

Abstract
We recently demonstrated that the LKB1 tumour suppressor kinase, in complex with the pseudokinase STRAD and the scaffolding protein MO25, phosphorylates and activates AMP-activated protein kinase (AMPK). A total of 12 human kinases (NUAK1, NUAK2, BRSK1, BRSK2, QIK, QSK, SIK, MARK1, MARK2, MARK3, MARK4 and MELK) are related to AMPK. Here we demonstrate that LKB1 can phosphorylate the T-loop of all the members of this subfamily, apart from MELK, increasing their activity >50-fold. LKB1 catalytic activity and the presence of MO25 and STRAD are required for activation. Mutation of the T-loop Thr phosphorylated by LKB1 to Ala prevented activation, while mutation to glutamate produced active forms of many of the AMPK-related kinases. Activities of endogenous NUAK2, QIK, QSK, SIK, MARK1, MARK2/3 and MARK4 were markedly reduced in LKB1-deficient cells. Neither LKB1 activity nor that of AMPK-related kinases was stimulated by phenformin or AICAR, which activate AMPK. Our results show that LKB1 functions as a master upstream protein kinase, regulating AMPK-related kinases as well as AMPK. Between them, these kinases may mediate the physiological effects of LKB1, including its tumour suppressor function.
Keywords: cancer, cell polarity, diabetes, PAR1/MARK kinase, Peutz–Jeghers syndrome, TOF–TOF mass spectrometry
Introduction
Mutations in the widely expressed LKB1 protein kinase in humans result in a disorder termed Peutz–Jeghers syndrome (PJS), which predisposes to a wide spectrum of benign and malignant tumours (Hemminki et al, 1998; Jenne et al, 1998). LKB1+/− heterozygous mice develop tumours resembling those found in PJS in humans (reviewed in Boudeau et al, 2003c). The overexpression of LKB1 in LKB1-deficient cancer cells induced a G1 cell cycle arrest (Tiainen et al, 1999,2002)l, and genetic studies in Caenorhabditis elegans (Watts et al, 2000), Drosophila (Martin and St Johnston, 2003) and Xenopus (Ossipova et al, 2003) indicated that the LKB1 homologue in these organisms plays a role in regulating cell polarity. Taken together, these findings support the notion that the LKB1 protein kinase functions as a tumour suppressor and that the benign and malignant tumours in PJS patients could result in defects in the ability of cells to regulate their proliferation and/or polarity.
Recently, we demonstrated that LKB1 is activated through its interaction with STRAD (Baas et al, 2003) and MO25 (Boudeau et al, 2003a). STRAD possesses a domain with high sequence homology to protein kinases but lacks the key catalytic residues required for catalysis and has therefore been classified a ‘pseudokinase'. MO25 bears no sequence homology to other proteins in the database, but recent studies indicate that it is structurally related to the Armadillo repeat domain (Milburn et al, 2004). In addition to activating LKB1, STRAD and MO25 anchor it in the cell cytoplasm (Baas et al, 2003; Boudeau et al, 2003a; Brajenovic et al, 2003), where LKB1 appears to exert its cell cycle-arresting function (Tiainen et al, 2002).
AMP-activated protein kinase (AMPK) is a sensor of cellular energy charge that regulates physiological processes that consume or regenerate ATP to restore the energy charge in the cell (Hardie et al, 2003). Both catalytic subunit isoforms of AMPK (AMPKα1 and AMPKα2) are activated by ATP-depleting processes such as exercise and cellular stress, through a rise in cellular AMP that accompanies the fall of ATP levels due to the reaction catalysed by adenylate kinase. AMPK is also activated by metformin, the drug most commonly employed for the treatment of type II diabetes (Zhou et al, 2001). The mechanism by which metformin, or its closely related analogue phenformin, activates AMPK is unknown but is not thought to involve changes in intracellular levels of AMP or ATP (Zhou et al, 2001; Hawley et al, 2002). The activation of AMPK by both ATP depletion and phenformin requires phosphorylation of the AMPK catalytic (α) subunit at its T-loop residue (Thr172 in AMPKα1) by an upstream kinase. Studies performed in Saccharomyces cerevisiae indicated that the T-loop residue of the yeast orthologue of AMPK (SNF1) was phosphorylated by a group of three related protein kinases bearing homology to LKB1 (Hong et al, 2003; Nath et al, 2003; Sutherland et al, 2003). We and others have shown that LKB1 functions to phosphorylate and activate AMPK in mammalian cells (Hawley et al, 2003; Woods et al, 2003a). Studies in cell-free systems demonstrated that LKB1 complexed to STRAD and MO25 activated AMPK by phosphorylating Thr172, and that the STRAD and MO25 subunits enhanced phosphorylation of AMPK by over 100-fold (Hawley et al, 2003). Moreover, AMPK could not be activated in mammalian cells that lacked LKB1 expression (Hawley et al, 2003) or in cells that were treated with Hsp90 inhibitors (Woods et al, 2003a), which decreases LKB1 expression (Boudeau et al, 2003b).
Inspection of the human kinome indicates that there are 12 protein kinases (BRSK1, BRSK2, NUAK1, NUAK2, QIK, QSK, SIK, MARK1, MARK2, MARK3, MARK4 and MELK) that are closely related to AMPKα1 and AMPKα2 (Figure 1A). The nomenclature we will employ for the ‘AMPK-related kinases' is based upon that used by Manning et al (2002). MARK3 is also known as PAR1A or C-TAK1 (Peng et al, 1998; Spicer et al, 2003), NUAK1 as ARK5 (Suzuki et al, 2003b), NUAK2 as SNARK (Lefebvre et al, 2001) and QIK as SIK2 (Horike et al, 2003). The MARK kinases have been proposed to play key roles in controlling cell polarity and are known to be regulated by phosphorylation of their T-loop Thr residue (Drewes and Nurse, 2003; Spicer et al, 2003). Little is known about the function or mechanism of regulation of the remaining AMPK-related enzymes. The T-loop Thr residue of AMPKα1 and AMPKα2 that is phosphorylated by LKB1, and many of the surrounding residues, is conserved in the AMPK-related kinases (Figure 1A). This indicates that LKB1 might also phosphorylate the T-loop Thr residue and hence regulate the AMPK-related kinases in the same way that PDK1 regulates the activity of a group of related AGC kinases (Alessi, 2001). In this study, we sought to investigate the role that LKB1 plays in regulating the activity of the AMPK-related kinases.
Figure 1.
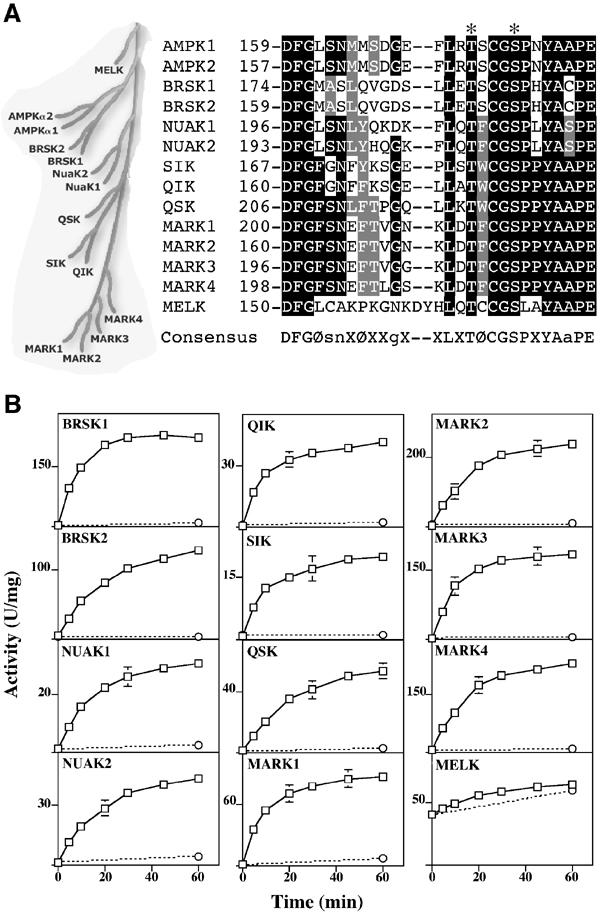
Activation of AMPK-related kinases by LKB1. (A) Dendrogram and T-loop sequences of AMPK subfamily of protein kinases (Manning et al, 2002). The identical residues are shaded black and the conserved residues in grey. The T-loop Thr and Ser are indicated with an asterisk. (B) The indicated AMPK-related kinases were incubated with wild-type LKB1:STRAD:MO25 (open squares) or catalytically inactive LKB1[D194A]:STRAD:MO25 (open circles) complexes in the presence of Mg2+ and ATP. At the indicated times, the activity of the AMPK-related kinases was assayed with the AMARA peptide, and the results are expressed as specific activity. Results shown are means±s.d. of assays carried out in triplicate and representative of two independent experiments. The error bars are only shown when larger than the size of the open squares. The suggested consensus sequence for optimal LKB1 phosphorylation is indicated. Ø represents a large hydrophobic residue; X, any amino acid; s, n, g and a preferences for Ser, Asn, Gly and Ala, respectively.
Results
Activation of AMPK-related kinases by the LKB1 complex
We cloned and expressed in Escherichia coli the full-length versions of all of the 12 AMPK-related kinases and developed assays for these enzymes employing the AMARA peptide substrate for AMPK (Dale et al, 1995), which is not phosphorylated by the LKB1 complex (JM Lizcano, data not shown). We found that all of the AMPK-related kinases purified from E. coli phosphorylated this peptide but possessed low basal activities of <1 U/mg (1 U=1 nmol peptide phosphorylated per minute), with the exception of MELK, which had an activity of ∼40 U/mg (Figure 1B). Following incubation of the AMPK-related kinases with the LKB1:STRAD:MO25 complex and MgATP, the activity of the AMPK-related kinases was increased 50- to 200-fold, except for MELK whose activity was hardly increased (Figure 1B). Catalytically inactive LKB1 complexed to STRADα and MO25α failed to increase the basal activity of the AMPK-related kinases, indicating that the kinase activity of LKB1 was required (Figure 1B).
Activation of AMPK-related kinases by LKB1 requires STRAD and MO25
To determine the importance of the STRAD and MO25 subunits in enabling LKB1 to activate the AMPK-related kinases, we expressed GST-tagged LKB1, FLAG-tagged STRADα/β and myc-tagged MO25α/β in various combinations in 293 cells, and affinity purified the complexes on glutathione–sepharose (Figure 2). The purified complexes were incubated in the presence of MgATP with the AMPK-related kinases and their activation was measured. LKB1 on its own did not activate any of the AMPK-related kinases significantly (Figure 2, compare lanes 1 and 14). The same result was obtained with LKB1 that had been coexpressed with MO25α or MO25β (Figure 2, lanes 4 and 5). This was as expected, as these proteins do not interact with LKB1 in the absence of STRADα/β (Boudeau et al, 2003a). An LKB1:STRADα complex gave a small activation (Figure 2, compare lanes 1 and 2), but a heterotrimeric complex containing LKB1, STRADα or STRADβ, and MO25α or MO25β was required to obtain a large activation (Figure 2, lanes 6–9). As previously reported for AMPKα1 (Figure 2; Hawley et al, 2003), all AMPK-related kinases were activated most efficiently by the LKB1:STRADα:MO25α complex, although the relative effectiveness of different heterotrimeric complexes varied from substrate to substrate (Figure 2). Heterotrimeric complexes containing a catalytically inactive mutant of LKB1 were unable to activate any enzyme (Figure 2, lanes 10–13). It should be noted that when STRAD isoforms were coexpressed with LKB1 in the absence of MO25, the amount of STRAD that co-precipitated with LKB1 was markedly reduced (Figure 2, compare lanes 2 and 3 with lanes 6 and 7), as MO25 is required to stabilise the LKB1:STRAD complex (Boudeau et al, 2003a).
Figure 2.
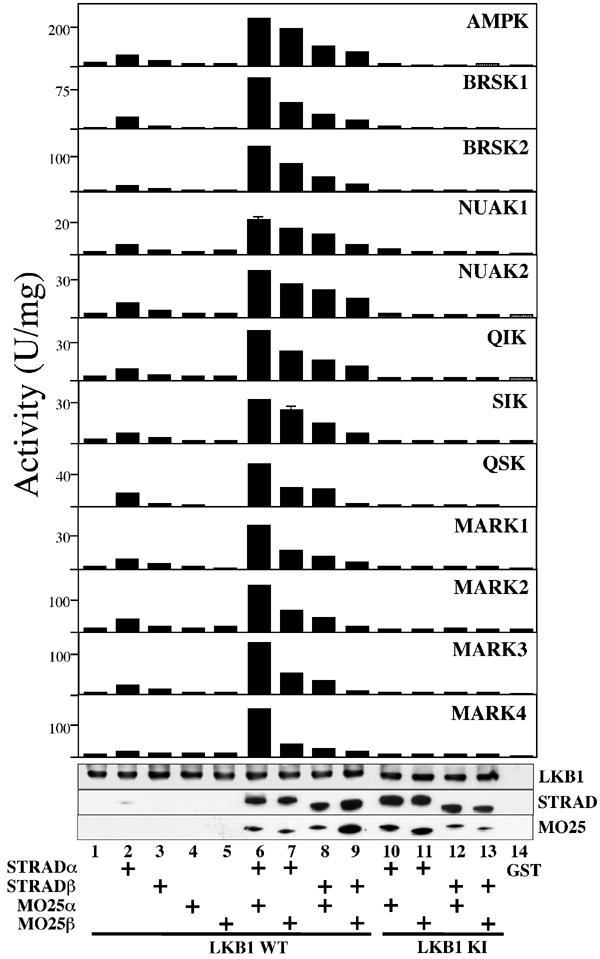
Efficient activation of AMPK-related kinases by LKB1 requires STRAD and MO25 subunits. The indicated combinations of GST-tagged wild-type LKB1 (WT, lanes 1–9), or catalytically inactive (KI, D194A, lanes 10–13) LKB1, or GST alone (lane 14), FLAG-tagged STRADα or STRADβ, and myc-tagged MO25α or MO25β were coexpressed in HEK-293T cells and purified on glutathione–sepharose. The complexes were tested for their ability to activate the catalytic domain of AMPKα1 or the indicated AMPK-related kinases. The results are expressed as specific activity employing the AMARA peptide as substrate. Results shown are means±s.d. of assays carried out in triplicate and representative of two independent experiments. Samples from each incubation were also analysed by Western blotting and probed using the indicated antibodies (from top to bottom): anti-GST to detect LKB1; anti-FLAG to detect STRADα and STRADβ; and anti-myc to detect MO25α and MO25β. All proteins migrated with the expected mobility, taking into account the epitope tags.
The LKB1 complex phosphorylates AMPK-related kinases at the activation loop
As the LKB1 complex was previously shown to phosphorylate AMPKα1 specifically at Thr172 in its activation loop, we first mutated the equivalent T-loop residue in the AMPK-related kinases to Ala, and tested how this affected phosphorylation of these enzymes by the LKB1 complex. We found that the LKB1 complex phosphorylated the wild-type AMPK-related kinases, and mutation of the T-loop Thr residue to Ala abolished or significantly reduced phosphorylation (Figure 3). We also performed detailed phosphorylation site analysis of catalytically inactive BRSK2 and NUAK2 mutants that had been phosphorylated in vitro by the LKB1 complex (Supplementary Figure 1. The 32P-labelled BRSK2 and NUAK2 proteins were digested with trypsin and the resulting peptides were separated by chromatography on a C18 column. The major 32P-labelled peptides were analysed by MALDI TOF–TOF mass spectrometry and solid-phase Edman sequencing, which demonstrated that these enzymes were phosphorylated at their T-loop Thr residue (Supplementary Figure 1).
Figure 3.
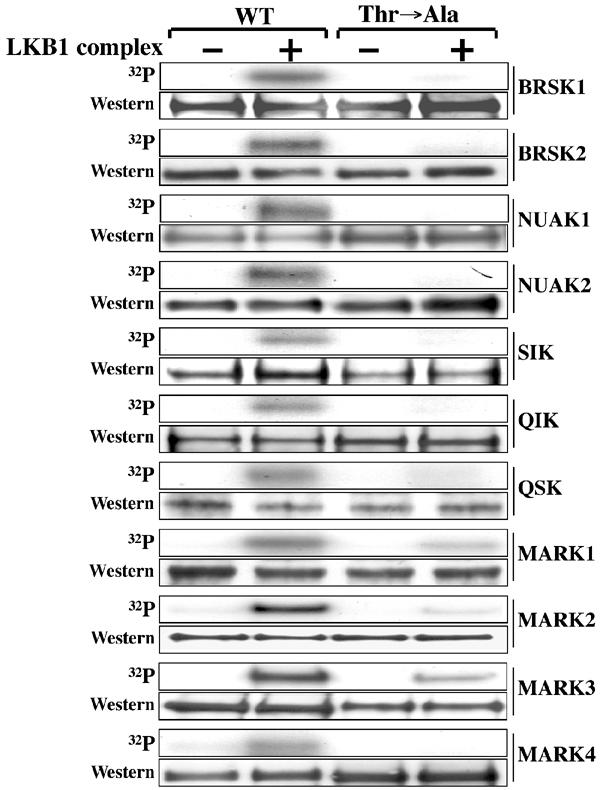
The T-loop Thr is the major site of LKB1 phosphorylation on the AMPK-related kinases. Wild-type (WT) and T-loop Thr to Ala (Thr → Ala) mutants of the indicated GST-AMPK-related kinases were incubated with the LKB1:STRAD:MO25 complex in the presence of Mg2+ and [γ32P]ATP. Phosphorylation of protein substrates was determined by electrophoresis on a polyacrylamide gel and subsequent autoradiography of the Coomassie blue-stained bands corresponding to each substrate. An aliquot of each incubation was also analysed by Western blotting probing with an anti-HA antibody to ensure equal loading of wild-type and mutant AMPK-related kinases (which all possess an HA epitope tag). All proteins migrated with the expected mobility, taking into account the epitope tags. Similar results were obtained in three separate experiments.
T-loop phosphorylation activates AMPK-related kinases
To study the role of T-loop phosphorylation of BRSK1, BRSK2, NUAK1, NUAK2, QIK, QSK, SIK and MELK in their regulation, the T-loop Thr residues of these enzymes were mutated to either Ala to prevent phosphorylation or Glu to mimic phosphorylation. In all cases, the basal activity of the Ala mutants was low and their activity was not increased by incubation with the LKB1 complex (Figure 4). In contrast, the basal activity of the Glu mutants was similar to that of the wild-type enzyme phosphorylated by LKB1, and was not further increased following incubation with the LKB1 complex and MgATP (Figure 4). In the case of MELK, the Ala mutant possessed low activity and the Glu mutant was of similar activity to the wild-type enzyme, suggesting that wild-type MELK (like other AMPK-related kinases) required T-loop phosphorylation for activity. As wild-type MELK expressed in E. coli was active, MELK may be able to catalyse phosphorylation of its own T-loop Thr residue. Consistent with this, MALDI TOF–TOF analysis of wild-type MELK expressed in E. coli established that the T-loop was indeed phosphorylated (Supplementary Figure 1C).
Figure 4.
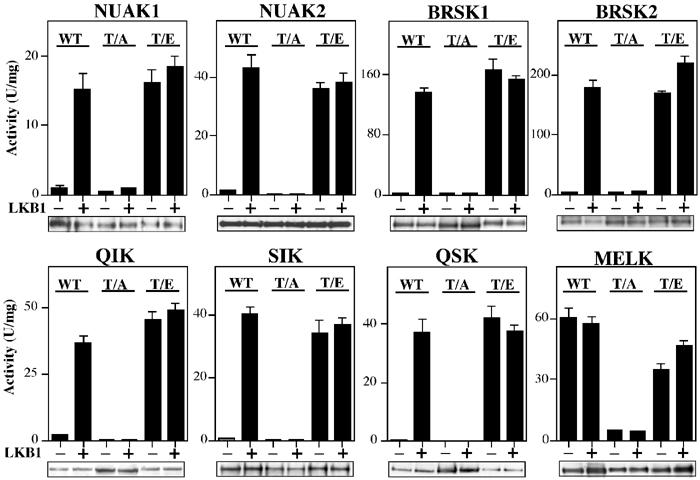
Effect of mutation of Thr in the T-loop on activation of AMPK-related kinases by LKB1. The indicated wild-type (WT) AMPK-related kinases or mutants of these enzymes in which the T-loop Thr was changed to either Ala (T/A) or Glu (T/E) were incubated in the absence (−) or presence (+) of wild-type LKB1:STRAD:MO25 in the presence of Mg2+ and ATP. After 30 min, the AMPK-related kinases were assayed with the AMARA peptide, and the results are expressed as specific activity. Results shown are means±s.d. of assays carried out in triplicate and representative of two independent experiments. An aliquot of each incubation was also analysed by Western blotting probing with an anti-HA antibody to ensure equal loading of wild-type and mutant AMPK-related kinases (which all possess an HA tag).
Phosphorylation and activation of the MARK kinases by LKB1
Previous studies have revealed that the MARK2 and MARK3 kinases, in addition to being phosphorylated at the T-loop Thr residue, are also phosphorylated at a nearby Ser residue (Figure 1A; Drewes et al, 1997; Timm et al, 2003). Peptide mapping of catalytically inactive MARK3 phosphorylated by the LKB1 complex revealed that both the Thr211 and Ser215 residues within the T-loop were phosphorylated (Figure 5A). Interestingly, catalytically inactive MARK3[T211A] mutant was not phosphorylated at Ser215 by LKB1, but the catalytically inactive MARK3[S215A] mutant was still phosphorylated at Thr211 (Figure 5A). We next investigated how mutation of the T-loop Thr or Ser residue affected activation of MARK3 by the LKB1 complex (Figure 5C). The MARK3[T211A] mutant could not be activated by the LKB1 complex, but the MARK3[S215A] mutant was activated to a small extent. The MARK3[T211E] mutant possessed only ∼15% of the activity of the wild-type enzyme activated by the LKB1 complex and could not be activated further by LKB1. The MARK3[S215E] mutant was inactive and could not be activated by LKB1 (Figure 5C). As expected, we also found that mutation of the T-loop Thr to Ala in MARK1, MARK2 and MARK4 prevented their activation by LKB1 (Figure 5D–F). Mutation of the T-loop Thr residue to Glu in MARK1, MARK2 and MARK3 resulted in either no activation or only a small activation of these enzymes (Figure 5D–F). In contrast, the equivalent mutation in MARK4 vastly increased activity (Figure 5F). Peptide-mapping studies and MALDI TOF–TOF mass spectrometry demonstrated that LKB1 phosphorylated the MARK4 T-loop at only the Thr residue and not the Ser residue (Figure 5G and Supplementary Figure 1C). Consistent with these findings, it has recently been reported that LKB1 immunoprecipitated from cell lysates can phosphorylate and activate MARK4 (Brajenovic et al, 2003).
Figure 5.
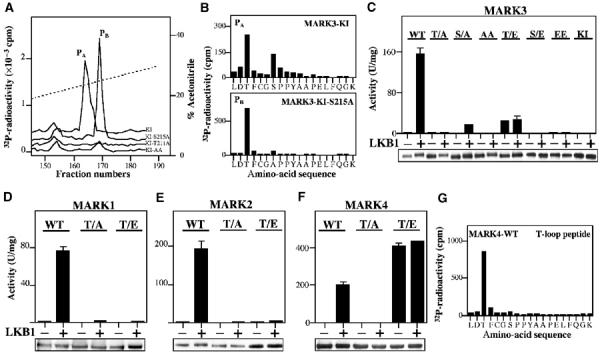
Analysis of phosphorylation and activation of MARK kinases. (A) Catalytically inactive MARK3[D196A] (KI), which cannot autophosphorylate, and the indicated mutants were incubated with the LKB1 complex for 30 min with Mg2+-[γ32P]ATP and separated by electrophoresis on a polyacrylamide gel, which was then autoradiographed. The 32P-labelled MARK3 proteins were digested with trypsin and the resulting 32P-labelled peptides were chromatographed on a C18 column. Fractions containing the 32P-labelled T-loop tryptic peptide (peptides PA and PB) are shown. (B) Peptide PA and PB were subjected to solid-phase sequencing and 32P-radioactivity was measured after each cycle of Edman degradation. In combination with MALDI TOF–TOF mass spectrometry, this enabled the identification of the sites phosphorylated in each peptide. Peptide PA comprise the MARK3 T-loop peptide phosphorylated at Thr211 and Ser215, and Peptides PB comprise the MARK3 T-loop peptide phosphorylated at Thr211. (C–F) The indicated wild-type (WT) or mutant forms of MARK kinases in which the the T-loop Thr or Ser was mutated to either Ala (T/A, S/A) or Glu to (T/E, S/E) were incubated in the absence (−) or presence (+) of wild-type LKB1:STRAD:MO25 in the presence of Mg2+ and ATP. After 30 min, the MARK kinases were assayed using the AMARA peptide, and the results are expressed as specific activity. An aliquot of each incubation was also analysed by Western blotting probing with an anti-HA antibody to ensure equal loading of wild-type and mutant MARK kinases (which all possessed an HA epitope tag). Results shown are average±s.d. of a triplicate assay and are representative of at least two independent experiments. (G) MARK4 was phosphorylated with the LKB1 complex as in (A) and the major 32P-labelled peptide was analysed by solid-phase Edman sequencing as in (B). In combination with MALDI TOF–TOF mass spectrometry (Supplementary Figure 1C), this peptide was shown to comprise the T-loop of MARK4 phosphorylated at only the Thr residue.
Evidence of differing substrate specificities of AMPK-related kinases
We investigated the substrate specificity of the AMPK-related kinases by comparing the rate at which these enzymes phosphorylated the AMARA peptide and two other peptides that are also efficiently phosphorylated by AMPK, namely the SAMS peptide (Davies et al, 1989) and the LNR peptide (Ross et al, 2002). While all AMPK-related kinases phosphorylated all three peptides, there were marked differences in the relative rates at which the enzymes phosphorylated them (Supplementary Figure 2 For example, the BRSK and NUAK enzymes had a marked preference for the LNR peptide, whereas QSK and QIK preferentially phosphorylated the AMARA peptide.
Identification of peptide substrates for LKB1
We next tested whether the LKB1 complex was able to phosphorylate peptides encompassing the activation loop of AMPKα1, BRSK2, NUAK2, SIK, MARK3 and MELK. All of the peptides were phosphorylated, but with differing efficiencies (Figure 6A). Solid-phase Edman sequencing confirmed that every peptide was specifically phosphorylated by LKB1 at the T-loop Thr residue (Figure 6B). The optimal peptide substrate derived from the T-loop of NUAK2 was phosphorylated by LKB1 with a Km value of 150 μM and was a better substrate than the peptide derived from the T-loop of AMPKα1 (Km, 1.4 mM) (Figure 6A). The MELK peptide was the poorest substrate. We also assayed the different combinations of LKB1 STRADα/β and MO25α/β complexes used in Figure 2 with the T-loop peptides as substrates, and the pattern of LKB1 activity mirrored that observed using the full-length proteins (compare Figures 2 and 6C). These results indicate that the T-loop peptides can be employed as bona fide substrates to measure LKB1 activity in a facile single-step assay. The NUAK2 T-loop peptide was termed LKBtide. Based on sequence alignments of the T-loop of AMPK-related kinases, an optimal substrate phosphorylation motif for LKB1 phosphorylation is proposed in Figure 1A.
Figure 6.
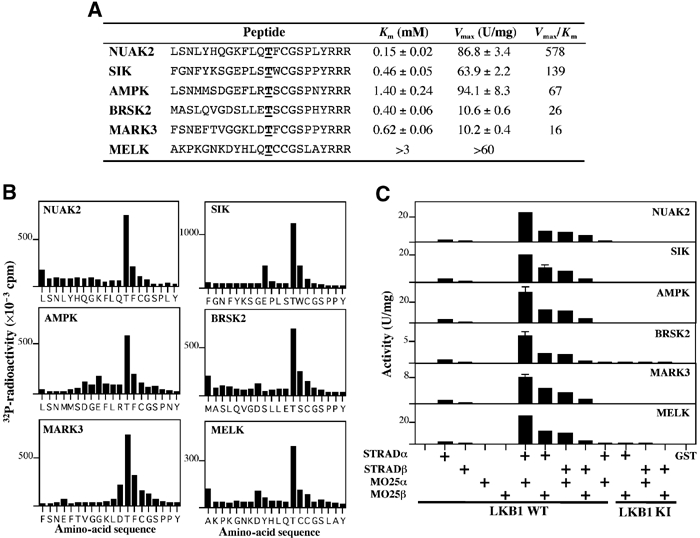
Identification of peptide substrates for LKB1. (A) Kinetic analysis of the phosphorylation of the indicated T-loop by the LKB1:STRAD:MO25 complex was performed. The T-loop Thr residue in each peptide is underlined and is in boldface type. Three Arg residues were added to the C-terminus of each T-loop peptide to enable their capture on phosphocellulose p81 paper. Km and Vmax values were determined from nonlinear regression. (B) An aliquot of each peptide phosphorylated by the LKB1 complex was subjected to solid-phase Edman sequencing and 32P-radioactivity was measured after each cycle of Edman degradation. A small proportion of each peptide can become coupled to the Sequelon arylamine membrane through acidic internal Asp and Glu residues rather than their C-terminal carboxyl group. This accounts for the apparent small releases of 32P-radiactivity that are observed at some Asp and Glu residues. (C) Same combinations of GST-tagged wild-type LKB1 (WT, lanes 1–9), or catalytically inactive (D194A, lanes 10–13) LKB1, or GST alone (lane 14), FLAG-tagged STRADα or STRADβ, and myc-tagged MO25α or MO25β employed in Figure 2 were tested for their ability to phosphorylate the indicated peptides (peptide concentration 200 μM). The results are expressed as the peptide kinase activity generated per mg of LKB1:STRAD:MO25 added to the assay. Results shown are average±s.d. of three assays and are representative of at least two independent experiments.
Role of LKB1 in regulating AMPK-related kinases in fibroblasts
In order to investigate the role of LKB1 in regulating AMPK-related kinases in intact cells, we generated peptide antibodies against specific sequences of the 12 AMPK-related kinases. The ability of these antibodies to immunoprecipitate the active forms of the AMPK-related kinases was assessed employing HEK-293 cell lysates in which the kinases had been overexpressed and found to be active (O Göransson, data not shown). Using this approach, we were able to develop antibodies that selectively immunoprecipitated all the AMPK-related kinases except for MARK2 and MARK3, whose activity was assessed using a commercial antibody that immunoprecipitated both of these kinases, but not MARK1 and MARK4 (O Göransson, data not shown). Using the specific antibodies, we were able to immunoprecipitate and assay endogenously expressed NUAK2, QIK, QSK, SIK MARK1, MARK2/3 and MARK4 in LKB1+/+ mouse embryonic fibroblasts (MEFs) (Figure 7). We compared the activities of these kinases immunoprecipitated from LKB1+/+ and LKB1−/− MEFs, and found that the activity of NUAK2, QIK, QSK, SIK, MARK1 and MARK4 was reduced from 7- to 35-fold in the LKB1−/− MEFs (Figure 7). The combined MARK2/3 activity was reduced ∼3-fold in the LKB1−/− MEFs (Figure 7G). Immunoblotting demonstrated that the reduced activity of AMPK-related kinases in LKB1−/− cells was not caused by a decrease in the expression of the enzymes, which were either present at the same or slightly reduced levels in the knockout cells.
Figure 7.
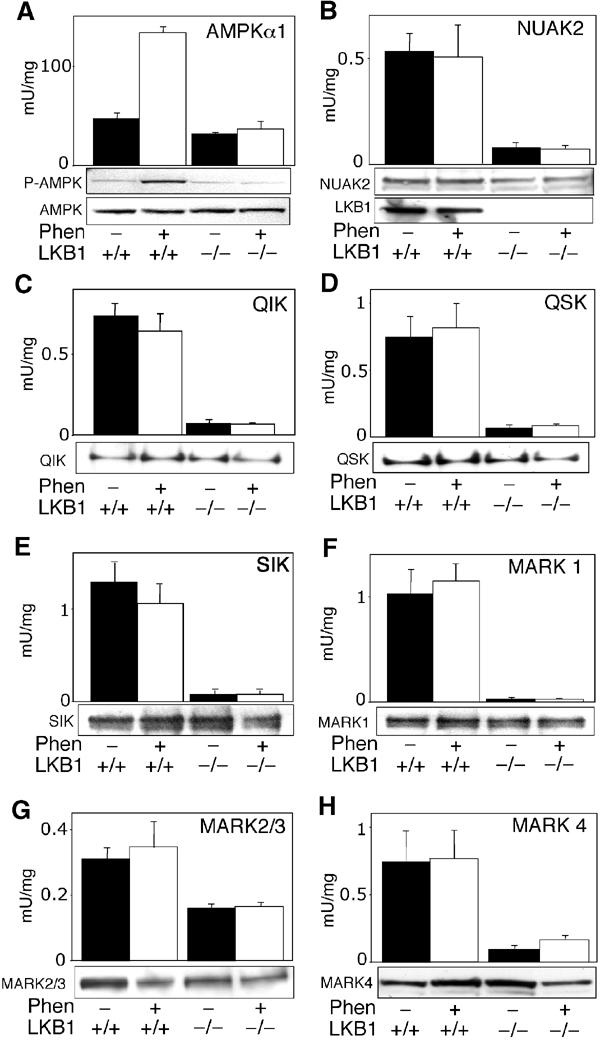
Activity of AMPK-related kinases in LKB1+/+ and LKB1−/− MEFs. LKB1+/+ or LKB1−/− MEFs were either left untreated (black bars) or stimulated with 10 mM phenformin (Phen, white bars) for 1 h. AMPKα1 and AMPK-related kinases were immunoprecipitated from the cell lysates, and in vitro kinase activity towards the AMARA peptide was measured as described in Materials and methods. To confirm equal expression of the kinases in each sample, cell lysates (AMPKα1, NUAK2, MARK2/3 and MARK4) or immunoprecipitated proteins (QIK, QSK, SIK and MARK1) were subjected to SDS–PAGE and Western blot analysis. All AMPK-related kinases migrated with the expected molecular mass. In the case of AMPKα1, cell lysates were immunoblotted with a phospho-Thr172 antibody (P-AMPK) that recognises the phosphorylated T-loop. A control immunoblot of LKB1 levels in cell lysates is also included in panel B. Results shown are average±s.d. of 2–4 assays and are representative of at least two independent experiments.
Interestingly, and in contrast to AMPKα1 (Figure 7A), the AMPK-related kinases assayed were not significantly stimulated by phenformin (Figure 7B–H). This suggested that phenformin was not triggering activation of AMPK by activating LKB1. Consistent with this, we demonstrated that increasing doses of phenformin that progressively activated AMPK (and increased the phosphorylation of Thr172) did not stimulate LKB1 activity (Figure 8A). The drug 5-aminoimidazole-4-carboxamide riboside (AICAR) activates AMPK in intact cells by being taken up and converted by adenosine kinase to AICAR monophosphate, which mimics the effect of AMP on the AMPK system (Corton et al, 1995). AICAR activated AMPK in LKB1+/+ MEFs, but failed to activate markedly any of the AMPK-related kinases (Figure 8C).
Figure 8.
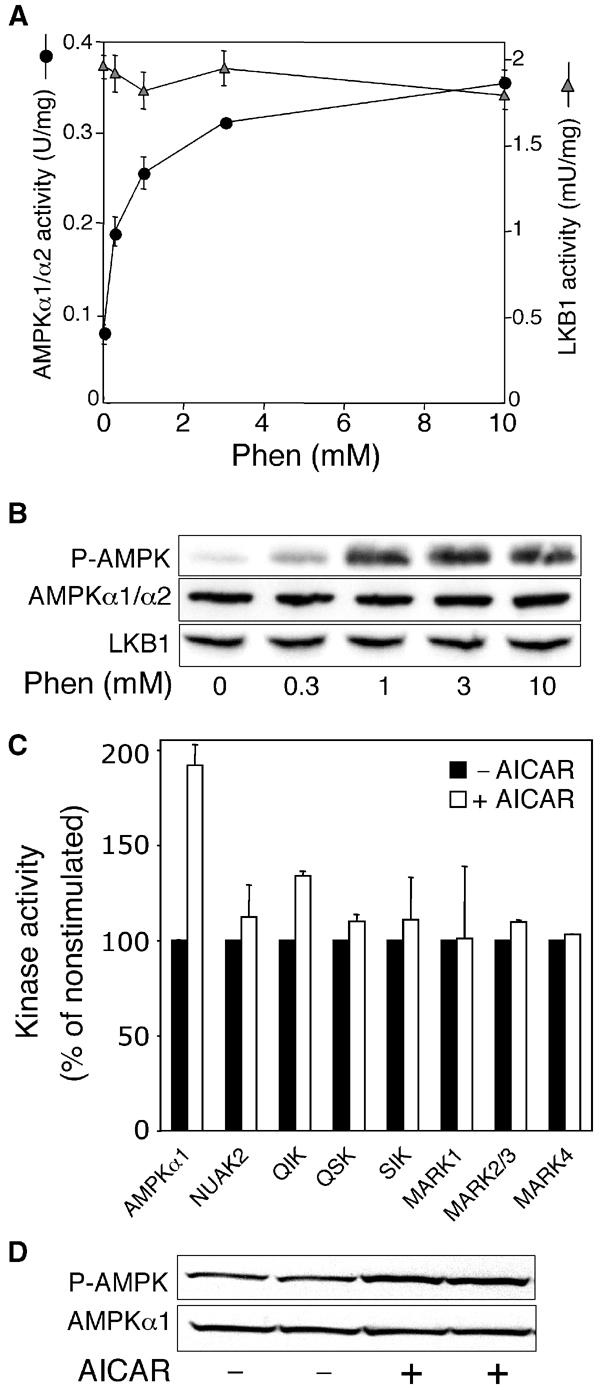
AICAR does not activate AMPK-related kinases. (A) LKB1+/+ MEFs were either left untreated or stimulated with the indicated concentrations of phenformin (Phen) for 1 h. AMPKα1/α2 and LKB1 were immunoprecipitated from the cell lysates, and in vitro kinase activity towards the AMARA and LKBtide peptides, respectively, was measured as described in Materials and methods. Results shown are average±s.d. of a triplicate assay and are representative of two independent experiments. (B) To confirm equal expression of the kinases in each sample, cell lysates were subjected to SDS–PAGE and Western blot analysis with the indicated antibodies. The phospho-Thr172 antibody (P-AMPK) recognises the phosphorylated T-loop of AMPKα1. (C) LKB1+/+ MEFs were either left untreated (black bars) or stimulated with 2 mM AICAR (white bars) for 1 h. AMPKα1 and the indicated AMPK-related kinases were immunoprecipitated from the cell lysates, and in vitro kinase activity towards the AMARA peptide was measured as described in Materials and methods. Results are presented as % relative to the activity observed in nonstimulated cells, and are averages±s.e.m. of two independent experiments. 100% corresponds to the following absolute activities: AMPKα1, 135 mU/mg; NUAK2, 0.25 mU/mg; QIK, 0.76 mU/mg; QSK, 1.9 mU/mg; SIK, 2.73 mU/mg; MARK1, 0.14 mU/mg; MARK2/3, 3.5 mU/mg; and MARK4, 1.6 mU/mg. (D) Immunoblotting of AMPK was performed as in (B).
Expression of LKB1 restores activation of AMPK-related kinases in HeLa cells
To obtain further genetic evidence that LKB1 acts as an upstream regulator of the AMPK-related kinases, we compared the activity of AMPK-related kinases in normal HeLa cells, which do not express LKB1, and in HeLa cells stably expressing either wild-type or catalytically inactive LKB1 (Sapkota et al, 2002; Hawley et al, 2003). In control HeLa cells not expressing LKB1, or cells expressing catalytically inactive LKB1, the activity of NUAK2, QIK, QSK and SIK was 20- to 40-fold lower than that observed in HeLa cells expressing wild-type LKB1 (Figure 9B–E). MARK1, combined MARK2/3 activity and MARK4 activity in control HeLa cells was about ∼2- to 3-fold lower in normal HeLa cells than that in cells expressing LKB1 (Figure 9F–H). In contrast to AMPKα1 (Figure 9A), none of the AMPK-related kinases were significantly activated when HeLa cells were stimulated with phenformin (Figure 9B–H).
Figure 9.
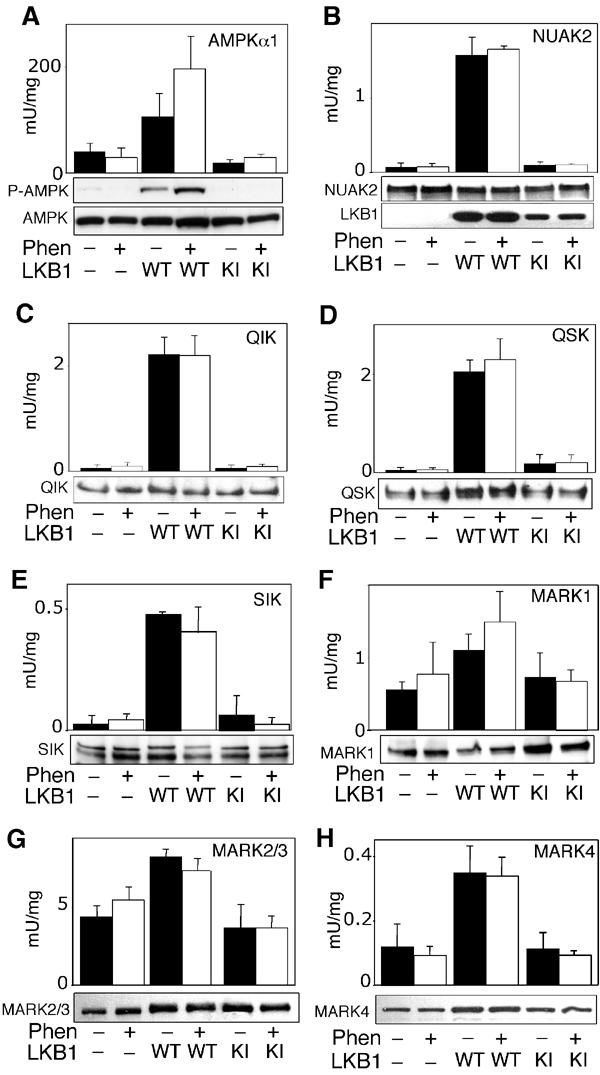
Activity of AMPK-related kinases in HeLa cells. Control HeLa cells lacking LKB1 expression (−), or HeLa cells stably expressing wild-type LKB1 (WT) or kinase inactive LKB1 (KI), were either left untreated (black bars) or stimulated with 10 mM phenformin (Phen, white bars) for 1 h. AMPKα1 and AMPK-related kinases were immunoprecipitated from the cell lysates, and in vitro kinase activity towards the AMARA peptide was measured as described in Materials and methods. To confirm equal expression of the kinases in each sample, cell lysates (AMPKα1, MARK2/3 and MARK4) or immunoprecipitated proteins (NUAK2, QIK, QSK, SIK and MARK1) were subjected to SDS–PAGE and Western immunoblot analysis. All AMPK-related kinases migrated with the expected molecular mass. In the case of AMPKα1, cell lysates were immunoblotted with a phospho-Thr172 antibody (P-AMPK) that recognises the phosphorylated T-loop. A control immunoblot of LKB1 levels in cell lysates is also included in panel B. Results shown are average±s.d. of 2–4 assays and are representative of at least two independent experiments.
Discussion
In this study, we provide evidence that LKB1 functions to regulate the activity of 11 of the 12 members of the AMPK-related family of protein kinases. In cell-free systems we established that all AMPK-related kinases tested, with the exception of MELK, are activated over 50-fold following the phosphorylation of their T-loop Thr residue, by the LKB1:STRAD:MO25 complex. In the case of MELK, our results indicate that this enzyme requires T-loop phosphorylation for activity, but that it is able to phosphorylate its own T-loop residue. To our knowledge this is the first demonstration that the BRSK1, BRSK2, NUAK1, NUAK2, QIK, QSK, SIK as well as MELK enzymes can be activated by phosphorylation. Mutation of the T-loop Thr residue to Ala prevented the activation of all of these enzymes by the LKB1 complex, whereas its mutation to Glu was sufficient to activate BRSK1, BRSK2, NUAK1, NUAK2, SIK, QIK, QSK and MARK4 to specific activities similar to that observed for the LKB1-phosphorylated forms of these enzymes (Figure 4). This latter finding could be exploited in future overexpression or knock-in studies to introduce constitutively active forms of these enzymes in cells to examine their cellular roles.
Previous work has indicated that MARK2 and MARK3 were phosphorylated at the T-loop Thr residue as well as at a nearby Ser residue (Drewes et al, 1997). The T-loop Ser residue phosphorylated in MARK2/MARK3 is conserved in AMPKα1, AMPKα2 and all AMPK-related kinases (Figure 1A and B), but at least for AMPKα1 there is no evidence that this residue is phosphorylated in vivo (Woods et al, 2003b). Our analysis (Figure 5A and B), as well as that of a recent study (Timm et al, 2003), showed that phosphorylation of the T-loop Thr residue of the MARK kinases played the most crucial role in regulating the activity of this class of kinase. The LKB1 complex phosphorylated a catalytically inactive MARK3 mutant at both the Thr and Ser T-loop residues (Figure 5A). However, the isolated MARK3 T-loop peptide was only phosphorylated by LKB1 at the Thr residue (Figure 6), signifying that the primary sequence of the peptide was not sufficient to enable LKB1 to phosphorylate the Ser residue. The finding that mutation of the T-loop Thr211 to Ala on MARK3 prevented phosphorylation of Ser215 by the LKB1 complex suggests that phosphorylation of Thr211 is required for Ser215 phosphorylation. MARK4 may be regulated differently, as peptide-mapping studies indicated that MARK4 was only phosphorylated at the T-loop Thr by LKB1 (Figure 5G), and that mutation of the T-loop Thr to Glu increased activity to a much greater level than observed for other MARK isoforms (Figure 5F). It was previously reported for MARK2 that mutation of the T-loop to Glu increased its specific activity three-fold (Timm et al, 2003), and we have made similar findings with this mutation increasing MARK2 activity from 2.2 to 6.7 U/mg (Figure 5D). However, in contrast to TAO1, which only activated MARK2 10-fold (Timm et al, 2003), the LKB1 complex increased MARK2 activation 200-fold, showing that in this case the Thr to Glu mutation only modestly activated MARK2 compared to phosphorylation by LKB1.
As observed for the activation of AMPKα1 and AMPKα2 (Hawley et al, 2003), the presence of STRAD and MO25 subunits was essential for LKB1 to activate all of the AMPK-related kinases in cell-free assays. There is considerable evidence that many protein kinases rely on sequences, termed ‘docking sites', lying outside of the catalytic core, which stabilise the interaction between the kinase and its substrate. It was possible that the STRAD and MO25 subunits played a similar role in docking the substrate to the kinase complex. However, the finding that STRAD and MO25 subunits stimulated phosphorylation of the short T-loop peptides and the whole kinase subunits in a very similar manner suggests that they activate LKB1 directly rather than just acting as docking sites. Nevertheless, these results do not rule out additional roles of the STRAD and MO25 subunits in targeting the complex or in substrate recognition.
Employing LKB1−/− MEFs and/or HeLa cells that lack LKB1, we were able to demonstrate that the activities of endogenously expressed NUAK1, NUAK2, QIK, QSK and SIK were 7- to 40-fold lower than in LKB+/+ MEFs or HeLa cells that stably express wild-type LKB1. This provides genetic evidence that LKB1 is rate limiting in the activation of these enzymes in intact cells. However, our data do not rule out the possibility that other kinases can regulate the activity of the AMPK-related kinases in vivo in addition to LKB1. Recently, the TAO1 kinase was purified from pig brain as the major activity that phosphorylated the T-loop Thr residue of MARK2, resulting in its activation (Timm et al, 2003). Although TAO1 also activated MARK2 in overexpression studies (Timm et al, 2003), so far no genetic evidence indicating how the lack of TAO1 affected the activity of MARK family kinases in vivo has been reported. Such evidence may be hard to acquire, as mammalian cells possess three closely related TAO isoforms (Manning et al, 2002). Lack of specific immunoprecipitating antibodies meant that we were unable to establish how a deficiency of LKB1 affected the individual activities of MARK2 or MARK3. However, the finding that the combined activity of MARK2 and MARK3 in LKB1−/− and control HeLa cells was substantial implies that other kinases, such as TAO1, may regulate the activities of these enzymes. It should be noted that LKB1 and TAO1 share no obvious amino-acid sequence homology and lie in distinct regions of the human kinase dendrogram (Manning et al, 2002). TAO1 belongs to the STE20 group of kinases and is therefore related to STRADα and STRADβ, but whether this has any significance is not clear.
The MARK (MAP/microtubule affinity regulating kinase) family of kinases are the most studied AMPK-related kinase family members and are thought to play key roles in establishing cell polarity. For example, genetic studies indicate that MARK homologues control partitioning of the C. elegans zygote (Guo and Kemphues, 1995) and embryonic axis formation in Drosophila (Shulman et al, 2000). In neuronal cells, MARK kinases phosphorylate the neuronal microtubule-associated protein Tau, resulting in destabilisation of microtubules (Drewes et al, 1997). Interestingly, the counterpart of mammalian LKB1 in C. elegans (Watts et al, 2000), termed PAR4, was originally identified as a member of the maternally expressed PAR (partitioning defective) gene family, required for establishing cell polarity during the first cycle of C. elegans embryogenesis (Kemphues et al, 1988). Maternal lethal mutations in the gene encoding C. elegans PAR4 have been shown to affect several aspects of cell polarity (Morton et al, 1992). These lead to phenotypes similar to those observed in C. elegans mutated in the PAR1 gene, which encodes the homologue of MARK3 (Guo and Kemphues, 1995). More recently, both the Drosophila (Martin and St Johnston, 2003) and Xenopus (Ossipova et al, 2003) homologues of human LKB1 were shown to play important roles in regulating cell polarity. Originally, it was suggested that Drosophila LKB1 functioned downstream of PAR1/MARK3, as overexpression of Drosophila LKB1 suppressed the polarity phenotype of PAR1/MARK3 mutants (Martin and St Johnston, 2003). However, in HeLa cells that do not express LKB1, MARK3 was found to exist in a dephosphorylated state and reintroduction of LKB1 promoted phosphorylation of MARK3 at its T-loop (Spicer et al, 2003), signifying that LKB1 was upstream of MARK3. One explanation that would reconcile the apparent discrepancy between the Drosophila and mammalian studies would be whether overexpression of Drosophila LKB1 suppressed the polarity phenotype of PAR1/MARK3 mutants, through the activation of other AMPK-related kinases, which might be able to compensate for the loss of PAR1/MARK3 function.
Much less is known regarding the function of other AMPK-related kinases, that is, BRSK1, BRSK2, NUAK1, NUAK2, QIK, QSK and SIK. Previous Northern blotting of NUAK1, NUAK2, QIK and SIK performed by other groups (see below) and analysis of EST clones (Supplementary Table I) indicated that the AMPK-related kinases with the exception of BRSK1, BRSK2 and MELK are likely to be expressed in many tissues. BRSK1 and BRSK2 EST clones were mainly derived from neuronal tissues and, after immunoblot analysis of a variety of rat tissues, these enzymes were only detected in the brain and at low levels in the testis (K Sakamoto, unpublished results). To date, MELK (maternal embryonic leucine zipper kinase) has only been reported to be expressed during mammalian embryogenesis, with the strongest expression detected during maturation of oocytes and preimplantation development (Heyer et al, 1999). A recent study indicates that MELK can inhibit spliceosome assembly by interacting with the splicing factor NIPP1 (Vulsteke et al, 2003). Although our antibodies raised against BRSK1, BRSK2 and MELK readily immunoprecipitated the recombinant enzymes, we were unable to detect their activity in MEFs or HeLa cells (O Göransson, data not shown). SIK (Salt-inducible kinase) was first cloned from the adrenal glands of rats fed a high-salt diet (Wang et al, 1999). SIK mRNA was also induced by membrane depolarisation in the brain (Feldman et al, 2000), and recent studies have indicated that when SIK is overexpressed in cells, it might play a role in steroidogenesis (Takemori et al, 2003). The mRNA expressing QIK (also termed SIK2) was highest in adipose tissue and, in overexpression studies, QIK was reported to phosphorylate human IRS1 at Ser794 (Horike et al, 2003), the residue equivalent to Ser789 in rat IRS1, shown to be phosphorylated by AMPK (Jakobsen et al, 2001). In other overexpression studies, NUAK1 (ARK5) was shown to suppress apoptosis induced by some stimuli, including nutrient starvation (Suzuki et al, 2003a). Furthermore, it has been claimed that Akt/PKB phosphorylated NUAK1 at a C-terminal site outside of the catalytic domain, leading to a three-fold activation of the enzyme (Suzuki et al, 2003b). NUAK2 (SNARK) was most highly expressed in the kidney, and its activity was reportedly stimulated by glucose starvation of cells (Lefebvre et al, 2001; Suzuki et al, 2003b). To our knowledge, no previous studies have addressed the roles of BRSK1, BRSK2 and QSK.
Clearly, further work needs to be carried out to characterise the mechanism of regulation and function of the AMPK-related kinases. Our studies indicate that, at least in MEFs and HeLa cells that stably express LKB1, the AMPK-related kinases were not activated in response to the drugs phenformin or AICAR in contrast to AMPK. This suggests that the beneficial antidiabetic effects of phenformin and metformin may be mediated through the activation of AMPK rather than the AMPK-related kinases. The finding that AMPK-related kinases are not activated by phenformin or AICAR is consistent with findings that these treatments do not activate LKB1 directly in LKB1+/+ MEFs (Figure 8A) or in COS7 cells (Woods et al, 2003a). Phenformin stimulates AMPK without affecting the AMP or the ADP/ATP ratio in cells (Fryer et al, 2002; Hawley et al, 2002). It is therefore currently unclear how phenformin activates AMPK. Possibilities include phenformin inhibiting an AMPK-specific phosphatase that does not target the AMPK-related kinases, although the related drug metformin did not affect dephosphorylation of AMPK by protein phosphatase-2C in vitro (Hawley et al, 2002). Alternatively, phenformin may somehow generate a metabolite or another molecule that inside the cell binds to AMPK and promotes its activation by the LKB1 complex. In future studies it will be important to define the physiological stimuli that do cause activation of the AMPK-related kinases, and establish whether these enzymes, like AMPK, possess regulatory subunits that control their activation. The finding that AMPK-related kinases phosphorylate peptide substrates at different relative rates (Supplementary Figure 2) indicates that these enzymes have distinct substrate specificity preferences and may thus phosphorylate different substrates in vivo.
A significant number of inherited forms of PJS found in certain families do not display mutations in the LKB1 gene (Buchet-Poyau et al, 2002), indicating that there is a second causative locus for PJS. It will clearly now be critical to find out whether PJS families that express the normal LKB1 protein have mutations in other AMPK-related kinases. In conclusion, we have shown that LKB1 functions as a master upstream protein kinase activating 11 AMPK-related kinases in addition to AMPKα1 and AMPKα2. It is possible that these enzymes mediate some of the physiological effects previously ascribed to LKB1 and that one or more of these kinases may themselves function as tumour suppressors.
Materials and methods
A detailed Materials and methods section is provided in the supplementary section.
Supplementary Material
Supplementary Figure 1 Analysis of phosphorylation of BRSK2, NUAK2 and MARK4 and MELK. (A) Catalytically inactive mutants of BRSK2[D159A] and NUAK2[D193A], that are unable to autophosphorylate, were incubated with LKB1 complex for 30 min with Mg2+ -[γ32P]ATP and separated by electrophoresis on a polyacrylamide gel which was then autoradiographed. The 32P-labelled proteins were digested with trypsin and the resulting peptides were chromatographed on a C18 column. Fractions containing the major 32P-labelled peptides are marked. (B) Peptides P1, P2 and P3 were subjected to solid phase sequencing and 32P-radioactivity was measured after each cycle of Edman degradation. We were unable to determine the identity of the NUAK2 peptide indicated with “?”. (C) The indicated peptides were analysed by MALDI TOF–TOF mass spectrometry as described above. The deduced amino acid sequence and the site of phosphorylation was indicated, together with the observed and theoretical mass. The peptide labelled “MARK4” was derived from MARK4 phosphorylated by LKB1 as described in the legend to Figure 5G. This peptide encompasses the T-loop motif of MARK4 phosphorylated at only the Thr residue. The peptide labelled “MELK” is derived from a tryptic digest of unlabelled MELK that had been expressed and purified from E. coli as described above. This peptide encompasses the T-loop of MELK phosphorylated at the Thr residue. Abbreviations: m, methionine sulphone; (p) indicates preceding Thr residue is phosphorylated.).
Supplementary Figure 2 Substrate specificity analysis of the AMPK-related kinases. The indicated AMPK-related enzymes in which the T-loop was mutated to Glu, in order to activate these enzymes, were assayed using either 0.1 mM or 1 mM of either the AMARA peptide (AMARAASAAALARRR (Dale et al., 1995), the SAMS peptide (HMRSAMSGLHLVKRR (Davies et al., 1989)) or the LNR peptide (KKLNRTLSFAEPG (Ross et al., 2002)). The activities are presented as average ± SD of two separate experiments with each determination performed in triplicate.).
Supplementary Table I
supplementary section
Acknowledgments
We thank Agnieszka Kieloch for assistance with cell culture, Moustapha Aoubala for preparation of antibodies, the Sequencing Service (School of Life Sciences, University of Dundee, Scotland) for DNA sequencing and the Post Genomics and Molecular Interactions Centre for Mass Spectrometry facilities. OG is supported by a Wenner-Gren Foundation fellowship. We thank the Association for International Cancer Research (DRA), Diabetes UK (DRA and DGH), the Medical Research Council (DRA and DGH), the Wellcome Trust (DGH), the European Commission (QLG1-CT-2001-01488, DGH) and the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck & Co. Inc., Merck KgaA and Pfizer) for financial support.
References
- Alessi DR (2001) Discovery of PDK1, one of the missing links in insulin signal transduction. Biochem Soc Trans 29: 1–14 [DOI] [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J 22: 3062–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR (2003a) MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J 22: 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Deak M, Lawlor MA, Morrice NA, Alessi DR (2003b) Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem J 370: 849–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Sapkota G, Alessi DR (2003c) LKB1, a protein kinase regulating cell proliferation and polarity. FEBS Lett 546: 159–165 [DOI] [PubMed] [Google Scholar]
- Brajenovic M, Joberty G, Kuster B, Bouwmeester T, Drewes G (2003) Comprehensive proteomic analysis of human Par protein complexes reveals an interconnected protein network. J Biol Chem [DOI] [PubMed] [Google Scholar]
- Buchet-Poyau K, Mehenni H, Radhakrishna U, Antonarakis SE (2002) Search for the second Peutz–Jeghers syndrome locus: exclusion of the STK13, PRKCG, KLK10, and PSCD2 genes on chromosome 19 and the STK11IP gene on chromosome 2. Cytogenet Genome Res 97: 171–178 [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG (1995) 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229: 558–565 [DOI] [PubMed] [Google Scholar]
- Dale S, Wilson WA, Edelman AM, Hardie DG (1995) Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett 361: 191–195 [DOI] [PubMed] [Google Scholar]
- Davies SP, Carling D, Hardie DG (1989) Tissue distribution of the AMP-activated protein kinase, and lack of activation by cyclic-AMP-dependent protein kinase, studied using a specific and sensitive peptide assay. Eur J Biochem 186: 123–128 [DOI] [PubMed] [Google Scholar]
- Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 89: 297–308 [DOI] [PubMed] [Google Scholar]
- Drewes G, Nurse P (2003) The protein kinase kin1, the fission yeast orthologue of mammalian MARK/PAR-1, localises to new cell ends after mitosis and is important for bipolar growth. FEBS Lett 554: 45–49 [DOI] [PubMed] [Google Scholar]
- Feldman JD, Vician L, Crispino M, Hoe W, Baudry M, Herschman HR (2000) The salt-inducible kinase, SIK, is induced by depolarization in brain. J Neurochem 74: 2227–2238 [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D (2002) The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277: 25226–25232 [DOI] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ (1995) par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell 81: 611–620 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Scott JW, Pan DA, Hudson ER (2003) Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546: 113–120 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG (2002) The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 51: 2420–2425 [DOI] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA (1998) A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature 391: 184–187 [DOI] [PubMed] [Google Scholar]
- Heyer BS, Kochanowski H, Solter D (1999) Expression of Melk, a new protein kinase, during early mouse development. Dev Dyn 215: 344–351 [DOI] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M (2003) Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 100: 8839–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, Sun XJ, Yamamoto H, Kasayama S, Muraoka M, Nonaka Y, Okamoto M (2003) Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J Biol Chem 278: 18440–18447 [DOI] [PubMed] [Google Scholar]
- Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE (2001) 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem 276: 46912–46916 [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M (1998) Peutz–Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18: 38–43 [DOI] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS (1988) Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell 52: 311–320 [DOI] [PubMed] [Google Scholar]
- Lefebvre DL, Bai Y, Shahmolky N, Sharma M, Poon R, Drucker DJ, Rosen CF (2001) Identification and characterization of a novel sucrose-non-fermenting protein kinase/AMP-activated protein kinase-related protein kinase, SNARK. Biochem J 355: 297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- Martin SG, St Johnston D (2003) A role for Drosophila LKB1 in anterior–posterior axis formation and epithelial polarity. Nature 421: 379–384 [DOI] [PubMed] [Google Scholar]
- Milburn CC, Boudeau J, Deak M, Alessi DR, van aalten DMF (2004) Crystal structure of MO25a in complex with the C-terminus of the pseudokinase STE20-Related ADaptor (STRAD). Nat Struct Biol 11: 193–200 [DOI] [PubMed] [Google Scholar]
- Morton DG, Roos JM, Kemphues KJ (1992) par-4, a gene required for cytoplasmic localization and determination of specific cell types in Caenorhabditis elegans embryogenesis. Genetics 130: 771–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N, McCartney RR, Schmidt MC (2003) Yeast Pak1 kinase associates with and activates Snf1. Mol Cell Biol 23: 3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipova O, Bardeesy N, DePinho RA, Green JB (2003) LKB1 (XEEK1) regulates Wnt signalling in vertebrate development. Nat Cell Biol 5: 889–894 [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Ogg S, Thoma RS, Byrnes MJ III, Wu Z, Stephenson MT, Piwnica-Worms H (1998) C-TAK1 protein kinase phosphorylates human Cdc25C on serine 216 and promotes 14-3-3 protein binding. Cell Growth Differ 9: 197–208 [PubMed] [Google Scholar]
- Ross H, Armstrong CG, Cohen P (2002) A non-radioactive method for the assay of many serine/threonine-specific protein kinases. Biochem J 366: 977–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapkota GP, Deak M, Kieloch A, Morrice N, Goodarzi AA, Smythe C, Shiloh Y, Lees-Miller SP, Alessi DR (2002) Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J 368: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Benton R, St Johnston D (2000) The Drosophila homolog of C. elegans PAR-1 organizes the oocyte cytoskeleton and directs oskar mRNA localization to the posterior pole. Cell 101: 377–388 [DOI] [PubMed] [Google Scholar]
- Spicer J, Rayter S, Young N, Elliott R, Ashworth A, Smith D (2003) Regulation of the Wnt signalling component PAR1A by the Peutz–Jeghers syndrome kinase LKB1. Oncogene 22: 4752–4756 [DOI] [PubMed] [Google Scholar]
- Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG (2003) Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol 13: 1299–1305 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Esumi H (2003a) ARK5 suppresses the cell death induced by nutrient starvation and death receptors via inhibition of caspase 8 activation, but not by chemotherapeutic agents or UV irradiation. Oncogene 22: 6177–6182 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Lu J, Ogura T, Lavin MF, Esumi H (2003b) Identification of a novel protein kinase mediating Akt survival signaling to the ATM protein. J Biol Chem 278: 48–53 [DOI] [PubMed] [Google Scholar]
- Takemori H, Doi J, Horike N, Katoh Y, Min L, Lin XZ, Wang ZN, Muraoka M, Okamoto M (2003) Salt-inducible kinase-mediated regulation of steroidogenesis at the early stage of ACTH-stimulation. J Steroid Biochem Mol Biol 85: 397–400 [DOI] [PubMed] [Google Scholar]
- Tiainen M, Vaahtomeri K, Ylikorkala A, Makela TP (2002) Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1). Hum Mol Genet 11: 1497–1504 [DOI] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Makela TP (1999) Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA 96: 9248–9251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm T, Li XY, Biernat J, Jiao J, Mandelkow E, Vandekerckhove J, Mandelkow EM (2003) MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. EMBO J 22: 5090–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulsteke V, Beullens M, Boudrez A, Keppens S, Van Eynde A, Rider MH, Stalmans W, Bollen M (2003) Inhibition of spliceosome assembly by the cell-cycle regulated protein kinase MELK and involvement of the splicing factor NIPP1. J Biol Chem [DOI] [PubMed] [Google Scholar]
- Wang Z, Takemori H, Halder SK, Nonaka Y, Okamoto M (1999) Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett 453: 135–139 [DOI] [PubMed] [Google Scholar]
- Watts JL, Morton DG, Bestman J, Kemphues KJ (2000) The C. elegans par-4 gene encodes a putative serine–threonine kinase required for establishing embryonic asymmetry. Development 127: 1467–1475 [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003a) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–2008 [DOI] [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH (2003b) Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem 278: 28434–28442 [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Analysis of phosphorylation of BRSK2, NUAK2 and MARK4 and MELK. (A) Catalytically inactive mutants of BRSK2[D159A] and NUAK2[D193A], that are unable to autophosphorylate, were incubated with LKB1 complex for 30 min with Mg2+ -[γ32P]ATP and separated by electrophoresis on a polyacrylamide gel which was then autoradiographed. The 32P-labelled proteins were digested with trypsin and the resulting peptides were chromatographed on a C18 column. Fractions containing the major 32P-labelled peptides are marked. (B) Peptides P1, P2 and P3 were subjected to solid phase sequencing and 32P-radioactivity was measured after each cycle of Edman degradation. We were unable to determine the identity of the NUAK2 peptide indicated with “?”. (C) The indicated peptides were analysed by MALDI TOF–TOF mass spectrometry as described above. The deduced amino acid sequence and the site of phosphorylation was indicated, together with the observed and theoretical mass. The peptide labelled “MARK4” was derived from MARK4 phosphorylated by LKB1 as described in the legend to Figure 5G. This peptide encompasses the T-loop motif of MARK4 phosphorylated at only the Thr residue. The peptide labelled “MELK” is derived from a tryptic digest of unlabelled MELK that had been expressed and purified from E. coli as described above. This peptide encompasses the T-loop of MELK phosphorylated at the Thr residue. Abbreviations: m, methionine sulphone; (p) indicates preceding Thr residue is phosphorylated.).
Supplementary Figure 2 Substrate specificity analysis of the AMPK-related kinases. The indicated AMPK-related enzymes in which the T-loop was mutated to Glu, in order to activate these enzymes, were assayed using either 0.1 mM or 1 mM of either the AMARA peptide (AMARAASAAALARRR (Dale et al., 1995), the SAMS peptide (HMRSAMSGLHLVKRR (Davies et al., 1989)) or the LNR peptide (KKLNRTLSFAEPG (Ross et al., 2002)). The activities are presented as average ± SD of two separate experiments with each determination performed in triplicate.).
Supplementary Table I
supplementary section