

Abstract
Purpose
The role of E-cadherin in carcinogenesis is of great interest, but few studies have examined its relevance to pancreatic carcinoma.
Experimental Design
We evaluated E-cadherin protein expression by immunohistochemistry in pancreatobiliary cancers having a noncohesive histologic phenotype (21 undifferentiated adenocarcinomas and 7 signet ring carcinomas), comparing the results with pancreatic cancers having a cohesive phenotype (25 moderately differentiated and 14 poorly differentiated adenocarcinomas).
Results
Twenty of 21 undifferentiated cancers had complete absence of E-cadherin expression, as did two signet ring carcinomas. In contrast, cohesive cancers (n = 39) had E-cadherin labeling at the plasma membrane (P < 0.001). Subsets of cancers were also evaluated for β-catenin expression. All of the cohesive lesions (n = 28) showed a membranous β-catenin expression pattern, whereas noncohesive foci (n = 7) were characterized by either cytoplasmic labeling or complete absence of β-catenin protein expression, suggestive of a deficient zonula adherens in noncohesive cancers. E-cadherin promoter hypermethylation was observed in an undifferentiated pancreatic cancer cell line, MiaPaCa-2, whereas two pancreatic cancer cell lines derived from differentiated lesions lacked any evidence of E-cadherin promoter methylation. No pattern of E-cadherin promoter methylation could be determined in three primary cancers having mixed histologic patterns (contained both cohesive and noncohesive foci). No somatic mutations in E-cadherin were identified in noncohesive pancreatic cancers having inactivated E-cadherin.
Conclusions
Noncohesive pancreatic cancers were characterized by the loss of E-cadherin protein expression. Promoter hypermethylation is a possible mechanism of E-cadherin gene silencing in a subset of these cancers.
It is a curious fact that most of the studies of pancreatic carcinogenesis have examined primary tumors as opposed to advanced disease. One explanation is the greater availability of surgically resected tissue compared with unresectable metastatic deposits. A natural consequence of the increased attention given to resectable lesions is that the molecular biology of advanced pancreatic cancer has been relatively understudied despite ~80% of all pancreatic cancers being unresectable at the time of diagnosis.
A recent study evaluated advanced pancreatic cancers for the types of genetic abnormalities previously observed in resectable disease (1). An increased rate of SMAD4 gene inactivation was found in advanced lesions compared with the rates reported for surgically resected invasive pancreatic cancers and its precursor lesions (2, 3). Another study found increased MKK4 gene inactivation in metastatic pancreatic cancer compared with resectable and locally unresectable disease (4). Nonetheless, molecular distinctions between localized and advanced pancreatic cancer have been poorly explored.
Similarly, relatively little is known of the molecular events leading to the emergence of rare but biologically aggressive pancreatic adenocarcinoma subtypes. These cancers offer a unique opportunity to examine potential molecular factors responsible for the invasive and lethal nature of pancreatic cancer. Several of the aggressive adenocarcinoma variants exhibit a noncohesive phenotype (defined here as a pancreaticobiliary adenocarcinoma having prominent regions lacking a ductal architecture and having a paucity of cell-cell contact) on light microscopy, raising the possibility that a molecular defect involving the adherens junction could contribute to the observed histologic pattern and, more importantly, aggressive biological behavior.
We evaluated expression of E-cadherin (a key component of adherens junctions) in a panel of noncohesive pancreatobiliary cancers, including undifferentiated and signet ring pancreatic carcinomas. Both primary and metastatic noncohesive pancreatic cancers were compared with differentiated cancers having a tubular (ductal) histologic pattern (i.e., cohesive pancreatic cancer). We also investigated potential molecular mechanisms of E-cadherin inactivation in noncohesive pancreatic cancer. Finally, we propose a tumor progression model that is based on the timing of E-cadherin inactivation during cancer development to account for the various noncohesive histologic patterns observed in this study.
Materials and Methods
Tissue specimens
This study evaluated histologic sections from 67 pancreaticobiliary cancers, including 60 primary and 7 metastatic lesions. Sixty-five were pancreatic and 2 were biliary in origin. Two non-cohesive adenocarcinomas of the distal common bile duct (a resected signet ring carcinoma and a resected anaplastic carcinoma) were included in the analysis because distal bile duct adenocarcinomas bear a striking similarity to adenocarcinomas of the pancreatic head with respect to acquired genetic abnormalities, histologic appearance, and clinical outcomes (5–7). Thirty-seven samples of paraffin-embedded primary pancreatic carcinoma were collected from resection specimens at the Johns Hopkins Hospital. Fourteen samples of advanced-stage and/or metastatic pancreatic carcinoma were collected from participants of the Johns Hopkins Gastrointestinal Cancer Rapid Medical Donation Program.4 Archival samples of primary and metastatic pancreatic cancer from an additional 16 patients were obtained from the Autopsy Pathology Files of The Johns Hopkins Hospital. When available, clinicopathologic data were obtained from the patient’s medical records or from the Johns Hopkins pancreaticoduodenectomy database. The collection and use of all paraffin-embedded surgical and autopsy samples and clinicopathologic information for this project were approved by the Johns Hopkins Institutional Review Board.
Immunohistochemistry
Unstained 4-μm sections were deparaffinized by routine techniques and placed in 200 mL Target Retrieval Solution, pH 6.0 (Envision Plus Detection kit, Dako) for 20 min at 100°C. After cooling for 20 min, the slides were quenched with 3% H2O2 for 5 min and then incubated at room temperature with an appropriate dilution of a primary antibody (1:10 dilution of mouse anti-human E-cadherin, clone HEDC-1, Zymed; 1:1,000 dilution of mouse anti-human β-catenin, Transduction Laboratories; 1:200 dilution of mouse anti-human Melan-A, Dako; and 1:4,000 dilution of mouse anti-human AE1/AE3, Chemicon). Labeling of antibodies was detected with the Dako Envision system following the manufacturer’s protocol. A negative control in each run substituted an equal volume of PBS in place of primary antibody.
Cancers were classified as cohesive if infiltrating glands were observed throughout the lesion and noncohesive if any cancerous foci lacked infiltrating glands. Pancreatic cancer subtypes characterized by a noncohesive pattern include anaplastic carcinoma, undifferentiated cancer with osteoclast-like giant cells, and signet ring carcinoma. Immunohistochemical analyses of noncohesive cancers were compared with analyses of cohesive pancreatic cancers. Immunohistochemical labeling of each tissue section was scored on an intensity scale of 0 to 3, with 0 corresponding to no labeling of neoplastic epithelial cells, 1 corresponding to weak labeling of neoplastic epithelium (labeling best seen at 10× objective or greater), 2 corresponding to unequivocal labeling of epithelial cells, and 3 corresponding to intense labeling. The percentage of labeled neoplastic epithelial cells was scored from 0 (complete absence) to 100 (all cells labeling). The labeling intensity and labeling percentage generated a histology score (H-score) ranging from 0 to 300, with H-score = intensity of immunolabel (range, 0–3) × the percentage of cancer cells that were reactive. Scoring was done by consensus of two authors (J.M.W. and C.I.D.) at a two-headed microscope.
PCR amplification and sequencing
Frozen tumor from selected noncohesive pancreatobiliary cancers was macrodissected to achieve at least 70% neoplastic cellularity. DNA was isolated using the QIAmp DNA Mini kit (Qiagen). PCR amplification of the entire protein-coding sequence (exons 1-16) from genomic DNA was done with primer pairs flanking each exon (sequences are available on request). PCR-amplified products were purified using QIAquick columns (Qiagen) and studied by automated sequencing using an ABI Prism model 3700 (Applied Biosystems) and Sequencher software (Gene Codes).
Bisulfite modification and methylation-specific PCR
Genomic DNA was prepared from pancreatic cancer cell lines or from frozen tumors of heterogeneous pancreatic cancers having both cohesive (i.e., undifferentiated) and noncohesive (i.e., differentiated) components. The DNA was bisulfite modified as previously described (8). Treated DNA was then subjected to PCR amplification using separate primer sets designed to specifically amplify unmethylated and methylated DNA, respectively (9). The amplification products were subjected to a second round of PCR in a nested fashion (primers available on request), thereby allowing semiquantitative assessment of methylation at the E-cadherin promoter. The reaction mixture for the first round of PCR included ~100 ng of bisulfite-treated DNA, 25 pmol of each primer, 100 pmol deoxynucleotide triphosphates, 10× PCR buffer, and 1 unit of JumpStart Red Taq Polymerase (Sigma) in a final reaction volume of 25 μL. Cycle conditions were 95°C × 5 min; 35 cycles × (95°C × 30 s, 58°C × 30 s, 72°C × 30 s); 72°C × 5 min. PCR product (1 μL) was used in the second round of PCR, which ran for six cycles, with an annealing temperature of 60°C. Methylation-specific PCR products were resolved using 2% agarose gel electrophoresis. Bisulfite-treated water was used as a negative control and in vitro–methylated DNA was used as a positive control. The investigator doing the methylation-specific PCR experiments was blinded to the differentiation status of the pancreaticobiliary tumors.
Statistical analysis
Categorical variables were analyzed using a χ2 test and continuous variables were analyzed by the Mann-Whitney ranks sum test. Long-term survival was analyzed using the Kaplan-Meier method and the log-rank test. The survival of patients having resected E-cadherin–negative cancers was compared with the survival of a control group of 1,229 patients with resected tubuloglandular ductal adenocarcinoma (cohesive pancreatic cancer) from the authors’ institution.
Results
Immunohistochemistry
Twenty-eight noncohesive pancreaticobiliary cancers were examined for E-cadherin protein expression, including 15 anaplastic carcinomas, 6 adenocarcinomas with osteoclast-like giant cells, and 7 signet ring carcinomas. Noncohesive cancers were compared with 39 cohesive pancreatic cancers, including 24 moderately differentiated ductal adenocarcinomas, 13 poorly differentiated ductal adenocarcinomas, 1 moderately differentiated colloid carcinoma, and 1 poorly differentiated colloid carcinoma. Results are summarized in Table 1.
Table 1.
E-cadherin expression detected by immunohistochemistry
| Histology | Proportion of cancers with E-cadherin loss | Median H-score in E-cadherin–positive cancers |
|---|---|---|
| Noncohesive carcinoma* | ||
| Undifferentiated carcinoma | ||
| Anaplastic | 14/15 (93%) | 10 (n = 1) |
| Osteoclast-like giant cells | 6/6 (100%) | – (n = 0) |
| Signet ring | 2/7 (29%) | 200 (n = 5) |
| Overall | 22/28 (79%) | 200 (n = 6) |
| Cohesive carcinoma | ||
| Ductal adenocarcinoma | ||
| Moderate | 0/24 (0%) | 200 (n = 24) |
| Poor | 0/13 (0%) | 200 (n = 13) |
| Colloid carcinoma | ||
| Moderate | 0/1 (0%) | 200 (n = 1) |
| Poor | 0/1 (0%) | 200 (n = 1) |
| Overall | 0/39 (0%) | 200 (n = 39) |
Heterogeneous cancers (i.e., having both cohesive and noncohesive components) were classified in the noncohesive group. The H-scores calculated from the heterogeneous tumors apply specifically to noncohesive foci.
All 39 cancers in the cohesive group had membranous E-cadherin expression. A median H-score of 200 (range, 50–300) was observed in the entire group of cohesive cancers as well as in the moderately (n = 25) and poorly differentiated (n = 14) subgroups. In contrast, 22 of 28 noncohesive cancers had complete E-cadherin loss in noncohesive foci (P < 0.001). All six undifferentiated cancers with osteoclast-like giant cells and 14 of 15 anaplastic carcinomas had complete absence of E-cadherin protein expression in the noncohesive regions of the tumor. The single anaplastic carcinoma with E-cadherin protein expression in a noncohesive area had a low H-score of 10. Two signet ring carcinomas also had complete loss of E-cadherin expression. Nineteen of the 22 noncohesive histologic sections with E-cadherin loss exhibited a good internal positive control, validating the immunolabeling reaction in these cases.
Representative histologic sections of cohesive and non-cohesive cancers stained with H&E appear in Fig. 1 along with corresponding sections labeled with an E-cadherin antibody. In Fig. 1A and B, a moderately differentiated pancreatic cancer shows a membranous E-cadherin labeling pattern, which is characteristic for cohesive lesions; in Fig. 1C and D, an anaplastic carcinoma of the pancreas reveals complete loss of E-cadherin protein expression.
Fig. 1.
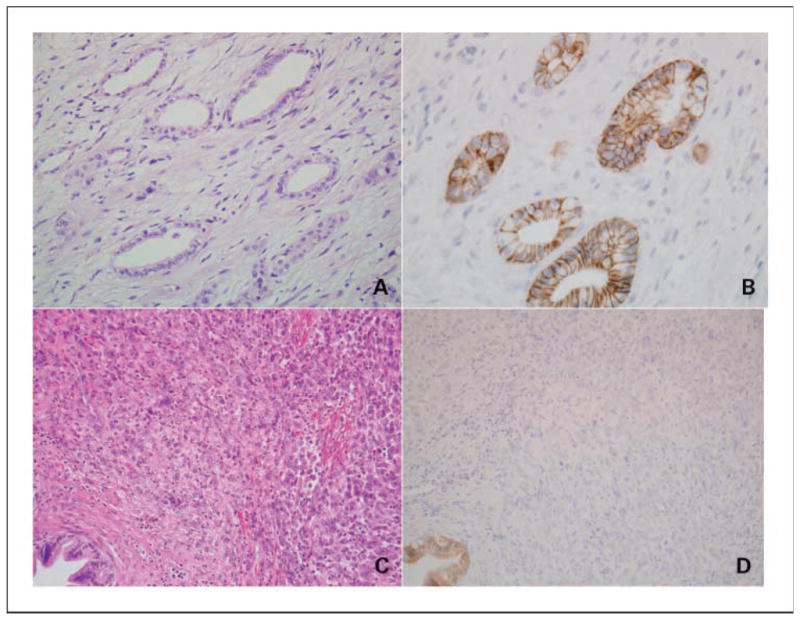
Representative histologic appearance (H&E) of a moderately differentiated adenocarcinoma (A) and an anaplastic carcinoma (C). The corresponding E-cadherin immunolabeling studies are provided in B and D, respectively. A normal pancreatic duct with membranous E-cadherin protein expression serves as an internal positive control for the noncohesive cancer (anaplastic carcinoma) shown in D.
Seven noncohesive pancreaticobiliary cancers with complete E-cadherin loss and 28 cohesive cancers with membranous E-cadherin expression were also immunolabeled for β-catenin. All 28 cancers in the cohesive group had membranous β-catenin labeling. The median H-score for these cases was 200 (range, 100–300). In contrast to the cohesive cancers, abnormal β-catenin expression was observed in each of the evaluated noncohesive cancers: five cancers had complete β-catenin loss in noncohesive foci and two cancers had a cytoplasmic distribution of β-catenin (as opposed to a membranous distribution observed in cohesive pancreatic cancers) in the areas of interest.
Representative tissues labeled with the β-catenin antibody are provided in Fig. 2. A typical membranous labeling pattern in a moderately differentiated cancer is presented in Fig. 2A, and an anaplastic carcinoma with cytoplasmic β-catenin expression appears in Fig. 2B. Figures 2C and D are separate regions from the same tumor; the moderately differentiated component in Fig. 2C has a membranous β-catenin distribution, whereas the anaplastic component in Fig. 2D shows a complete loss of β-catenin protein expression.
Fig. 2.
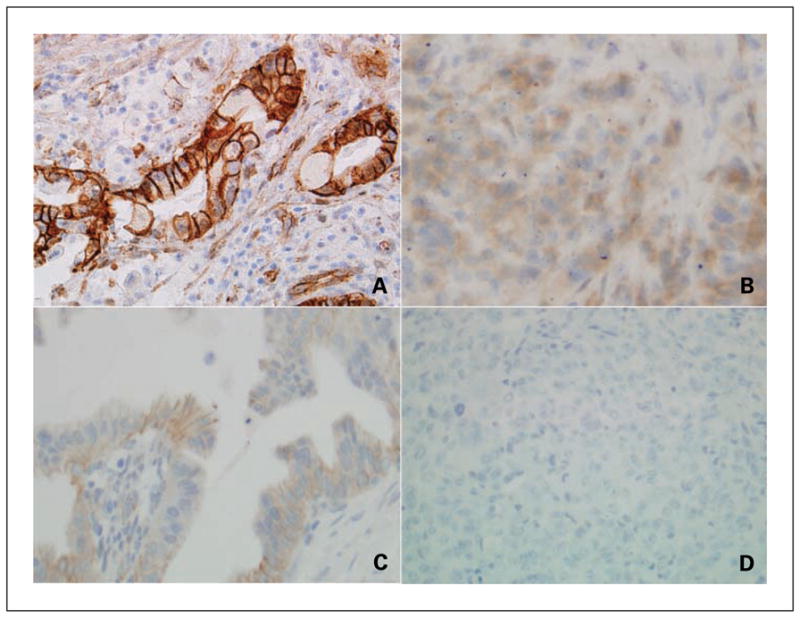
Representative β-catenin immunolabeling studies of a moderately differentiated adenocarcinoma (A) and an anaplastic carcinoma (B). β-Catenin protein expression in moderately differentiated (C) and anaplastic (D) components of a heterogeneous cancer is also provided.
Ten cancers in the present series, including the one described above in Fig. 2C and D, contained noncohesive and cohesive components within the same tumor. Cohesive foci in these tumors expressed E-cadherin at the plasma membrane (median H-score of 200), whereas noncohesive foci completely lost E-cadherin protein expression with just one exception; the non-cohesive area in the exceptional case showed weak E-cadherin labeling (H-score of 10), as previously noted in Table 1.
A subset of noncohesive pancreaticobiliary cancers with E-cadherin loss was tested with additional immunohistochemical markers to exclude alternative pathologic diagnoses. Three anaplastic carcinomas and one signet ring carcinoma having E-cadherin loss were tested for Melan-A expression (a melanosome-associated protein present in melanomas), and none of them were reactive. Four anaplastic carcinomas, one undifferentiated cancer with osteoclast-like giant cells, and one signet ring carcinoma with E-cadherin loss were tested for AE1/AE3 (a pancytokeratin marker present in epithelial-derived cancers). Although the undifferentiated carcinoma with osteoclast-like giant cells was unreactive, the remaining five noncohesive cancers were reactive with the pancytokeratin antibody. This finding suggests that E-cadherin inactivation in these cases was not only a function of a global loss of epithelial markers.
Genetic and epigenetic analyses
Somatic E-cadherin gene mutations occur commonly in lobular breast cancer and diffuse gastric cancer, but to our knowledge, none have been reported in pancreatic cancer. We entertained the possibility that the lack of reported mutations in pancreatic cancer could have only been a reflection of the rareness of resectable noncohesive pancreatic cancer. We therefore surveyed four noncohesive cancers from the present series for E-cadherin gene mutations. No mutations were identified.
We next explored an epigenetic alternative for E-cadherin gene inactivation using nested methylation-specific PCR to screen for abnormal methylation at the E-cadherin promoter. MiaPaca-2 is an undifferentiated pancreatic cancer cell line that has been noted to be deficient in E-cadherin mRNA and protein expression (10, 11). To our knowledge, the mechanism for E-cadherin loss in this cell line has not been elucidated. We also evaluated E-cadherin promoter methylation in two other pancreatic cancer cell lines derived from differentiated lesions: AsPC-1 and BxPC-3 (12, 13). Total methylation at the E-cadherin promoter was observed in the noncohesive pancreatic cancer, whereas the two cohesive cancers had no detectable methylation (Fig. 3A). The analysis was expanded to include frozen samples from primary tissue. Three heterogeneous pancreatic cancers were macrodissected to separate cohesive foci with membranous E-cadherin expression and noncohesive foci showing E-cadherin loss. A convincing pattern of methylation was not found in these samples (Fig. 3B).
Fig. 3.

Unmethylated (U)-specific (lanes1, 3, 5, 7, and 9) and methylation (M)-specific (lanes 2, 4, 6, 8, and10) PCR amplification of the CpG island in the E-cadherin promoter. Bisulfite-treated water and in vitro – methylated DNA served as negative and positive controls, respectively. A, MiaPaca-2 [undifferentiated (Undiff.) pancreatic cancer], AsPC-1 [differentiated (Diff.) pancreatic cancer], and BxPC-3 (differentiated pancreatic cancer). B, noncohesive (i.e., undifferentiated) and cohesive (i.e., differentiated) foci from three different pancreatic cancers were separated by macrodissecting frozen tumor according to the microscopic appearance of a histologic section from each frozen tumor sample. Neg., negative; Pos., positive.
Additional clinicopathologic details were available for nine of the patients with resected noncohesive pancreaticobiliary cancer and inactivated E-cadherin. The long-term survival of these patients was significantly less (P = 0.002) than the long-term survival of a reference group of patients with tubular (ductal) adenocarcinoma resected at our institution (Fig. 4A). In subgroup analyses (Fig. 4B), patients with E-cadherin loss had decreased survival compared with patients with well-differentiated (P < 0.001), moderately differentiated (P < 0.001), and poorly differentiated pancreatic cancer (P = 0.03).
Fig. 4.

A, Kaplan-Meier survival curves of 9 patients with noncohesive pancreaticobiliary cancers and inactivated E-cadherin (6 anaplastic carcinomas, 1 undifferentiated cancer with osteoclast-like giant cells, and 2 signet ring carcinomas) and 1,229 resected tubuloglandular ductal adenocarcinomas (i.e., cohesive pancreatic cancer; P = 0.002, log-rank test). B, the 1,229 cohesive pancreatic cancers are subgrouped according to histologic grade (well, moderately, and poorly differentiated) and compared with the 9 noncohesive cancers. The P values resulting from comparisons between the nine noncohesive cancers and the subgroups of differentiated pancreatic cancer are P < 0.001, P = 0.0001, and P = 0.03, respectively.
Discussion
Undifferentiated pancreatic cancers are rare variants of pancreatic adenocarcinoma and can be further subclassified as anaplastic, sarcomatoid, carcinosarcomatoid (i.e., includes both epithelial and mesenchymal features), or undifferentiated with osteoclast-like giant cells (14, 15). Anaplastic carcinoma is probably the most common of the different subtypes and is characterized by large, bizarre-appearing, pleomorphic cells with abundant eosinophilic cytoplasm. The cells are often multinucleated and lack a supporting stroma. Several reports suggest that anaplastic carcinoma is even more aggressive than tubuloglandular ductal adenocarcinoma (16, 17). The survival data from the present series agree with prior studies on this point. Undifferentiated pancreatic cancer with osteoclast-like giant cells also carries a grave prognosis and can be distinguished histologically from anaplastic carcinoma by an associated nonneoplastic population of large and multinucleated cells resembling osteoclasts (15, 18).
Undifferentiated pancreatic cancers are reported to constitute between 1% and 23% of pancreatic cancers (16, 17, 19–21), although a survey of the literature reveals that autopsy series generally report higher percentages (17, 20, 21) than surgical series. The experience at our institution mirrors these findings. Nearly 20% of advanced pancreatic cancers observed in a rapid autopsy series had a significant undifferentiated component.5 In contrast, <1% of surgically resected pancreatic cancers were undifferentiated (19). This difference suggests that pancreatic cancer dedifferentiation is an important factor in the transition from locoregional to advanced disease.
We hypothesized that E-cadherin inactivation may be involved in the process of dedifferentiation in pancreatic cancer based on its known function as a transmembrane protein within the zonula adherens (a protein complex that connects the actin cytoskeleton between cells) and its central role in the epithelial-mesenchymal transition model of tumor progression (22). The data from previous studies on the role of E-cadherin in pancreatic carcinogenesis have been inconsistent. A few groups have observed E-cadherin inactivation in poorly differentiated pancreatic cancers (23, 24), yet a separate study disputed this finding (25). Two studies observed E-cadherin expression in poorly differentiated cancers but noted that undifferentiated cancers had lost E-cadherin protein expression (26, 27).
In this study, the absence of E-cadherin expression distinguished undifferentiated pancreatic cancer from the more commonly observed ductal adenocarcinoma. Every case of differentiated ductal adenocarcinoma, including poorly differentiated cases, had membranous E-cadherin labeling. In contrast, undifferentiated foci in 20 of 21 cancers (14 anaplastic and 6 undifferentiated with osteoclast-like giant cells) completely lacked E-cadherin expression.
Although signet ring cancers of the pancreatobiliary tract are not an undifferentiated subtype, they also display an inability to form ductal structures. In some tumor systems, signet ring morphology has been linked with E-cadherin inactivation. This has been well described in breast and stomach cancer (28–33). Additionally, individuals with inherited mutations in the E-cadherin gene are at substantial risk for developing signet ring carcinomas of the breast and stomach (28, 34, 35). Indeed, we observed a total loss of E-cadherin expression in two signet ring cancers of the pancreatobiliary tract. Alternative mechanisms of signet cell change (unrelated to E-cadherin inactivation) have also been described, such as tissue hypoxia, and could account for the morphologic changes observed in five E-cadherin–positive signet ring cancers in this study (36, 37).
We sequenced the E-cadherin gene in noncohesive cancers with available high-template DNA (i.e., frozen tumor) in an attempt to elucidate the mechanism of E-cadherin loss in these tumors. We were unable to identify any alterations within the E-cadherin coding sequence, thereby ruling out truncating and missense mutations in these samples. This mutation survey, however, does not exclude the possibility of large in-frame or homozygous deletions involving the E-cadherin locus because contaminating wild-type DNA in our samples would have masked these findings.
Promoter hypermethylation was perhaps a more attractive mechanism for E-cadherin inactivation because loss of heterozygosity at 16q (the E-cadherin locus) is a rare event in pancreatic cancer (38). Furthermore, epigenetic events are the primary mechanism responsible for E-cadherin inactivation in virtually every tumor system, except breast and stomach cancer (39, 40). In this study, we attempted to separate noncohesive and cohesive foci from heterogeneous pancreatic cancers containing both histologic patterns by macrodissecting frozen tumors, but no consistent patterns of E-cadherin expression were observed in these samples. However, we observed complete methylation at the E-cadherin promoter in the undifferentiated pancreatic cancer cell line, MiaPaCa-2, in contrast to two differentiated pancreatic cancer cell lines. These data suggest that E-cadherin promoter hypermethylation may explain E-cadherin gene silencing in some undifferentiated cancers.
We also examined the role of the E-cadherin transcriptional repressor Snail (41) as a potential regulator of E-cadherin expression in noncohesive pancreatic cancers lacking E-cadherin protein expression. We did quantitative PCR to assay for Snail expression in both noncohesive and cohesive foci of heterogeneous pancreatic cancers. Again, we could not identify a consistent pattern linking Snail expression to E-cadherin gene silencing in the frozen tumors (data not shown). Cell lines may prove to be more appropriate for mechanistic studies because they are a renewable population of cells that can be manipulated on a molecular level and are free of any non-cancerous contaminants. We are currently in the process of developing cell lines from undifferentiated pancreatic cancers for this purpose.
Based on the observations from this study, and our understanding of the E-cadherin literature in solid tumors, we propose a theoretical tumor progression model (Fig. 5) that accounts for the different noncohesive histologic patterns (e.g., anaplastic carcinoma, undifferentiated cancer with osteoclast-like giant cells, or signet ring carcinoma). Our model is an adaptation of the classic tumor progression model for pancreatic cancer described by Hruban et al. (42) and factors in the timing of E-cadherin loss during the transition of normal ductal epithelium to invasive cancer.
Fig. 5.

Proposed model to explain the different noncohesive histologic phenotypes of pancreaticobiliary cancer based on the timing of E-cadherin inactivation during the pancreatic intraepithelialneoplasia (PanIN) adenocarcinoma sequence. OCGT, osteoclast-like giant cell tumor.
The E-cadherin literature supports the idea that early E-cadherin loss (before the late pancreatic intraepithelial neoplasia stage) could lead to the development of a signet ring carcinoma. E-cadherin inactivation has been observed, for instance, in in situ signet ring carcinomas of the breast and stomach (30, 32–34). Furthermore, individuals with germ-line mutations in the E-cadherin gene typically develop gastric cancers with signet ring morphology (34).
Pure undifferentiated pancreatic cancers (either undifferentiated cancer with osteoclast-like giant cells or anaplastic carcinomas) may lose E-cadherin expression at a later stage once the tumor has sustained a sufficient number of background genetic hits necessary for the high-grade appearance of these lesions. Most anaplastic carcinomas also contained a prominent differentiated (cohesive) component. In these mixed lesions, E-cadherin inactivation was likely a late event, occurring only after an invasive cancer has emerged. Thus, a subpopulation of cancer cells lacks functional E-cadherin, resulting in a heterogeneous cancer with both cohesive and noncohesive foci.
Additional studies of noncohesive pancreatobiliary cancers are needed to better understand the mechanisms of E-cadherin gene silencing as well as the downstream molecular sequelae of inactivated E-cadherin. These pursuits may one day yield novel strategies to treat pancreatic cancer.
Acknowledgments
Grant support: NIH Specialized Program of Research Excellence grant CA 62924 and training grantT32-DK007713.
Footnotes
C.A. Iacobuzio-Donahue et al., in preparation.
References
- 1.Embuscado EE, Laheru D, Ricci F, et al. Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther. 2005;4:548–54. doi: 10.4161/cbt.4.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21. 1. Science. 1996;271:350–3. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 3.Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]
- 4.Xin W, Yun KJ, Ricci F, et al. MAP2K4/MKK4 expression in pancreatic cancer: genetic validation of immunohistochemistry and relationship to disease course. Clin Cancer Res. 2004;10:8516–20. doi: 10.1158/1078-0432.CCR-04-0885. [DOI] [PubMed] [Google Scholar]
- 5.Bortolasi L, Burgart LJ, Tsiotos GG, Luque-De Leon E, Sarr MG. Adenocarcinoma of the distal bile duct. A clinicopathologic outcome analysis after curative resection. Dig Surg. 2000;17:36–41. doi: 10.1159/000018798. [DOI] [PubMed] [Google Scholar]
- 6.Rijken AM, Hu J, Perlman EJ, et al. Genomic alterations in distal bile duct carcinoma by comparative genomic hybridization and karyotype analysis. Genes Chromosomes Cancer. 1999;26:185–91. [PubMed] [Google Scholar]
- 7.Zerbi A, Balzano G, Leone BE, Angeli E, Veronesi P, Di Carlo V. Clinical presentation, diagnosis and survival of resected distal bile duct cancer. Dig Surg. 1998;15:410–6. doi: 10.1159/000018654. [DOI] [PubMed] [Google Scholar]
- 8.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of Cp Gislands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–9. [PubMed] [Google Scholar]
- 10.Caca K, Kolligs FT, Ji X, et al. β- and g-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ. 1999;10:369–76. [PubMed] [Google Scholar]
- 11.Frixen UH, Behrens J, Sachs M, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173–85. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen WH, Horoszewicz JS, Leong SS, et al. Human pancreatic adenocarcinoma: in vitro and in vivo morphology of a new tumor line established from ascites. In Vitro. 1982;18:24–34. doi: 10.1007/BF02796382. [DOI] [PubMed] [Google Scholar]
- 13.Tan MH, Nowak NJ, Loor R, et al. Characterization of a new primary human pancreatic tumor line. Cancer Invest. 1986;4:15–23. doi: 10.3109/07357908609039823. [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Klimstra DS, Pitman MB. Tumors of the pancreas. 4. Washington (DC): Armed Forces Institute of Pathology; 2006. [Google Scholar]
- 15.Peters K, Kloppel G. Undifferentiated pancreatic carcinomas. Leap into chaos. Pathologe. 2005;26:18–21. doi: 10.1007/s00292-004-0731-4. [DOI] [PubMed] [Google Scholar]
- 16.Paal E, Thompson LD, Frommelt RA, Przygodzki RM, Heffess CS. A clinicopathologic and immunohistochemical study of 35 anaplastic carcinomas of the pancreas with a review of the literature. Ann Diagn Pathol. 2001;5:129–40. doi: 10.1053/adpa.2001.25404. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Baithun SI. Morphological study of 391 cases of exocrine pancreatic tumours with special reference to the classification of exocrine pancreatic carcinoma. J Pathol. 1985;146:17–29. doi: 10.1002/path.1711460103. [DOI] [PubMed] [Google Scholar]
- 18.Wilentz RE, Hruban RH. Pathology of cancer of the pancreas. Surg Oncol Clin N Am. 1998;7:43–65. [PubMed] [Google Scholar]
- 19.Winter JM, Cameron JL, Campbell KC, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single institution experience. J Gastrointest Surg. 2006;10:1199–211. doi: 10.1016/j.gassur.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 20.Ogutu EO. The pattern of pancreatic carcinoma at Kenyatta National Hospital. East Afr Med J. 1989;66:105–8. [PubMed] [Google Scholar]
- 21.Tschang TP, Garza-Garza R, Kissane JM. Pleomorphic carcinoma of the pancreas: an analysis of 15 cases. Cancer. 1977;39:2114–26. doi: 10.1002/1097-0142(197705)39:5<2114::aid-cncr2820390528>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 22.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Karayiannakis AJ, Syrigos KN, Chatzigianni E, et al. Aberrant E-cadherin expression associated with loss of differentiation and advanced stage in human pancreatic cancer. Anticancer Res. 1998;18:4177–80. [PubMed] [Google Scholar]
- 24.Pignatelli M, Ansari TW, Gunter P, et al. Loss of membranous E-cadherin expression in pancreatic cancer: correlation with lymph node metastasis, high grade, and advanced stage. J Pathol. 1994;174:243–8. doi: 10.1002/path.1711740403. [DOI] [PubMed] [Google Scholar]
- 25.Weinel RJ, Neumann K, Kisker O, Rosendahl A. Expression and potential role of E-cadherin in pancreatic carcinoma. Int J Pancreatol. 1996;19:25–30. doi: 10.1007/BF02788372. [DOI] [PubMed] [Google Scholar]
- 26.Sedivy R, Peters K, Kloppel G. Osteopontin expression in ductal adenocarcinomas and undifferentiated carcinomas of the pancreas. Virchows Arch. 2005;446:41–5. doi: 10.1007/s00428-004-1142-x. [DOI] [PubMed] [Google Scholar]
- 27.Yonemasu H, Takashima M, Nishiyama KI, et al. Phenotypical characteristics of undifferentiated carcinoma of the pancreas: a comparison with pancreatic ductal adenocarcinoma and relevance of E-cadherin, a catenin and β catenin expression. Oncol Rep. 2001;8:745–52. doi: 10.3892/or.8.4.745. [DOI] [PubMed] [Google Scholar]
- 28.Chan JK, Wong CS. Loss of E-cadherin is the fundamental defect in diffuse-type gastric carcinoma and infiltrating lobular carcinoma of the breast. Adv Anat Pathol. 2001;8:165–72. doi: 10.1097/00125480-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 29.De Leeuw WJ, Berx G, Vos CB, et al. Simultaneous loss of E-cadherin and catenins in invasive lobular breast cancer and lobular carcinoma in situ. J Pathol. 1997;183:404–11. doi: 10.1002/(SICI)1096-9896(199712)183:4<404::AID-PATH1148>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Fuchs CS, Mayer RJ. Gastric carcinoma. N Engl J Med. 1995;333:32–41. doi: 10.1056/NEJM199507063330107. [DOI] [PubMed] [Google Scholar]
- 31.Merino MJ, Livolsi VA. Signet ring carcinoma of the female breast: a clinicopathologic analysis of 24 cases. Cancer. 1981;48:1830–7. doi: 10.1002/1097-0142(19811015)48:8<1830::aid-cncr2820480821>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Muta H, Noguchi M, Kanai Y, Ochiai A, Nawata H, Hirohashi S. E-cadherin gene mutations in signet ring cell carcinoma of the stomach. Jpn J Cancer Res. 1996;87:843–8. doi: 10.1111/j.1349-7006.1996.tb02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vos CB, Cleton-Jansen AM, Berx G, et al. E-cadherin inactivation in lobular carcinoma in situ of the breast: an early event in tumorigenesis. Br J Cancer. 1997;76:1131–3. doi: 10.1038/bjc.1997.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huntsman DG, Carneiro F, Lewis FR, et al. Early gastric cancer in young, asymptomatic carriers of germ-line E-cadherin mutations. N Engl J Med. 2001;344:1904–9. doi: 10.1056/NEJM200106213442504. [DOI] [PubMed] [Google Scholar]
- 35.Keller G, Vogelsang H, Becker I, et al. Diffuse type gastric and lobular breast carcinoma in a familial gastric cancer patient with an E-cadherin germline mutation. Am J Pathol. 1999;155:337–42. doi: 10.1016/S0002-9440(10)65129-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimet S, Lazure T, Bedossa P. Signet-ring cell change in acute erosive gastropathy. Am J Surg Pathol. 2004;28:1111–2. doi: 10.1097/01.pas.0000126638.73795.1a. [DOI] [PubMed] [Google Scholar]
- 37.Wang K, Weinrach D, Lal A, et al. Signet-ring cell change versus signet-ring cell carcinoma: a comparative analysis. Am J Surg Pathol. 2003;27:1429–33. doi: 10.1097/00000478-200311000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004;64:871–5. doi: 10.1158/0008-5472.can-03-2756. [DOI] [PubMed] [Google Scholar]
- 39.Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12:226–37. doi: 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 40.Strathdee G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol. 2002;12:373–9. doi: 10.1016/s1044-579x(02)00057-3. [DOI] [PubMed] [Google Scholar]
- 41.Cano A, Perez-Moreno MA, Rodrigo I, et al. Thetranscription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 42.Hruban RH, Wilentz RE, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol. 2000;156:1821–5. doi: 10.1016/S0002-9440(10)65054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]