exposed to DNA damage and hence many cell cycles targeted, but was only observed to a limited extent when given an acute dose of DNA damage targeting a single cell cycle.
Figure 1.
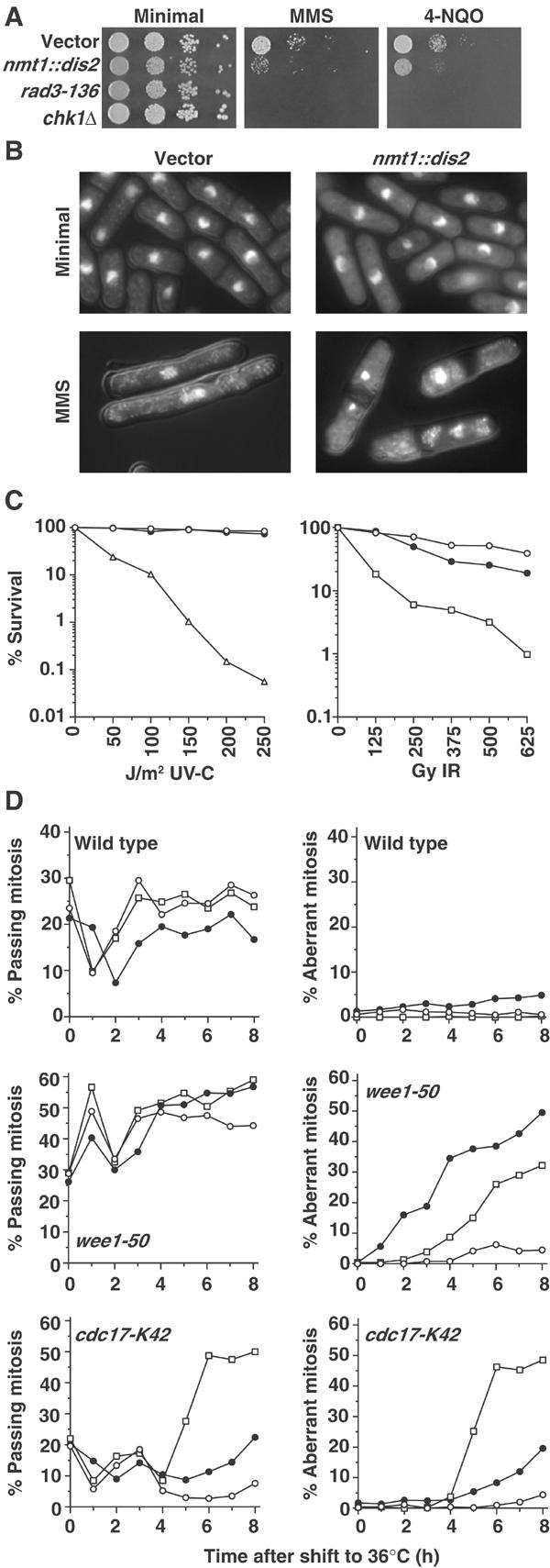
Dis2 overexpression sensitises cells to DNA damage. (A) Wild-type cells transformed with vector or nmt1::dis2 plasmid were grown in the absence of thiamine for 16 h at 30°C and, together with checkpoint mutant strains rad3-136 and chk1Δ, serial dilutions were spotted onto minimal medium agar containing no drug, 0.013% MMS or 62 ng/ml 4-NQO. (B) DAPI-stained cells of strains grown as described in (A), and treated with 0.01% MMS for 16 h. (C) Wild-type cells transformed with vector (○) or nmt1::dis2 plasmid (•), together with chk1Δ (□) and rad3-136 (▵), were treated with the indicated doses of UV-C and ionising radiation, and per cent survival compared with unirradiated controls was determined. (D) Wild-type and t.s. mutant strains transformed with vector (○) or nmt1::dis2 (•), or crossed with chk1Δ (□), were grown in the absence of thiamine for 24 h at 25°C, then shifted to the restrictive temperature of 36°C, and the per cent of cells passing mitosis or with aberrant mitoses was followed over time.
To corroborate these findings, we used a temperature-sensitive (t.s.) wee1-50 strain, which inactivates Wee1 at 36°C, resulting in synthetic lethality in all checkpoint mutants due to mitotic catastrophe (Al-Khodairy and Carr, 1992; Walworth et al, 1993). wee1-50 cells overexpressing Dis2, together with vector and chk1Δ controls, were shifted to 36°C, and the percentage of cells passing normal or aberrant mitoses was assayed over time. The percentage of cells passing mitosis was similar for each strain throughout the time course, with an initial increase at 1 h characteristic of the first ‘wee' cell cycle in which Wee1 is inactivated (Figure 1D). However, while the per cent of aberrant mitoses was negligible in vector control cells, ∼50% of wee1-50 Dis2-overexpressing cells underwent aberrant mitosis within 8 h of Wee1 inactivation. This was not seen in a wee1+ background. As expected, aberrant mitoses were also detected in chk1Δwee1-50 cells at 36°C.
Checkpoint mutants also undergo mitotic catastrophe in the absence of functional DNA ligase, entering mitosis with a large number of replication-induced DNA strand breaks (Al-Khodairy and Carr, 1992). Therefore, the effect of Dis2 overexpression in the t.s. cdc17-K42 DNA ligase mutant was also examined in a similar experiment. There was an initial dip at 1 h in cells passing mitosis for all strains, presumably due to a heat shock-induced cell cycle delay. In the vector control strain, the proportion of cells passing mitosis decreased to ⩽5% at 4–7 h due to checkpoint initiation. This decrease in mitotic cells was significantly diminished in Dis2-overexpressing cells and, moreover, there was an increase in the number of aberrant mitoses, albeit to a level somewhat lower than that observed for the chk1Δcdc17-K42 strain, which is consistent with the semidominance of Dis2 overexpression.
dis2 null cells have a prolonged checkpoint arrest in response to DNA damage
If dis2 were involved in release from the G2 DNA damage checkpoint, then deletion of the dis2 gene would be expected to result in hypersensitivity to DNA damage due to an inability to recover from checkpoint arrest. Indeed, dis2Δ cells were found to be hypersensitive to both MMS and 4-NQO (Figure 2A). As previously reported (Ohkura et al, 1989), dis2Δ cells were indistinguishable from wild-type cells when grown in the absence of DNA-damaging drugs (Figure 2B), but in the presence of MMS were hyperelongated compared with wild-type cells, indicative of a prolonged checkpoint arrest. Additionally, a small percentage of cells showed hypercondensed nuclei. Similar results were obtained for the cold-sensitive dis2-11 mutant, which was hypersensitive to MMS at the semirestrictive temperature of 22°C, and became hyperelongated in the presence of MMS at the restrictive temperature of 20°C. Cells null for the other S. pombe PP1, sds21, were not hypersensitive to or hyperelongated in the presence of MMS (Supplementary Figure 1). The sensitivity of dis2Δ cells to an acute dose of UV-C or IR was also examined by survival assays, and was found to be comparable with that of wild-type cells (Figure 2C). These data suggest that dis2Δ cells are able to recover from an acute dose of DNA damage, perhaps following a single prolonged checkpoint (see below). These effects are not extensive on cell viability, but do have a significant effect on cell proliferation. This physiology is accentuated by chronic exposure to low doses of DNA-damaging agents, resulting in additive prolonged checkpoint arrests across several cell cycles in the absence of dis2, eventually leading to cell death.
Figure 2.
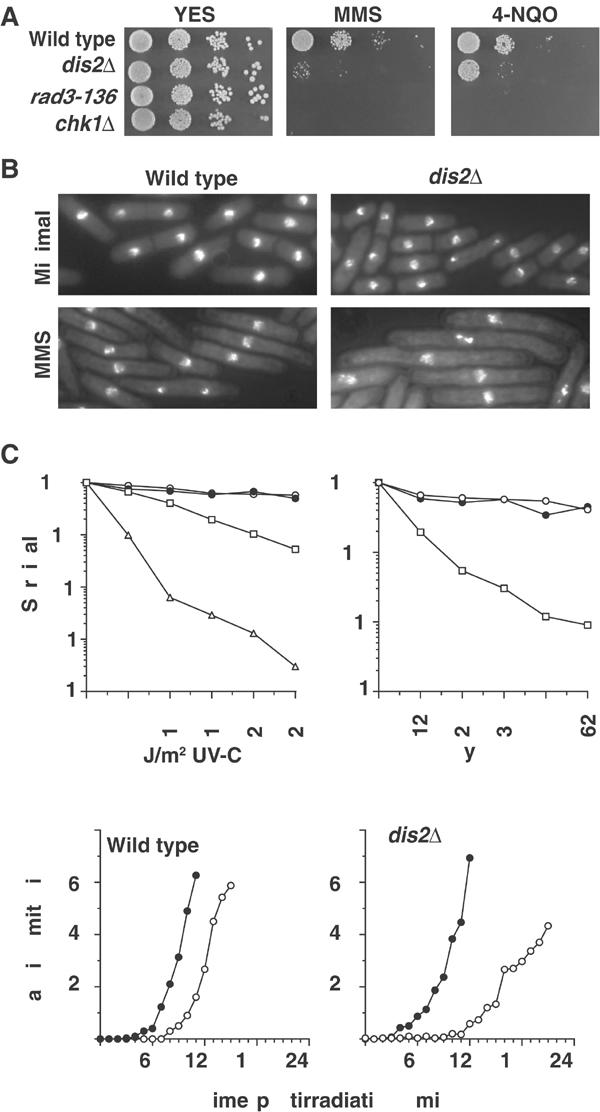
dis2Δ cells have a prolonged G2 DNA damage checkpoint. (A) Serial dilutions of the indicated strains were spotted onto YES agar containing no drug, 0.01% MMS or 62 ng/ml 4-NQO. (B) DAPI-stained cells of strains grown in the presence of 0.008% MMS for 16 h. (C) Wild type (○), dis2Δ (•), chk1Δ (□) and rad3-136 (▵) strains were treated with the indicated doses of UV-C and ionising radiation, and per cent survival compared with unirradiated controls was determined. (D) Synchronous G2-phase cultures of wild-type and dis2Δ strains, obtained by centrifugal elutriation, were treated with 150 J/m2 UV-C radiation (○) or left untreated (•), and the per cent of cells passing mitosis was followed over time.
Next, we directly analysed the checkpoint kinetics of dis2Δ cells using synchronous cultures. G2 populations of wild type and dis2Δ cells were obtained by centrifugal elutriation, and were either left untreated or given 150 J/m2 of UV-C radiation, and mitotic indices were analysed at 10 min intervals. In a wild-type strain, we observed a 30 min checkpoint arrest (Figure 2D), while a dis2Δ strain had a damage-induced arrest of ∼90 min. We conclude that deletion of dis2 results in a substantially prolonged G2 DNA damage checkpoint, consistent with a role for dis2 in checkpoint release.
DNA damage repair in dis2Δ cells
Given the precocious entry into mitosis observed for cells overexpressing Dis2 in the presence of MMS or 4-NQO, together with the lack of hypersensitivity to acute DNA-damaging regimes for either dis2Δ or Dis2-overexpressing cells, it is highly unlikely that the prolonged G2 DNA damage checkpoint arrest observed in dis2Δ cells results from compromised DNA damage repair. However, to confirm this, we examined the DNA damage sensitivity of dis2Δ combined with mutants defective in nucleotide excision (rad13-A), recombination (rhp51Δ) and postreplication (rhp18Δ) repair with the appropriate DNA-damaging drugs 4-NQO, MMS and 4-NQO/MMS, respectively. In such epistasis experiments, two genes are considered to function in different DNA damage sensitivity pathways when a double mutant is more sensitive than either parent to an appropriate lesion. In each case, we found that the dis2Δ double mutants were more sensitive than either parent (Figure 3A). Thus, dis2 does not appear to be affecting any single DNA damage repair pathway.
Figure 3.
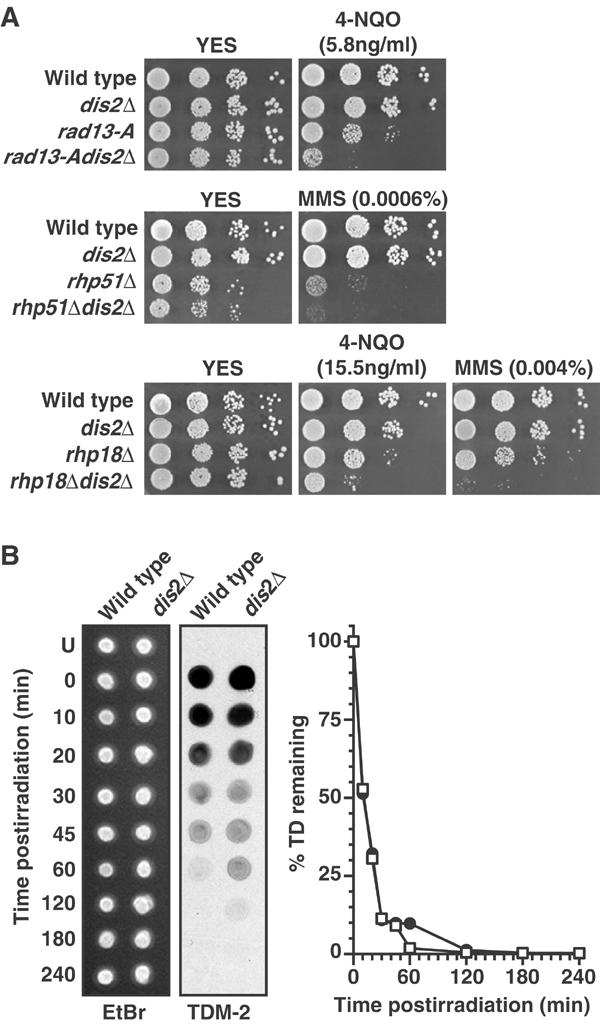
Analysis of DNA repair in dis2Δ strains. (A) Serial dilutions of strains were spotted onto YES agar containing no drug or the indicated concentrations of MMS or 4-NQO. (B) Synchronous G2-phase cultures of wild-type and dis2Δ strains, obtained by centrifugal elutriation, were treated with 150 J/m2 UV-C radiation, and the presence of thymidine dimers was monitored over time by immunoblotting dot blots of genomic DNA with an antibody against thymidine dimers (TDM-2). Ethidium bromide staining of genomic DNA was used as a loading control. The intensity of TDM-2 staining over time for wild-type DNA (□) and dis2Δ DNA (•) was quantified and expressed as a percentage of that at time 0 (U=unirradiated).
We also directly assayed the repair rate of thymidine dimers in wild type and dis2Δ strains following 150 J/m2 of UV-C radiation. The rate of repair in dis2Δ and wild-type cells was indistinguishable, and both strains excised >90% of dimers by 45 min postirradiation (Figure 3B). dis2Δ cells showed a modest retention of thymidine dimers for one time point at 60 min postirradiation, perhaps accounted for by a subpopulation of cells, but the dimers fell below levels of detection before wild type and dis2Δ cells entered mitosis in these elutriations (see Figure 2D).
dis2 affects chk1-dependent but not chk1-independent checkpoint pathways
In addition to their role in the G2 DNA damage checkpoint, rad3 and the other checkpoint rad genes are also required for the S-phase checkpoint induced by hydroxyurea (HU), which inhibits ribonucleotide reductase and results in the depletion of dNTPs and stalling of replication forks early in the S-phase. chk1 and crb2, however, are not required for the HU checkpoint (O'Connell et al, 2000). If Dis2 were affecting checkpoint release via Rad3 or the other checkpoint Rad proteins, then Dis2-overexpressing or dis2Δ cells would be expected to be hypersensitive to HU. However, neither strain was hypersensitive to HU (Figure 4A).
Figure 4.
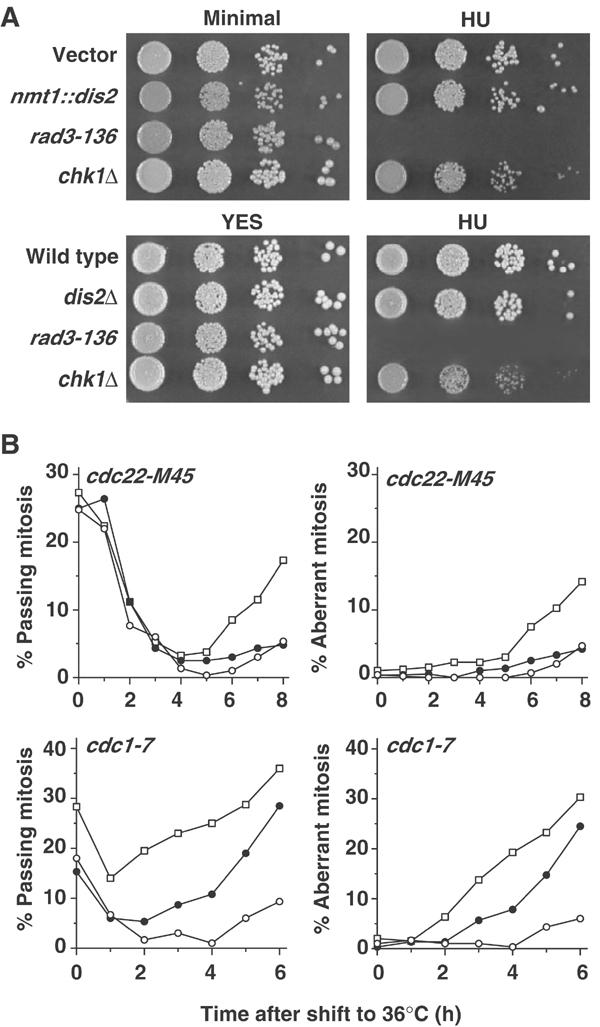
Dis2 does not affect the dNTP-depletion checkpoint. (A) Serial dilutions of the indicated strains were spotted onto minimal medium or YES agar±HU. Wild-type cells transformed with vector or nmt1::dis2 were grown in the absence of thiamine for 16 h at 30°C prior to plating. The pictures shown are of the highest HU concentrations at which chk1Δ cells were not sensitive (4 mM for minimal medium agar and 3 mM for YES agar). (B) t.s. mutant strains transformed with vector (○) or nmt1::dis2 (•), or crossed with chk1Δ (□), were grown in the absence of thiamine for 24 h at 25°C, then shifted to the restrictive temperature of 36°C, and the per cent of cells passing mitosis or with aberrant mitoses was followed over time.
The t.s. cdc22-M45 ribonucleotide reductase mutant also results in dNTP depletion and stalled replication forks at 36°C. Therefore, the effect of Dis2 overexpression on this strain was analysed, together with vector and chk1Δ controls. Consistent with their lack of HU hypersensitivity, Dis2-overexpressing cells behaved like vector control cells and had negligible aberrant mitoses when Cdc22 was inactivated at 36°C (Figure 4B).
The cdc1-7 DNA polδ t.s. mutant strain results in stalled replication forks and checkpoint arrest in the mid–late S-phase at 36°C. Unlike the dNTP-depletion checkpoint, this checkpoint requires Chk1 (O'Connell et al, 2000). Therefore, we examined the effects of Dis2 overexpression in a cdc1-7 background at 36°C. Cultures overexpressing Dis2 had a significantly higher proportion of cells passing mitosis than the vector control strain from 3 h onwards, and by 6 h had 27% aberrant mitoses compared with only 7% in control cells. By 6 h, 30% of chk1Δcdc1-7 cells had aberrant mitoses, consistent with the reported requirement of chk1 for this checkpoint.
We conclude that Dis2 overexpression over-rides chk1-dependent checkpoints, but has no effect on the chk1-independent early S-phase checkpoint.
Dis2 does not affect a Rad3-independent delay caused by Chk1 overexpression
Two key observations indicate that Dis2 is unlikely to cause checkpoint release downstream of Chk1 by altering the phosphorylation of Cdc25 and Wee1: (i) Dis2 has no discernible effect on the cell cycle in the absence of DNA damage (Ohkura et al, 1989; Yamano et al, 1994) (Figures 1 and 2); and (ii) checkpoint arrest in response to stalled replication forks, which acts via Cdc25 and Wee1 (O'Connell et al, 2000), but in a manner independent of Chk1, is not affected by Dis2 levels. To verify this assertion by yet another means, we utilised the ability of Chk1, when overexpressed, to induce a cell cycle arrest via phosphorylation of Cdc25 and Wee1. This is independent of Chk1 S345 phosphorylation and of all other checkpoint proteins. When expressed from a multicopy plasmid, Chk1 causes a sustained G2 arrest and cell death (Walworth et al, 1993), whereas from an integrated single copy plasmid, Chk1 overexpression results in a G2 delay with minimal effects on cell viability (Figure 5A). We reasoned, therefore, that if Dis2 were causing checkpoint release by dephosphorylating cell cycle proteins downstream of Chk1, then dis2Δ should lead to further cellular elongation in response to Chk1 overexpression from a single integrated copy, reducing cell viability. Conversely, Dis2 overexpression would be expected to force Chk1-overexpressing cells into mitosis, suppressing the G2 delay and accompanying cellular elongation.
Figure 5.
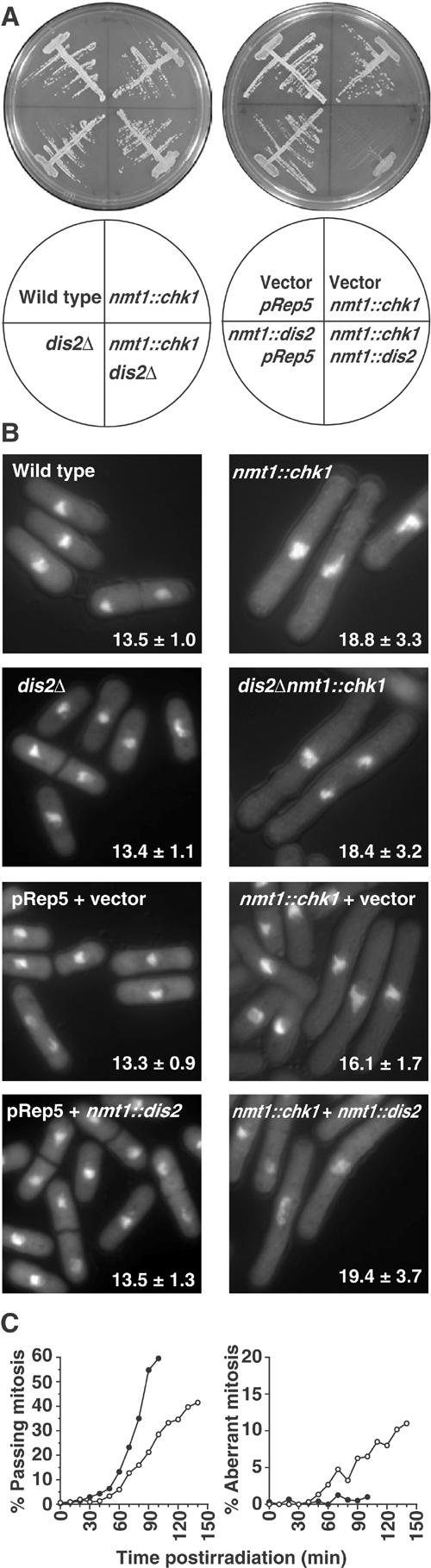
Effect of Dis2 on the G2 checkpoint requires Rad3-dependent phosphorylation of Chk1. (A) Strains were streaked onto minimal medium agar containing phloxin B and lacking thiamine, and were grown at 30°C for 3 days to determine cell viability. nmt1::chk1 refers to strains where a single copy of nmt1::chk1 was integrated into the genome, and integrated pRep5 acted as a control for these strains. Vector refers to cells transformed with pRep1, which acted as a control for cells overexpressing Dis2 from an nmt1::dis2 plasmid. (B) Strains shown in (A) were grown in the absence of thiamine for 16 h at 30°C, DAPI-stained and measured for average cell length±standard deviation (μm, n=25). (C) Synchronous G2-phase chk1Δdis2Δ cells, obtained by centrifugal elutriation, were treated with 150 J/m2 UV-C radiation (○) or left untreated (•), and the per cent of cells passing mitosis or with aberrant mitoses was followed over time (compare with Figure 2D).
As can be seen in Figure 5, dis2Δ had no effect on cell elongation or viability in response to Chk1 overexpression. Surprisingly, cooverexpression of both Dis2 and Chk1 was lethal, corresponding to a moderately enhanced cell cycle arrest. Thus, while there appeared to be a genetic interaction between combined Dis2 and Chk1 overexpression, we saw no evidence for Dis2 overexpression forcing Chk1-overexpressing cells into mitosis.
As the effects of Chk1 overexpression are independent of its phosphorylation by Rad3, any effect of Dis2 on the duration of chk1-dependent arrests is confined to circumstances where the Rad3-Chk1 cascade is intact. Considering these data together with the lack of effect on HU-induced checkpoints, our observations link Dis2 to Chk1 regulation by Rad3-mediated phosphorylation. Indeed, the prolonged checkpoint arrest of dis2Δ cells was found to be dependent on Chk1 activation, as it was completely suppressed in a chk1Δdis2Δ strain, resulting in the induction of DNA damage-dependent aberrant mitoses (Figure 5C).
Regulation of Chk1 phosphorylation and activity by Dis2
Chk1 becomes activated when phosphorylated by Rad3 on S345, an event that retards its mobility in SDS–PAGE (Capasso et al, 2002). We therefore examined the effect of Dis2 overexpression on Chk1 phosphorylation and kinase activity in response to 150 J/m2 of UV-C radiation. In response to UV radiation, Chk1 kinase activity increased ∼7-fold in an asynchronously growing chk1::HA strain (Figure 6A). Note that Chk1 appeared to be phosphorylated and activated in asynchronous wild-type cultures long after cells enter mitosis. This is likely to be because, following transient dephosphorylation upon mitotic entry, Chk1 is rephosphorylated in the G1/S-phase (see below, Figure 8B). Importantly, in Dis2-overexpressing cells, Chk1 activation was substantially attenuated, being only ∼40% that of wild type. This corresponded to a marked reduction in the levels of phosphorylated Chk1 in Dis2-overexpressing cells (Figure 6B), suggesting that Dis2 was antagonising Rad3 phosphorylation of Chk1. Dis2 did not appear to dephosphorylate globally Rad3 substrates at random. Crb2, which migrates as multiple species that are further retarded by DNA damage-induced phosphorylation (Smeets et al, 2003), showed a small reduction in phosphorylation in Dis2-overexpressing cells, but to a significantly lesser degree than that observed for Chk1 (Figure 6C). It should be noted that hyperphosphorylated Crb2 binds to phosphorylated Chk1 (Mochida et al, 2004), and so it is not clear whether the large decrease in DNA damage-induced Chk1 phosphorylation in Dis2-overexpressing cells could indirectly affect Crb2 phosphorylation. Moreover, Dis2 overexpression had no effect on the UV-induced phosphorylation of Rad9, which is also catalysed by Rad3 (Chen et al, 2001) (Figure 6D), a result consistent with the lack of effect of Dis2 on HU-induced checkpoint arrest, which is Rad9-dependent.
Figure 6.
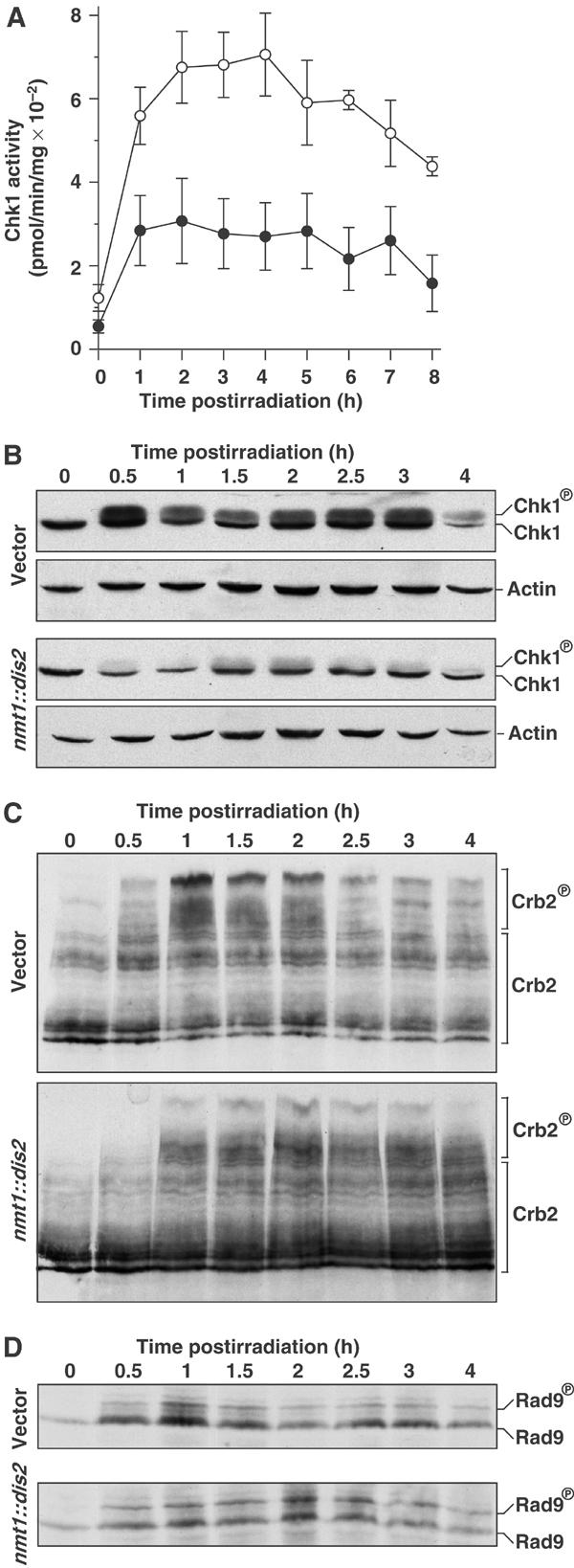
Dis2 overexpression reduces Chk1 phosphorylation and activation in response to DNA damage. (A) chk1::HA cells transformed with vector (○) or nmt1::dis2 (•) were grown in the absence of thiamine for 16 h at 30°C, treated with 150 J/m2 UV-C radiation and in vitro Chk1 kinase activity was assayed each hour. Kinase activity averaged from three independent assays is shown and error bars represent standard error. (B) Western blot of samples prepared as in (A). Chk1 was detected with anti-HA antibody, and actin was used as a loading control. (C) Western blot of crb2::18myc cells treated with 150 J/m2 UV-C radiation. Crb2 was detected with anti-myc antibody. (D) Western of rad9::HA cells treated with 150 J/m2 of UV-C radiation. Rad9 was detected with anti-HA antibody.
Figure 8.
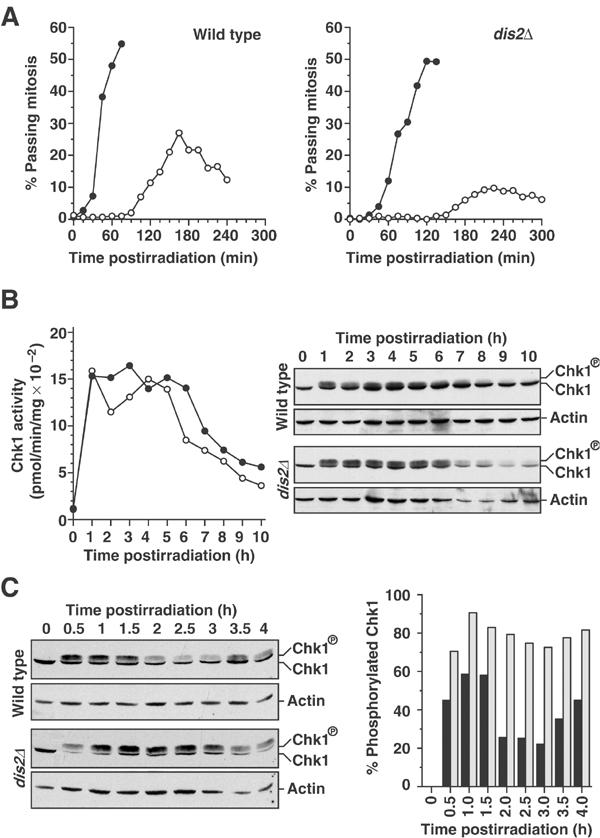
Chk1 dephosphorylation and inactivation are abrogated in dis2Δ cells. (A) Synchronous G2-phase cultures of chk1::HA and chk1::HAdis2Δ strains, obtained by centrifugal elutriation, were treated with 150 J/m2 UV-C radiation (○) or left untreated (•), and the percentage of cells passing mitoses was followed over time. (B) Chk1 kinase activity and phosphorylation of the irradiated cultures in (A) were analysed at hourly intervals by in vitro kinase assay and immunoblotting with anti-HA antibody, respectively. Data shown are representative of three such experiments. (C) Chk1 phosphorylation of samples from an experiment identical to (A), but in which samples were taken at 30 min intervals, was monitored by immunoblotting with anti-HA antibody. The percentage of Chk1 in the phosphorylated form at each time point for chk1::HA cells (solid bars) and chk1::HAdis2Δ cells (open bars) was quantified. Data shown are representative of two such experiments.
To determine whether Dis2 could dephosphorylate Chk1 directly, phosphorylated Chk1 immunoprecipitated (IPed) from a UV-C-irradiated dis2Δchk1::HA strain was incubated with Dis2 IPed from UV-C-irradiated sds21Δ cells using the D2C antibody, which recognises both Dis2 and Sds21 proteins (Stone et al, 1993). Chk1 dephosphorylation was assayed by the loss of the slower-migrating phosphorylated Chk1 band on SDS–PAGE gels. Despite the reduced mobility of a double solid-phase assay, Dis2 could readily dephosphorylate Chk1 in vitro, reducing the amount of phosphorylated Chk1 within 30 min (Figure 7A). Chk1 dephosphorylation by Dis2 was abolished by inhibitor-2 (I-2), which specifically inhibits PP1. Sds21 IPed from UV-C-irradiated dis2Δ cells had no discernible effect on Chk1 phosphorylation. The weaker band for Sds21 in Westerns of D2C antibody IPs mimicked results for whole-cell extracts (data not shown), and is consistent with published observations (Stone et al, 1993). Recombinant PP1α also dephosphorylated Chk1 within 30 min (Figure 7B).
Figure 7.
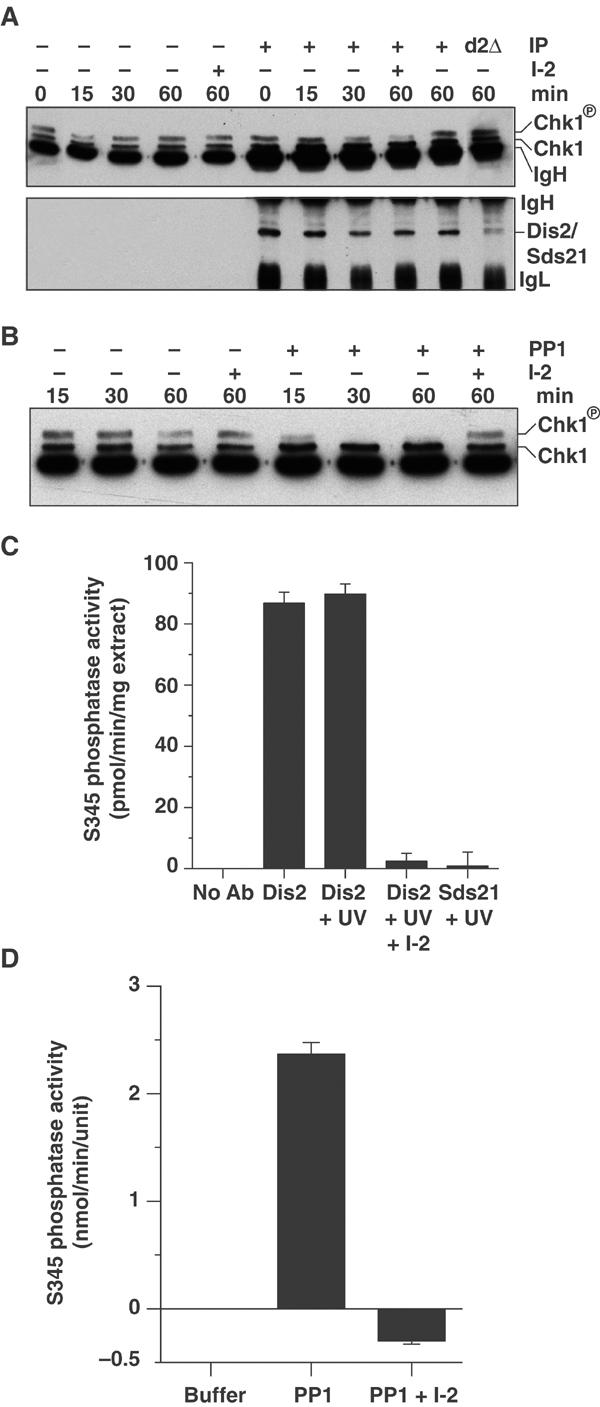
Dis2 and PP1α dephosphorylate Chk1 and phospho-S345 Chk1 peptide in vitro. (A) Dis2 IPed from sds21Δ cells and Sds21 IPed from dis2Δ cells (d2Δ), both 60 min post-treatment with 150 J/m2 UV-C radiation, were incubated at 30°C with IPs of phosphorylated Chk1-HA from dis2Δchk1::HA cells 60 min post-treatment with 150 J/m2 UV-C radiation for the indicated periods±1 μg I-2. Westerns of reactions were probed for Chk1 using anti-HA antibody and Dis2/Sds21 using D2C antibody. (B) Phosphatase assay as in (A), but using 0.05 U of recombinant rabbit PP1α in place of Dis2/Sds21 IPs. (C) Dis2 IPed from sds21Δ cells pre- or 60 min post-treatment with 150 J/m2 UV-C radiation and Sds21 IPed from dis2Δ cells 60 min post-treatment with 150 J/m2 UV-C radiation were incubated at 30°C with a 15-amino-acid phospho-S345 Chk1 peptide±1 μg I-2. Phosphate release was detected using malachite green. (D) Recombinant rabbit PP1α (0.05 U)±1 μg I-2 was incubated at 30°C with a 15-amino-acid phospho-S345 Chk1 peptide, and phosphate release was detected using malachite green.
The mobility shift of phosphorylated Chk1 in SDS–PAGE is dependent on phosphorylation of S345 (Capasso et al, 2002). Therefore, the removal of this phosphorylated band by Dis2 and PP1α suggests that they can dephosphorylate phospho-S345. To test this, phosphatase activity towards a 15-amino-acid phospho-S345 Chk1 peptide was determined for Dis2 IPed from sds21Δ cells and recombinant PP1α. Dis2 dephosphorylated the phospho-S345 Chk1 peptide at a rate of ∼90 pmol/min/mg of cell extract (Figure 7C), comparable to rates published for IPed human PP1 isoforms on phosphorylase α and a substrate peptide KRpTIRR (Puntoni and Villa-Moruzzi, 1997; Guo et al, 2002). Recombinant PP1α dephosphorylated phospho-S345 Chk1 peptide at a rate of 2.4 nmol/min/U of PP1α, where 1 U hydrolyses 1 nmol of p-nitrophenyl phosphate/min (Figure 7D). Phosphatase activity of Dis2 and PP1α was abolished by treatment with I-2, and Sds21 IPed from dis2Δ cells had negligible activity towards phospho-S345 Chk1 peptide.
To ensure that the in vivo effect of Dis2 on Chk1 phosphorylation and activity was not an artefact of overexpression, we also examined Chk1 phosphorylation and kinase activity in dis2Δ cells. Synchronous G2 populations of chk1::HA and chk1::HAdis2Δ cells were obtained by centrifugal elutriation, and either left untreated or irradiated with 150 J/m2 of UV-C. Checkpoint kinetics and Chk1 activity and phosphorylation were assayed over a time course at hourly intervals. In these experiments, where large populations of cells were required for biochemical analyses, synchrony was somewhat compromised, resulting in broader mitotic peaks and a checkpoint delay of ∼90 min in chk1::HA cultures (Figure 8A). Irradiated chk1::HAdis2Δ cells again had an extended checkpoint delay, and entered mitosis over a broad period beginning 105 min after unirradiated controls.
As previously reported for chk1::HA, 150 J/m2 of UV-C radiation resulted in a rapid increase in Chk1 activity (Figure 8B) (Capasso et al, 2002). The magnitude of this induction was always greater in extracts prepared from synchronous cultures than in those from asynchronous cultures. This increase in Chk1 activity was reproducibly followed by a decrease of 30–50% at the beginning of mitotic entry (2 h), and an increase in activity at 3–5 h, presumably due to activation of a DNA damage checkpoint in the G1- or S-phase. The decrease in activity at mitotic entry coincided with a decrease in the proportion of Chk1 in the phosphorylated form (Figure 8B, 2 h time point). The transient nature of Chk1 dephosphorylation in synchronous cultures can explain why we do not observe Chk1 dephosphorylation in asynchronous time courses. As asynchronous cultures contain <10% mitotic cells, decreases in Chk1 phosphorylation/activity during mitosis would be masked by phosphorylated Chk1 in interphase-arrested cells. The transient decrease in Chk1 phosphorylation and activity during mitosis, together with the reduced degree of synchrony in these large elutriations, means that the 30–50% decrease in Chk1 activity upon mitotic entry observed here is likely to be an underestimate. Importantly, in synchronous chk1::HAdis2Δ cultures, no decrease in Chk1 activity was observed 2–5 h after UV radiation, and there was no concomitant decrease in the level of phosphorylated Chk1.
To analyse the effect of dis2 deletion on Chk1 phosphorylation in response to UV radiation more precisely, we repeated the above experiment for chk1::HA and chk1::HAdis2Δ cultures, and quantified the proportion of Chk1 that was phosphorylated at 30 min intervals. Clearly, while there was a decrease in the proportion of Chk1 in the phosphorylated form in chk1::HA cells from 2 h, this decrease was not observed in chk1::HAdis2Δ cells, which also generated a greater proportion of Chk1 in the phosphorylated form following irradiation (Figure 8C).
Together, these data convincingly show that levels of Dis2 phosphatase affect the in vivo level of Chk1 phosphorylation and resultant kinase activity in response to DNA damage.
Discussion
The G2 DNA damage checkpoint prevents mitotic entry in the presence of DNA damage. Just as the establishment of this checkpoint is essential for viability in the presence of DNA damage, so too release from the checkpoint must be tightly controlled such that it is delayed until repair is completed. While many components involved in activation of the G2 DNA damage checkpoint have been identified, the means by which the checkpoint is turned off has remained an important gap in our understanding of this signalling network. Given that the DNA damage checkpoint consists of an S/T phosphorylation cascade, one mechanism of shutting off the pathway would be through dephosphorylation by an S/T protein phosphatase. However, equally possible are proteolytic degradation, interaction with other regulatory proteins or alterations in the localisation of checkpoint proteins. We have shown here that, like classical checkpoint mutants, overexpression of S. pombe PP1 Dis2 sensitises cells to DNA damage, resulting in mitotic catastrophe. Consistent with a role for dis2 in release from G2 DNA damage checkpoint arrest, dis2Δ and dis2-11 cells have a prolonged delay in response to DNA damage in the G2-phase. Moreover, we have shown that DNA damage-induced Chk1 phosphorylation and activation are compromised in cells overexpressing Dis2, while in cells lacking Dis2, the dephosphorylation and inactivation of Chk1 upon release of the G2 DNA damage checkpoint are abrogated. This, together with our recent finding that inactivating a t.s. Chk1 mutant following checkpoint initiation prematurely terminates G2 checkpoint arrest (Latif et al, in prep), clearly implicates the inactivation of Chk1 by Dis2 in release from the G2 DNA damage checkpoint.
The effect of Dis2 on Chk1 phosphorylation is direct and specific. It was shown both by means of overexpression and gene disruption in vivo, and by the direct dephosphorylation of Chk1 and a phospho-S345 Chk1 peptide by Dis2 and recombinant PP1α in vitro. Further, Dis2 does not affect the function or phosphorylation of other checkpoint components: Rad3 and downstream Rad proteins function normally irrespective of Dis2 levels, shown by the ability of cells to arrest normally in response to stalled replication forks; and the phosphorylation of the Rad3 substrate Rad9 is not affected by Dis2 overexpression. Dis2 overexpression also only modestly reduces the DNA damage-induced phosphorylation of Crb2, which may be due to an indirect effect involving Crb2's association with Chk1. The lack of effect of Dis2 on phosphorylated Rad3 substrates other than Chk1, which in other systems have been shown to bind to sites of DNA damage (Carr, 2003), may be because they are not accessible to Dis2 in that context.
During the revision of this manuscript, Margolis et al (2003) reported a role for PP1 in the M-phase entry in Xenopus oocytes via dephosphorylation of Cdc25 on Ser287. However, there is no evidence for the regulation of Cdc25 by Dis2 in S. pombe. The cell cycle is normal in dis2Δ and Dis2-overexpressing cells (Ohkura et al, 1989; Yamano et al, 1994); checkpoint arrest in response to stalled replication forks, which is dependent on Cdc25 and Wee1, is unaffected by Dis2 levels; and Dis2 does not affect a Chk1 overexpression-induced cell cycle arrest, which is also via Cdc25 and Wee1 regulation. This apparent discrepancy is likely to be explained by differences between species or between meiosis versus mitosis, including the absence of a functional G2 DNA damage checkpoint in intact oocytes.
Finally, we show that Dis2 is unique in its ability to cause Chk1 dephosphorylation and release of the G2 DNA damage checkpoint because deletion of the other S. pombe PP1, sds21, does not affect DNA damage checkpoint arrest, and no other S/T phosphatases were isolated in our screen. This is consistent with the finding that Sds21 overexpression actually causes a cell cycle delay (Ohkura et al, 1989; Yamano et al, 1994), and that overexpression of either of the S. pombe type IIA phosphatases, Ppa1 and Ppa2, results in an interphase arrest, whereas ppa2 deletion gives a semi-wee phenotype (Kinoshita et al, 1993). The other characterised S. pombe S/T phosphatases have been implicated in diverse cellular functions, but none in DNA damage responses.
Observations regarding PP1 isozymes from other organisms relate to its role here in release from the G2 DNA damage checkpoint. In S. cerevisiae, the PP1 homologue, Glc7, is implicated in release from the pachytene checkpoint, a meiosis-specific checkpoint that prevents chromosome segregation during first meiotic division until recombination and chromosome synapsis are completed (Bailis and Roeder, 2000). A downstream target of this checkpoint is the S. cerevisiae homologue of S. pombe Wee1, Swe1, which is hyperphosphorylated and increases in abundance in the pachytene checkpoint (Leu and Roeder, 1999), reminiscent of the increase in stability of Wee1 in the S. pombe G2 DNA damage checkpoint (Raleigh and O'Connell, 2000). In mammalian cells, PP1α has been shown to dephosphorylate a fragment of BRCA1 in vitro, and PP1α and BRCA1 coimmunoprecipitate (Liu et al, 2002). Furthermore, PP1C is activated following ionising radiation in Jurkat cells, in an ATM-dependent manner (Guo et al, 2002).
We propose a model whereby, in response to DNA lesions, the G2 DNA damage checkpoint kinase Rad3 is activated, resulting in the phosphorylation and activation of the effector kinase Chk1, and resultant inhibition of the mitosis-promoting cyclin B/Cdc2 kinase complex. Chk1 phosphorylation, and thus checkpoint arrest, may be maintained either by continued phosphorylation of Chk1 by Rad3 or another kinase, protection of Chk1 phosphorylation by another protein (e.g. 14-3-3 or Crb2, which associate preferentially with the phosphorylated form of Chk1 (Chen et al, 1999; Mochida et al, 2004)) and/or by prevention of Dis2-mediated Chk1 dephosphorylation. Upon completion of DNA repair, Dis2-mediated dephosphorylation and inactivation of Chk1 would allow cell cycle re-entry. With respect to the maintenance of Chk1 phosphorylation during DNA repair, it is noteworthy that BRCT domains, of which Crb2 contains two, and 14-3-3 proteins both bind to phospho-proteins, and that 14-3-3 proteins have been implicated in the protection of a phosphorylated motif of Cdc25 (Muslin et al, 1996; Manke et al, 2003; Margolis et al, 2003; Yu et al, 2003). Although we do not detect any change in Dis2 activity following UV irradiation (Figure 7C), PP1s have been shown to be regulated in vivo through association with an assortment of regulatory subunits and through varied subcellular localisations (Cohen, 2002). Also, while Dis2 activity has been shown to be downregulated by Cdc2 kinase (Yamano et al, 1994), this is not inconsistent with a role for Dis2 in checkpoint release, as Cdc2 is inactive prior to checkpoint release. Dis2 is unlikely to be the only protein responsible for cell cycle re-entry following DNA damage checkpoint arrest because the majority of dis2Δ cells do eventually recover, albeit several hours later than wild-type cells.
Dis2 is the first identified component of a checkpoint release pathway upstream of Cdc2 activation in S. pombe. With the conservation of the G2 DNA damage checkpoint among S. pombe and higher eukaryotes, PP1 enzymes are likely to play a conserved role in recovery from the G2 DNA damage checkpoint in eukaryotic cells. Future studies will determine how these events are regulated in time and space.
Materials and methods
S. pombe methods
All strains are derivatives of wild-type 972h− and 975h+. Standard methods were used for growth and genetic manipulation (Moreno et al, 1991). For sensitivity assays, exponentially growing cultures were serially diluted to 106, 105, 104 and 103 cells/ml, and 5 μl of each dilution was spotted onto agar plates containing the appropriate drugs. Cells containing nmt1 promoter plasmids were maintained in thiamine, and cDNA expression was derepressed by three washes in minimal medium and growth in minimal medium for six cell cycles prior to experimentation. An S. pombe cDNA library in pRep1 was used for an overexpression screen. The nmt1::dis2 plasmid used for subsequent experiments consisted of 179 base pairs of the 5′ untranslated region (UTR), the dis2 open reading frame and the entire 3′ UTR. Synchronous, early G2-phase cells were collected by centrifugal elutriation and irradiated as described (Verkade et al, 2001). Passage through mitosis was monitored by the septation index for asynchronous cultures. For synchronous cultures and experiments with cell cycle mutants, mitosis was monitored using fixed cells stained with 1 μg/ml 4′,6-diamino-2-phenylindole (DAPI). Cells passing mitosis included those cells with dividing nuclei, and cells joined by a septum (equivalent to G1- and early S-phase cells). Total and aberrant mitoses were averaged from at least three counts of 100 cells and each expressed as a percentage of total cells. UV-C and IR irradiation of S. pombe cultures were performed as described (Verkade et al, 1999).
Detection of thymidine dimers
G2-synchronised cells were either untreated or given 150 J/m2 UV-C radiation, and cell samples were snap-frozen in liquid nitrogen at the following time points. Cells were disrupted in a bead beater, and DNA was extracted in 50 mM Tris·Cl, pH 8, 5 mM EDTA, 1% SDS, followed by phenol–chloroform extraction and ethanol precipitation, RNase A treatment, and a further round of phenol–chloroform extraction and ethanol precipitation. DNA (1 μg) was dot blotted onto nitrocellulose, and an aliquot was stained with ethidium bromide and dot blotted to a duplicate filter to visualise DNA on a transilluminator. Thymidine dimers were detected with the TDM-2 monoclonal antibody and horseradish peroxidase (HRP)-linked anti-mouse antibody (Amersham), followed by chemiluminescence (Amersham).
Western blotting
Pellets of ∼2 × 108 cells were snap-frozen in liquid nitrogen. For Chk1-HA, Rad9-HA and Crb2-18myc proteins, cells were lysed as described for Chk1-HA (Harvey et al, 2004). For Dis2 and Sds21 proteins, cells were lysed in 50 mM Tris·Cl, pH 7.5, 400 mM NaCl, 1 mM EDTA, 1 mM 2-mercaptoethanol and 1 mM PMSF. Debris was removed by centrifugation at 5000 g for Crb2-18myc and 16 000 g for other proteins. Protein extracts (10–50 μg) and IPs were run on 6–10% SDS–PAGE gels and transferred to nitrocellulose in 25 mM Tris, 192 mM glycine, 20% methanol or 10 mM CAPS, pH 11 and 10% methanol. Chk1-HA and Rad9-HA were detected using 12CA5 monoclonal antibody; Dis2 and Sds21 were detected with rabbit polyclonal D2C antibody (Stone et al, 1993); and Crb2-18myc was detected using 9E10 anti-myc antibody. Actin, detected using a monoclonal anti-actin antibody (Chemicon), was used as a loading control. HRP-linked secondary antibodies (Amersham) were followed by chemiluminescence. Quantification of bands was performed using a BioRad GS-800 densitometer and Quantity One software.
Chk1 IPs and kinase assays
Chk1-HA IPs and in vitro kinase assays were performed as described (Harvey et al, 2004), using 1 mg of extract per reaction. For comparison of Chk1 kinase activity in different strains, kinase assays were performed simultaneously. Background, measured by incubating kinase reaction buffer in the absence of any protein A-sepharose-IP, was found to be equivalent to background from IPs using isotype control IgG antibody or from anti-HA IPs of untagged Chk1 strains. Thus, the former was used to remove background from kinase activity measurements.
Dis2 IPs and phosphatase assays
Chk1-HA protein extracts were prepared from dis2Δchk1::HA cultures 60 min post-treatment with 150 J/m2 UV-C radiation. Dis2 and Sds21 protein extracts were prepared from sds21Δ and dis2Δ strains, respectively, either pre- or 60 min post-treatment with 150 J/m2 UV-C radiation. Chk1-HA and Dis2/Sds21 were IPed with 2.5 μg of 12CA5 (Roche) and 3 μl of D2C antibody per milligram of protein, respectively, on a rotator for 2 h at 4°C, followed by incubation with 20 μl (Chk1) or 30 μl (Dis2/Sds21) of 50% protein A-sepharose (Amersham) per mg for 1 h at 4°C. Protein A-sepharose was washed six times with extraction buffer and four times with phosphatase assay buffer (50 mM Tris, pH 7.5, 30 mM NaCl, 5 mM DTT, 0.5 mM PMSF and 1 mM MnCl2). Recombinant rabbit PP1α (New England Biolabs) was assayed in the buffer supplied (50 mM Tris, pH 7.0, 0.1 mM EDTA, 5 mM DTT, 0.025% Tween-20, 1 mM MnCl2). For phosphatase assays, a 15 μl bed volume of protein A-sepharose-Dis2/Sds21 IP (equivalent to 0.5 mg of original extract) or 0.05 U of PP1α was incubated at 30°C for 15 min in 80 μl of assay buffer±1 μg I-2 (New England Biolabs). In all, 20 μl of 50% protein A-sepharose-Chk1-HA IP (equivalent to 0.5 mg of original extract) was then added, and reactions were incubated at 30°C. Reactions were stopped by the addition of SDS–PAGE loading dye, and run on SDS–PAGE gels, followed by Western analysis.
For phosphatase assays using the phospho-S345 Chk1 peptide VEVYGALpSQPVQLNK (Auspep), Dis2 and Sds21 were IPed as above, but were washed and assayed in PP1α assay buffer. A 15 μl bed volume of Dis2/Sds21 IP or 0.05 U PP1α was incubated at 30°C for 15 min in 25 μl of assay buffer±1 μg I-2. In all, 25 μl of assay buffer containing 10 nmol of peptide was added, and reactions were incubated at 30°C for 7.5 or 15 min. Protein A-sepharose was pelleted by centrifugation at 4°C, and 40 μl of each reaction was added to 100 μl of malachite green (Upstate). Phosphate release was determined by comparing absorbance at 650 nm to a standard curve according to the manufacturer's instructions. Appropriate incubation times were used to ensure that phosphate release was linear at the time point measured. Protein A-sepharose, anti-D2C antibody, PP1α and I-2 had no effect on A650 values when incubated in the absence of peptide. For IP-phosphatase assays, A650 measurements obtained for IPs without D2C antibody were used as background and subtracted from all measurements. For PP1α phosphatase assays, assay buffer containing peptide but lacking enzyme was used as background.
Supplementary Material
Supplementary Figure 1 (A) The indicated strains were grown at 30°C, and serial dilutions were spotted onto YES agar containing no drug, 0.01% MMS or 60 ng/ml 4-NQO, and incubated at 22°C or 30°C; (B) Cultures were grown at 30°C, then shifted to 20°C and incubated in the absence or presence of 0.01% MMS for 16 h. Cells were fixed and measured for average cell length±standard deviation (μm, n=25).
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Acknowledgments
We thank C Norbury for the cDNA library, M Yanagida for strains and the D2C antibody, T Carr and S Francesconi for strains, T Matsunaga for the TDM-2 antibody, J Hagekyriakou for his assistance with IR experiments, and C Latif and S Harvey for experimental assistance and helpful discussions. NRdE is an NHMRC Peter Doherty Fellow, and MJO'C is a Scholar of the Leukemia and Lymphoma Society. This research was supported by grants from the NHMRC (114229) and the NIH/NCI (CA100076-01).
References
- Al-Khodairy F, Carr AM (1992) DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J 11: 1343–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis JM, Roeder GS (2000) Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell 101: 211–221 [DOI] [PubMed] [Google Scholar]
- Capasso H, Palermo C, Wan S, Rao H, John UP, O'Connell MJ, Walworth NC (2002) Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci 115: 4555–4564 [DOI] [PubMed] [Google Scholar]
- Carr AM (2003) Molecular biology. Beginning at the end. Science 300: 1512–1513 [DOI] [PubMed] [Google Scholar]
- Chen L, Liu TH, Walworth NC (1999) Association of Chk1 with 14-3-3 proteins is stimulated by DNA damage. Genes Dev 13: 675–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY (2001) ATM-dependent phosphorylation of human Rad9 is required for ionizing radiation-induced checkpoint activation. J Biol Chem 276: 16580–16586 [DOI] [PubMed] [Google Scholar]
- Cohen PT (2002) Protein phosphatase 1—targeted in many directions. J Cell Sci 115: 241–256 [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Koshland D (1997) The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci USA 94: 14361–14366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG (1994) The decision to enter mitosis. Trends Cell Biol 4: 202–207 [DOI] [PubMed] [Google Scholar]
- Guo CY, Brautigan DL, Larner JM (2002) Ionizing radiation activates nuclear protein phosphatase-1 by ATM-dependent dephosphorylation. J Biol Chem 277: 41756–41761 [DOI] [PubMed] [Google Scholar]
- Harvey SH, Sheedy DM, Cuddihy AR, O'Connell MJ (2004) Coordination of DNA damage responses via the Smc5–Smc6 complex. Mol Cell Biol 24: 622–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N, Yamano H, Niwa H, Yoshida T, Yanagida M (1993) Negative regulation of mitosis by the fission yeast protein phosphatase ppa2. Genes Dev 7: 1059–1071 [DOI] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbien F, Guerois R, Haber JE, Marsolier-Kergoat MC (2003) PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell 11: 827–835 [DOI] [PubMed] [Google Scholar]
- Leu JY, Roeder GS (1999) The pachytene checkpoint in S. cerevisiae depends on Swe1-mediated phosphorylation of the cyclin-dependent kinase Cdc28. Mol Cell 4: 805–814 [DOI] [PubMed] [Google Scholar]
- Liu Y, Virshup DM, White RL, Hsu LC (2002) Regulation of BRCA1 phosphorylation by interaction with protein phosphatase 1alpha. Cancer Res 62: 6357–6361 [PubMed] [Google Scholar]
- Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P (2001) Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci USA 98: 11289–11294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, Yaffe MB (2003) BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302: 636–639 [DOI] [PubMed] [Google Scholar]
- Margolis SS, Walsh S, Weiser DC, Yoshida M, Shenolikar S, Kornbluth S (2003) PP1 control of M phase entry exerted through 14-3-3-regulated Cdc25 dephosphorylation. EMBO J 22: 5734–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida S, Esashi F, Aono N, Tamai K, O'Connell MJ, Yanagida M (2004) Regulation of checkpoint kinases through dynamic interaction with Crb2. EMBO J 23: 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Method Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84: 889–897 [DOI] [PubMed] [Google Scholar]
- O'Connell MJ, Walworth NC, Carr AM (2000) The G2-phase DNA-damage checkpoint. Trends Cell Biol 10: 296–303 [DOI] [PubMed] [Google Scholar]
- Ohkura H, Adachi Y, Kinoshita N, Niwa O, Toda T, Yanagida M (1988) Cold-sensitive and caffeine-supersensitive mutants of the Schizosaccharomyces pombe dis genes implicated in sister chromatid separation during mitosis. EMBO J 7: 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkura H, Kinoshita N, Miyatani S, Toda T, Yanagida M (1989) The fission yeast dis2+ gene required for chromosome disjoining encodes one of two putative type 1 protein phosphatases. Cell 57: 997–1007 [DOI] [PubMed] [Google Scholar]
- Puntoni F, Villa-Moruzzi E (1997) Association of protein phosphatase-1delta with the retinoblastoma protein and reversible phosphatase activation in mitotic HeLa cells and in cells released from mitosis. Biochem Biophys Res Commun 235: 704–708 [DOI] [PubMed] [Google Scholar]
- Raleigh JM, O'Connell MJ (2000) The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J Cell Sci 113: 1727–1736 [DOI] [PubMed] [Google Scholar]
- Shiozaki K, Russell P (1995) Counteractive roles of protein phosphatase 2C (PP2C) and a MAP kinase homolog in the osmoregulation of fission yeast. EMBO J 14: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets MF, Francesconi S, Baldacci G (2003) High dosage Rhp51 suppression of the MMS sensitivity of DNA structure checkpoint mutants reveals a relationship between Crb2 and Rhp51. Genes Cells 8: 573–586 [DOI] [PubMed] [Google Scholar]
- Stone EM, Yamano H, Kinoshita N, Yanagida M (1993) Mitotic regulation of protein phosphatases by the fission yeast sds22 protein. Curr Biol 3: 13–26 [DOI] [PubMed] [Google Scholar]
- Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell 10: 2905–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkade HM, Teli T, Laursen LV, Murray JM, O'Connell MJ (2001) A homologue of the Rad18 postreplication repair gene is required for DNA damage responses throughout the fission yeast cell cycle. Mol Genet Genom 265: 993–1003 [DOI] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D (1993) Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature 363: 368–371 [DOI] [PubMed] [Google Scholar]
- Yamano H, Ishii K, Yanagida M (1994) Phosphorylation of dis2 protein phosphatase at the C-terminal cdc2 consensus and its potential role in cell cycle regulation. EMBO J 13: 5310–5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science 302: 639–642 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 (A) The indicated strains were grown at 30°C, and serial dilutions were spotted onto YES agar containing no drug, 0.01% MMS or 60 ng/ml 4-NQO, and incubated at 22°C or 30°C; (B) Cultures were grown at 30°C, then shifted to 20°C and incubated in the absence or presence of 0.01% MMS for 16 h. Cells were fixed and measured for average cell length±standard deviation (μm, n=25).
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure
Supplementary Figure