

This review was developed by the Mitochondrial Liver Diseases Working Group of the Childhood Liver Disease Research and Education Network (ChiLDREN), supported by the National Institute of Digestive, Diabetes and Kidney Diseases, NIH, to guide evaluation of children with suspected mitochondrial liver disease. Data informing the evaluation guideline was supported by Medline searches of published English language literature and expert opinion from a committee of pediatric hepatologists and a mitochondrial metabolism specialist.
Mitochondrial respiratory chain defects can affect any tissue, with the most energy-dependent organs being most vulnerable.(1) In general, clinical manifestations include multisystem involvement such as brain, muscle, heart, or kidney, with acute or chronic liver dysfunction, sometimes in the presence of lactic acidosis, a biomarker of limited sensitivity.(2, 3) Heterogeneous clinical presentations can be explained by the fact that the mitochondrial quantity and function are uniquely influenced by both nuclear and mitochondria DNA (mtDNA) or by the fact that cells in various tissues can contain different mixtures of normal and abnormal mitochondrial genomes (heteroplasmy). Most mitochondrial proteins and enzymes are coded by nuclear genes with Mendelian inheritance, while some respiratory chain subunits, ribosomal and transfer RNAs are encoded by mitochondrial genes that are maternally inherited.(4) Mutations, deletions, or duplications in either of these classes can cause disease, and mutations in nuclear genes that control mitochondrial DNA replication, transcription, and translation may lead to mtDNA depletion syndrome or to a translational disorder.(5-7)
The respiratory chain, consisting of 5 multimeric complexes (I-V) in the mitochondrial inner membrane, generates energy as ATP via electron transport and oxidative phosphorylation (Figure 1). Defects in the respiratory chain enzymes or mitochondrial membrane transport proteins result in injury to energy-dependent organs, especially brain, retina, muscle, heart, and liver.(8) In addition, hepatic mitochondria oxidize fatty acids forming ketone bodies, an important source of energy for the brain in the fasting state. Fatty acid oxidation defects, an important group of primary bioenergetic defects, can present similarly with hepatopathy or encephalopathy, often with nonketotic hypoglycemia, acidosis and hyperammonemia, and are thus included in the differential diagnosis and should be simultaneously evaluated.(9)
Figure 1.

The respiratory chain, consisting of 5 multimeric complexes (I-V) in the mitochondrial inner membrane, generates energy as ATP via electron transport and oxidative phosphorylation.
Establishing the diagnosis of primary mitochondrial bioenergetic defects in patients with liver disease requires a high index of suspicion in specific clinical scenarios. A tiered diagnostic evaluation is useful (Table 1). Although mitochondrial hepatopathies are a heterogeneous group of disorders, there are several general laboratory investigations in blood and urine which can reveal an altered redox status suggestive of respiratory-chain defects (lactate:pyruvate molar ratios and ketone body ratios). Specific laboratory tests are considered in those with unique clinical presentations as well, and either tissue analysis or genotyping is used to identify the etiology. Other typically involved organ systems should be evaluated when mitochondrial hepatopathy is suspected (Table 2). Several important management issues should be addressed during this evaluation process. These guidelines outline the evaluation of the infant or child with suspected mitochondrial hepatopathy. Two summary tables (Table 3 and 4) describing each genetic etiology follow; reference clinical laboratories for the genetic tests can be found on www.genetests.org
Table 1.
Tiered Approach to Evaluation of Suspected Mitochondrial Disease
| Tier 1 | Early Screening |
| Comprehensive metabolic profile, INR, alpha fetoprotein, CPK (creatine phosphokinase), phosphorus, complete blood count, and ammonia. |
|
| Lactate/pyruvate, ideally obtained 1 hour after feeding (normal molar ratio is <20, normal postprandial lactate <2.8 mM). |
|
| Serum ketone bodies: both quantitative 3-hydroxybutyrate and quantitative acetoacetate (3-hydroxybutyrate/acetoacetate ratio <4 is normal) and total free fatty acids to calculate ketone bodies: free fatty acid ratio.(13) |
|
| Serum acylcarnitine profile; serum free and total carnitines. | |
| Urine organic acids (look for elevated lactate, succinate, fumarate, malate, 3- methyl-glutaconic or 2-hydroxyglutaric, 2-ketoglutaric, methylmalonic acid) |
|
| Serum amino acids. (Look for elevation of alanine (abnormal > 500 μM, but more specific if > 600 μM). |
|
| Consider: | Quantitative 3-methylglutaconic acid (serum or urine).(14) |
| Urine acylglycines and 2-ethylmalonic quantification (if multiple acyl-CoA dehydrogenase deficiency is suspected). |
|
| Thymidine (plasma) (especially in cases with coexistent intestinal dysmotility concerning for MNGIE syndrome). |
|
| Coenzyme-Q levels in leukocytes, not serum (for CoQ deficiency; leukocyte levels correlate better with tissue coenzyme Q levels, whereas serum levels reflect nutritional status). |
|
| Quantitative serum methylmalonic acid (elevated in SUCLA and SUCLG1 deficiencies). |
|
| CSF analysis: Lactate and pyruvate (if blood lactate is normal but evidence of CNS involvement), amino acids (especially elevated CSF alanine), and protein concentration (note CSF protein can be elevated in POLG1 disease early on, even when lactate is normal). |
|
| Tier 2 | Genotyping for More Common Genes |
| Most common with liver involvement: POLG1 (15, 16), DGUOK (17-19), MPV17 (20, 21) , SUCLG1 (22), C10ORF2/Twinkle (23), TRMU (see Tables 3 and 4). |
|
| If neuromuscular features suggest MELAS (Mitochondrial Myopathy Encephalopathy, Lactic Acidosis and Stroke-like Episodes) or pancreatic insufficiency suggests Pearson’s marrow/pancreas syndrome: mitochondrial DNA point mutations/deletions.(24) |
|
| If methylmalonic acid is elevated: SUCLG1.(22) If acylcarnitines and/or urine organic acids suggest specific fatty acid oxidation (FAO) defects: Genotyping for Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency (LCHAD) (25), Carnitine Palmitoyl Transferase Deficiency (CPTI & II deficiency) (26, 27), or Multiple Acyl-CoA Dehydrogenase Deficiency (MADD = Glutaric acidemia II = ETF & ETF-DH deficiency) (28), SLC25A20 for carnitine– acylcarnitine translocase (CACT deficiency).(29) |
|
| For recurrent acute liver failure: TRMU, ACAD9, CPTI, SUCLGI.(22, 26, 30, 31) Identification of TRMU mutations is urgent in infants with acute liver failure since these patients frequently recover without the need for transplantation.(31, 32) |
|
|
Note: Next Generation Sequencing (e.g., exome sequencing) will allow for simultaneous evaluation of panels of all nuclear genes encoding mitochondrial proteins and all mitochondrial DNA genes at considerably lower cost in the future, encompassing the above gene tests, and will eventually replace single gene sequencing. It is already available in some countries. |
|
| Tier 3 | Tissue Evaluation |
| Liver biopsy: 1. Light microscopy, electron microscopy (place specimen in glutaraldehyde); consider oil red O stain for fat on frozen section; consider iron stain if DGUOK or BCS1L are suspected (Figure 2). 2. Frozen tissue for respiratory chain enzyme activity analysis. 3. Frozen tissue for DNA quantification (mitochondrial DNA depletion analysis). 4. Consider storing frozen tissue for future studies if amount adequate. Consider blue native PAGE gel analysis with in-gel activity staining (Figure 3). |
|
| Skin biopsy for fibroblast culture. Can be used for fatty acid oxidation enzyme activity, respiratory chain enzyme activities, blue native PAGE (BNP) testing.(33) FAO probe studies (especially if carnitine profile is abnormal), or high resolution respirometry.(34) Note: Because of heteroplasmy of mitochondrial genes or due to differential tissue expression of nuclear genes, abnormalities in patients with mitochondrial hepatopathy can sometimes only be confirmed in liver tissue. |
|
| Muscle biopsy, especially if muscle involvement is present: light and electron microscopy. Consider histochemistry for respiratory chain complexes, respiratory chain enzyme assays, blue native PAGE (BNP) analysis, mtDNA depletion analysis, mtDNA whole genome sequencing and/or deletion analysis. |
|
| Tier 4 | Further Molecular and Biochemical Evaluation |
| Additional genes to consider: TRMU (31) BCS1L (35), SCO1 (36), TSFM (37), TWINKLE (5, 23), ACAD9 (30) (the latter especially if episodes of liver failure and fatty acid oxidation defect) (see Tables 3 and 4). Not all of these tests may be clinically available. A microarray is available to evaluate for large deletions or duplications in nuclear or mitochondrial genes.(38) Note: Next Generation Sequencing (e.g., exome sequencing) will allow for simultaneous evaluation of panels of all nuclear genes encoding mitochondrial proteins and many or all mitochondrial DNA genes at considerably lower cost in the future, and will eventually replace single gene sequencing. At present, its efficacy is highest in clinically and biochemically well characterized patients. |
|
| Targeted molecular analyses based on the results of tissue-based respiratory chain enzyme assays and primary liver disease as presentation: 1. Isolated complex I deficiency: ACAD9 (30) 2. Isolated complex III deficiency: BCS1L (35) 3. Isolated complex IV deficiency: SCO1 (36) 4. Combined complex I, III, and IV deficiency with incompletely assembled complex V bands on blue native PAGE: (This signifies a generic defect in the processing of mtDNA encoded subunits.(39) It can be caused by a deficiency of mtDNA (mtDNA depletion syndromes) or by a defect in transcription or translation. A) If mtDNA content is <5% of normal in liver: mtDNA depletion syndrome: POLG1, POLG2, TWINKLE, DGUOK, and TYMP.(40) B) With normal or mildly decreased mtDNA but with multiple deletions (assay with long range PCR or with NextGen mtDNA analysis): Same as above, but more often with POLG or TWINKLE or TYMP. C) With normal or (more common) elevated mtDNA: translation defects such as EF-Tu, EGF-1, TSFM, TRMU, FARS2 (and other tRNA synthase genes, tRNA modification genes, ribosomal genes, translation initiation, elongation and termination factors).(31, 37, 41) Consider exome or mito exome sequencing for the many genes associated with the translational machinery. D) Deficiency of combined complex II-III assay but normal isolated assay II and normal III indicates a likely coenzyme Q deficiency. Obtain coenzyme Q levels, and review for causes of coenzyme Q deficiency.(42) |
Table 2.
Evaluation for Disease in Other Organ Systems
| Organ System | Testing |
|---|---|
| Brain | Neurologic exam, MR/MR spectroscopy, EEG, CSF |
| Heart | Echocardiogram, EKG |
| Muscle | Muscle biopsy |
| Eyes | Detailed ophthalmologic exam |
| Kidney | Electrolytes, serum and urine phosphorus and creatinine, urine amino acids, urinalysis, urine protein |
| Endocrine | HbA1c, morning cortisol |
| Pancreas (exocrine) | Fecal pancreatic elastase, endoscopic pancreatic stimulation test |
| Hearing | Hearing testing |
Table 3.
Genetic Etiologies of Mitochondrial Hepatopathies Presenting in Neonates or Infants
| Mutation/ Syndrome |
Defect | Onset/Age at Presentation |
Hepatic Presentation | Neurological Features | Other Features | Diagnostic Tests |
|---|---|---|---|---|---|---|
| DGUOK (17-19) | mtDNA depletion Complex I, III, IV |
Acute/early neonatal |
Neonatal liver failure, progressive, cholestasis, mimic neonatal hemochromatosis, hepatocellular carcinoma risk, can have isolated liver disease |
Hypotonia, developmental regression, nystagmus |
Lactic acidosis, hypoglycemia |
DGUOK sequence analysis |
| POLG (15, 16) | mtDNA depletion Complex I, III, IV |
Acute/neonatal | Neonatal liver failure, micro- or macro-vesicular steatosis, cirrhosis |
Encephalopathy, seizures,, myopathy, neuropathy, blindness, developmental regression |
Vomiting, GERD |
POLG sequence analysis or panel |
|
MPV17/Navajo neuro-hepatopathy (20, 21) |
mtDNA depletion Complex I, III, IV |
Acute/neonatal | Isolated neonatal/infant liver failure or in multisystem syndrome |
Sensorimotor neuropathy, progressive CNS white matter lesions |
Acidosis, FTT, corneal anesthesia,/abrasions, acral mutilation |
MPV 17 sequence analysis or panel |
|
TWINKLE(PEO1) (C10ORF2) (5, 23) |
mtDNA depletion DNA helicase |
Acute/neonatal | Neonatal liver failure, cirrhosis, elevated liver enzymes |
Encephalopathy (athetosis, ataxia, seizures), sensory neuropathy, deafness |
C10ORF2 TWINKLE sequence analysis |
|
| TRMU (31, 32) | Decreased mito translation (tRNA modifying enzyme) Complex I, III, IV |
Acute/neonatal | Neonatal liver failure, some recover, possibly with cirrhosis |
TRMU sequence analysis |
||
|
TSFM, EFG1, EF- Tu, MRPS16 (37) |
Decreased mito translation (elongation) |
Acute/neonatal | Liver dysfunction in infancy, hepatomegaly |
Hypotonia, dystonia | Hypertrophic cardiomyopathy, tubulopathy |
Sequencing individual genes e.g., TSFM or exome sequencing |
| SUCLG1 (22) | mtDNA depletion abnormal succinate synthesis Complex I, III, IV |
Acute/neonatal | Neonatal liver failure, episodic liver failure |
Hypotonia/myopathy (progressive), hearing loss |
Acidosis, elevated methylmalonic acid |
SUCLG1 sequence analysis |
| BCS1L (35) | Complex III assembly deficiency |
Acute/neonatal | Neonatal liver failure, cholestasis, hepatic iron overload |
Growth failure, amino- aciduria, lactic acidosis, early death (GRACILE syndrome) |
BCS1L sequence analysis |
|
| SCO1 (36) | Complex IV deficiency |
Acute/neonatal | Neonatal liver failure, hepatomegaly |
Hypotonia | Acidosis | SCO1 sequence analysis |
| FARS2 (41) | Phenylalanyl tRNA synthetase |
Acute/neonatal | Neonatal liver failure (Alper’s like) |
Intractable seizures, encephalopathy |
FARS2 sequence analysis on Nexgen |
|
| SLC25A20 (29) | Carnitine acylcarnitine translocase deficiency CACT deficiency (carnitine, FAO) |
Acute/neonatal | Neonatal liver failure, steatohepatitis |
Myopathy | Hypoglycemia, cardiomyopathy |
SLC25A20 sequence analysis |
| HADHA/ LCHAD or tri-functional protein deficiency (25) |
FAO defect | Acute | Hepatomegaly, fatty liver, elevated LFTs, cholestasis, liver failure, AFLP |
Encephalopathy, peripheral neuropathy |
Acidosis, hypoglycemia, pigmentary retinopathy |
HADHA sequence analysis |
| CPTI deficiency (26) |
Carnitine cycle FAO defect |
Acute/infancy | Hepatomegaly, liver failure episodes |
Reye’s-like episodes of encephalopathy |
Hypoketotic hypoglycemia |
CPT IA sequence analysis |
| CPT II deficiency (26, 27) |
Carnitine cycle FAO defect |
Acute/severe neonatal |
Liver failure in infancy | Seizures | Hypoketotic hypoglycemia, cardiomyopathy, myopathy |
CPT II sequence analysis |
Table 4.
Genetic Etiologies of Mitochondrial Hepatopathies Presenting as Chronic Liver Disease or With Later Onset
| Mutation/ Syndrome |
Defect | Onset/Age at Presentation |
Hepatic Presentation |
Neurological Features |
Other Features | Diagnostic Tests |
|---|---|---|---|---|---|---|
| DGUOK (18) | mtDNA depletion Complex I, III, IV |
Late presentation |
Progressive cholestasis, may have iron overload, HCC risk |
Hypotonia, developmental regression, nystagmus |
Lactic acidosis |
DGUOK sequence analysis |
|
MPV17/Navajo neurohepatopathy (21) |
mtDNA depletion Complex I, III, IV |
Chronic/ neonatal to childhood |
Progressive liver disease or in multisystem syndrome |
Sensorimotor neuropathy, progressive CNS white matter lesions |
Acidosis, FTT, corneal anesthesia, abrasions, acral mutilation |
MPV 17 sequence analysis |
|
POLG/Alper’s Disease (15, 16) |
mtDNA depletion Complex I, III, IV Deficiency |
Sub-acute: toddlers, young adults |
Later onset liver failure, macro- or micro-vesicular steatosis, cirrhosis |
Intractable seizures and developmental regression, blindness, neuropathy, ataxia |
Vomiting, GERD |
POLG sequence analysis |
|
TWINKLE (C10ORF2) (23) |
mtDNA depletion DNA helicase |
Chronic | Neonatal liver failure, cirrhosis, elevated liver enzymes |
Encephalopathy (athetosis, ataxia, seizures), sensory neuropathy, deafness |
C10ORF2 TWINKLE sequence analysis |
|
| TRMU (31) | Decreased mito translation (tRNA modifying enzyme) |
Chronic | Chronic liver disease/cirrhosis, recurrent acute liver failure |
TRMU sequence analysis |
||
| SUCLG (22) | mtDNA depletion abnormal succinate synthesis (ATPgen) |
Chronic or episodic |
Episodes liver failure |
Hypotonia/myopath y (progressive), hearing loss |
Acidosis |
SUCLG1 sequence analysis |
| ACAD9 (30) | FAO defect Complex I assembly |
Episodic | Episodic liver failure |
ACAD9 sequencing and deletion analysis |
||
|
TYMP/MNGIES (40) |
Thymidine phosphorylase (TP) deficiency, Complex IV |
Chronic | Liver dysfunction, macrovesicular steatosis, cirrhosis |
Leuko- encephalopathy, ophthalmoplegia, ptosis, peripheral neuropathy, hearing loss |
Pseudo-obstruction, GERD |
Plasma thymidine and deoxyuridine levels, TP enzyme activity, TYMP sequencing |
| Villous atrophy syndrome (50) |
Complex III defect |
Chronic/early childhood |
Hepatomegaly, raised liver enzymes, steatosis |
Cerebellar ataxia, sensorineural deafness, seizures |
Vomiting, anorexia, chronic diarrhea (resolved later in life), villous atrophy, lactic acidosis, DM |
|
| Pearson’s syndrome (24) |
mtDNA deletion Complex I, III, VI |
Chronic/ infancy |
Cirrhosis with hepatomegaly, cholestasis, raised liver enzymes, progressive liver failure, death in early childhood |
Refractory sideroblastic anemia, vacuolization of marrow precursors, variable neutropenia and thrombo- cytopenia, lactic acidosis, pancreatic insufficiency |
Mt DNA common mutations and deletions screening |
|
| HADHA/LCHA D or trifunctional protein deficiency (25) |
FAO defect | Chronic, infancy or later |
Hepatomegaly, fatty liver, elevated LFTs, cholestasis, liver failure, acute fatty liver of pregnancy |
Encephalopathy, peripheral neuropathy |
Acidosis, hypoglycemia, pigmentary retinopathy |
HADHA sequence analysis |
| CPT II/ CPT2 deficiency (27) |
Carnitine cycle FAO defect |
Infantile cardio- hepatopathy, adult myopathy |
Liver failure episodes |
Seizures | Hypoketotic hypoglycemia, cardiomyopathy, myopathy, rhabdomyolysis |
CPT II sequence analysis |
| MADD (glutaric acidemia II) (28) |
FAO defect Complex II, III |
Chronic, infancy up to adulthood |
Hepatomegaly, steatosis |
Neurologic symptoms, myopathy |
Hypoglycemia, acidosis |
Sequence of ETF A, B, or ETF-DH |
I. Clinical Scenarios Suggesting Possible Mitochondrial Liver Disease
Mitochondrial liver disease can present acutely in a child with no history of hepatic dysfunction, or with chronic liver and CNS disease. Fulminant or acute liver failure is one important presentation of mitochondrial disease.(10) Especially in a young child or in one with pre-existing or disproportionate central nervous system (CNS) involvement, mitochondrial disease is in the differential diagnosis of acute liver failure. Importantly, if liver transplant is being considered, careful attention must be paid to potential extrahepatic manifestations of mitochondrial dysfunction. Another clinical presentation is chronic liver disease, manifested by elevated aminotransferases, hepatomegaly, cholestasis, cirrhosis and especially steatohepatitis; these may be accompanied by other indicators of mitochondrial disease including hypoglycemia or lactic acidosis. Thirdly, liver disease accompanied by chronic neuromuscular disease or disease in other organ systems may be a sign of mitochondrial disease.
II. Tiered Diagnostic Evaluation
A wide array of tests which are useful in establishing the diagnosis of mitochondrial hepatopathies is available. These tests range from simple, inexpensive, easily available screening tests to very expensive widely ranging genetic studies. In the child who is suspected of having a mitochondrial disease, a tiered approach to diagnostic testing is recommended. Early screening tests (Tier 1) might provide clues to abnormalities in energy metabolism and results of these tests might guide subsequent confirmatory testing to establish a molecular diagnosis. Genotyping is available clinically for the more common mitochondrial diseases (Tier 2); the clinical scenario or results of screening tests can inform the choice of genetic tests. For example, a panel screening for specific gene mutations in DGUOK, POLG1, and MPV17 responsible for infantile liver failure with lactic acidosis and mitochondrial DNA depletion is readily available and might be useful early in evaluation (see Table 3). The diagnostic role of Next-Generation Sequencing (NGS), which is now allowing sequencing of more than 100 genes involved in mitochondrial diseases with a single blood test and at relatively low cost, (11) or even whole exome or genome sequencing, will become increasingly important and will eventually replace genotyping single genes or small panels of genes in Tier 2; however, the identification of multiple gene variants of uncertain significance will require detailed clinical and biochemical confirmation for interpretation. Tissue may also be needed to make a specific biochemical diagnosis, particularly if the liver is the major or sole affected organ (tier 3; Figure 2). Occasionally, diagnostic findings will only be revealed in liver tissue rather than in blood, muscle, or skin fibroblasts. When further clarification is needed, genotyping for less common disease-causing genes may be required (Tier 4); however, the use of NGS earlier on in the evaluation process in the future (in Tier 2) may obviate the need for this step in the evaluation paradigm. At present, the diagnostic yield of NGS of all mitochondrial genes is high in patients with well characterized mitochondrial disease, in particular with biochemical evidence of mitochondrial enzymatic dysfunction (11), but is low in patients with only a clinical suspicion.(12) Biochemical studies evaluating the structure and function of mitochondrial subunits in the affected tissue can be performed as needed, specifically to determine if new genetic variants have a functional effect (Figure 3). Thus, a combination of biochemical and molecular studies may be needed to confirm the pathologic nature of new gene variants to be described in the future. Table 1 below outlines a tiered approach to diagnostic evaluation.
Figure 2.
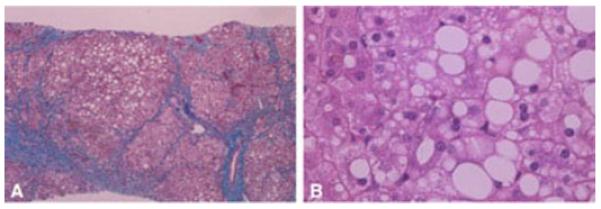
A) Micronodular cirrhosis, trichome stain, in a child with mutations in POLG1. B) Micro- and macrovesicular steatosis, hematoxylin and eosin stain.
Figure 3.
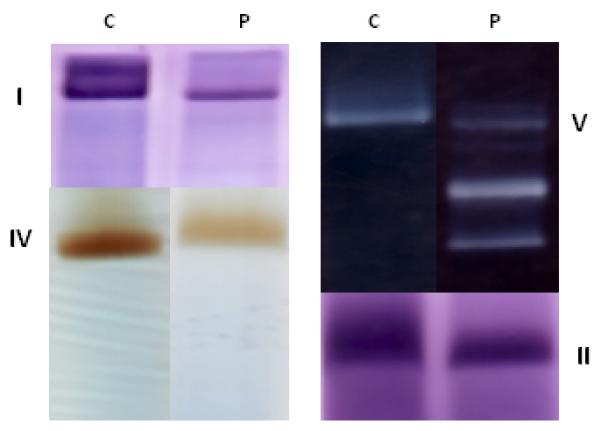
A blue native PAGE gel analysis with in gel activity staining of the respiratory chain components in the liver of a patient with DGUOK mtDNA depletion disease. C = control. P= patient. I, II, IV, V are the respective mitochondrial complexes. The added bands of lower molecular weight for complex V in the patient reflect incomplete assembly of complex V due to missing mtDNA encoded subunits. The intensities of the bands for complex I and complex IV are also reduced in the patient. This profile is seen in generalized defects of the synthesis of mitochondrial DNA encoded subunits such as in mtDNA depletion, or in defects of transcription or translation of mtDNA encoded subunits.
The following Tables 3 and 4 catalog known mitochondrial hepatopathies and briefly describe the mutation, defect, clinical description, and diagnostic testing. The typical hepatic presentation, ranging from hepatic failure to cholestasis to steatohepatitis to cirrhosis is briefly outlined; neurologic symptoms and other systems involved are briefly reviewed and references are provided. The disorders are separated into those with a neonatal or infantile presentation (Table 3) and those with later or more chronic onset (Table 4). There is, however, overlap between these two, and new diseases and presentations are recognized frequently.
III. Evaluation for Disease in Other Organ Systems
As part of the evaluation for mitochondrial hepatopathies, a systematic approach also needs to be instituted to search for involvement of other affected organ systems (Table 2). This takes on particular significance when liver failure occurs in a child with suspected mitochondrial disease, as the decision to consider liver transplantation is especially challenging. Because mitochondrial disease usually involves multiple organ systems and is generally progressive in other organs even following liver transplantation, there are many uncertainties regarding liver transplantation. Possible post-transplant appearance of new progressive symptoms in organs uninvolved prior to liver transplantation also needs to be considered.(43-46). Establishing criteria for liver transplantation in mitochondrial hepatopathies is beyond the scope of this report. However, in the evaluation for transplantation, meticulous evaluation for disease in other organ systems is paramount, especially since results of testing for specific disorders can be delayed by weeks. Evaluation of the central nervous system is critical. Besides a careful neurologic examination, MRI of the brain is done to evaluate for Leigh disease, cerebellar atrophy, leukodystrophy, and cerebral atrophy. MR spectroscopy can be especially helpful, but blood lactate >3 mmol/L may affect interpretation. Evaluation can also include EEG and CSF examination (see above). To evaluate for cardiomyopathy, EKG and echocardiogram should be done. A detailed ophthalmologic exam may reveal ophthalmoplegia in DGUOK deficiency, retinopathy in LCHAD or respiratory chain defects, corneal abrasions in MPV17, or optic atrophy in POLG disease. Serum electrolytes, serum and urine phosphorus and creatinine, urine amino acids, urinalysis, and urine protein are measured to evaluate renal function since abnormal tubular function may suggest a defect in BCS1L. Because diabetes and even adrenal insufficiency can be seen in mitochondrial disorders, HbA1c and morning cortisol level should be considered. Pancreatic insufficiency is seen in some mitochondrial diseases and may be detected by measuring fecal pancreatic elastase. Hearing screening should be performed.
IV. Management during Evaluation for Possible Mitochondrial Disease
The child with mitochondrial disease can be vulnerable to metabolic perturbations such as hypoglycemia or acidosis; close monitoring is important. It is important to discontinue or avoid medications that may exacerbate hepatopathy or impair mitochondrial function or mtDNA translation or transcription, including sodium valproate, tetracycline and macrolide antibiotics, reverse transcriptase inhibitors (particularly AZT), chloramphenicol, quinolones, and linezolid.(47) Use of Ringer’s lactate intravenous solution should be avoided because the liver may not be able to metabolize lactate; propofol should be avoided during anesthesia or sedated procedures since the drug can interfere with mitochondrial function.(48) The goal is to maintain anabolism using a balanced intake of fat and carbohydrates with at least 75% of normal caloric intake while avoiding unbalanced intakes (e.g., glucose only at high intravenous rate) or fasting for >12 hours.(49) In patients with preexisting lactic acidosis, lactate levels should be monitored around procedures to avoid excessive lactic acidosis.
Conclusion
Mitochondrial disease can present from infancy to adulthood with varying degrees of hepatic and extrahepatic involvement. In the last decade, there has been a rapid expansion of newly recognized mitochondrial diseases and their molecular bases. Available technology to aid in diagnosis has improved substantially. Nonetheless, diagnosis of suspected mitochondrial disease in children is complicated; a systematic clinical, biochemical, and molecular approach can aid in making a timely, accurate, and cost-effective diagnosis.
Acknowledgements
Vicki Haviland-Wilhite for expert secretarial assistance. Marisa Friederich, PhD for technical assistance.
Supported by NIH Grant: 1. Indiana University School of Medicine, U01 DK084536
2. University of Colorado Denver/Children’s Hospital Colorado, U01 DK062453
3. Johns Hopkins University School of Medicine, U01 DK062503
4. Cincinnati Children’s Hospital Medical Center, U01 DK062497
5. Seattle Children’s Hospital, U01 DK084575
6. University of Michigan Medical School, U01 DK062456
7. University of Pittsburgh School of Medicine, U01 DK062466
8. Emory University School of Medicine, U01 DK084585
Abbreviations
- MNGIE
mitochondrial neuro-gastrointestinal encephalomyopathy
- MADD
multiple acyl-CoA dehydrogenase deficiency
- FAO
fatty acid oxidation defect
- LCHAD
long chain 3-hydroxyacyl-CoA dehydrogenase deficiency
- ETF
electron transport flavoprotein
- HADHA
hydroxyacyl-CoA deHase/3-Ketoacyl-CoA thiolase/enoyl-CoA hydratase alpha subunit
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–68. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Cazorla A, De Lonlay P, Rustin P, et al. Mitochondrial respiratory chain deficiencies expressing the enzymatic deficiency in the hepatic tissue: A study of 31 patients. J Pediatr. 2006;149:401–05. e3. doi: 10.1016/j.jpeds.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 3.Haas RH, Parikh S, Falk MJ, et al. The in-depth evaluation of suspected mitochondrial disease. Mol Genet Metab. 2008;94:16–37. doi: 10.1016/j.ymgme.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koopman WJH, Willems PHGM, Smeitink JAM. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–41. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 5.Spinazzola A, Invernizzi F, Carrara F, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009;32:143–58. doi: 10.1007/s10545-008-1038-z. [DOI] [PubMed] [Google Scholar]
- 6.Wong L-JC. Molecular genetics of mitochondrial disorders. Dev Disabil Res Rev. 2010;16:154–62. doi: 10.1002/ddrr.104. [DOI] [PubMed] [Google Scholar]
- 7.Kemp JP, Smith PM, Pyle A, et al. Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain. 2011;134:183–95. doi: 10.1093/brain/awq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee WS, Sokol RJ. Mitochondrial hepatopathies: Advances in genetics and pathogenesis. Hepatology. 2007;45:1555–65. doi: 10.1002/hep.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett MJ. Pathophysiology of fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:533–7. doi: 10.1007/s10545-010-9170-y. [DOI] [PubMed] [Google Scholar]
- 10.Fearing MK, Israel EJ, Sahai I, et al. Case records of the Massachusetts General Hospital. Case 12-2011. A 9-month-old boy with acute liver failure. N Engl J Med. 2011;364:1545–56. doi: 10.1056/NEJMcpc1013928. [DOI] [PubMed] [Google Scholar]
- 11.Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci Transl Med. 2012;4:118ra10. doi: 10.1126/scitranslmed.3003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieber DS, Calvo SE, Shanahan K, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013 doi: 10.1212/WNL.0b013e3182918c40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonnefont JP, Specola NB, Vassault A, et al. The fasting test in paediatrics: application to the diagnosis of pathological hypo- and hyperketotic states. Eur J Pediatr. 1990;150:80–85. doi: 10.1007/BF02072043. [DOI] [PubMed] [Google Scholar]
- 14.Wortmann SB, Rodenburg RJT, Jonckheere A, et al. Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy. Brain. 2009;132:136–46. doi: 10.1093/brain/awn296. [DOI] [PubMed] [Google Scholar]
- 15.Saneto RP, Naviaux RK. Polymerase gamma disease through the ages. Dev Disabil Res Rev. 2010;16:163–74. doi: 10.1002/ddrr.105. [DOI] [PubMed] [Google Scholar]
- 16.Tang S, Wang J, Lee N-C, et al. Mitochondrial DNA polymerase γ mutations: an ever expanding molecular and clinical spectrum. J Med Genet. 2011;48:669–81. doi: 10.1136/jmedgenet-2011-100222. [DOI] [PubMed] [Google Scholar]
- 17.Dimmock DP, Zhang Q, Dionisi-Vici C, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum Mutat. 2008;29:330–31. doi: 10.1002/humu.9519. [DOI] [PubMed] [Google Scholar]
- 18.Labarthe F, Dobbelaere D, Devisme L, et al. Clinical, biochemical and morphological features of hepatocerebral syndrome with mitochondrial DNA depletion due to deoxyguanosine kinase deficiency. J Hepatol. 2005;43:333–41. doi: 10.1016/j.jhep.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Pronicka E, Weglewska-Jurkiewicz A, Taybert J, et al. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure. J Appl Genet. 2011;52:61–6. doi: 10.1007/s13353-010-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–75. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 21.Wong LJ, Brunetti-Pierri N, Zhang Q, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology. 2007;46:1218–27. doi: 10.1002/hep.21799. [DOI] [PubMed] [Google Scholar]
- 22.Van Hove JL, Saenz MS, Thomas JA, et al. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. 2010;68:159–64. doi: 10.1203/PDR.0b013e3181e5c3a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hakonen AH, Isohanni P, Paetau A, et al. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain. 2007;130:3032–40. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- 24.Rötig A, Cormier V, Blanche S, et al. Pearson’s marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest. 1990;86:1601–08. doi: 10.1172/JCI114881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.den Boer MEJ, Wanders RJA, Morris AAM, et al. Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency: Clinical Presentation and Follow-Up of 50 Patients. Pediatrics. 2002;109:99–104. doi: 10.1542/peds.109.1.99. [DOI] [PubMed] [Google Scholar]
- 26.Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77–85. doi: 10.1002/ajmg.c.30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olpin SE, Afifi A, Clark S, et al. Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J Inherit Metab Dis. 2003;26:543–57. doi: 10.1023/a:1025947930752. [DOI] [PubMed] [Google Scholar]
- 28.Schiff M, Froissart R, Olsen RKJ, et al. Electron transfer flavoprotein deficiency: Functional and molecular aspects. Mol Genet Metab. 2006;88:153–58. doi: 10.1016/j.ymgme.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 29.Lopriore E, Reinoud JBJG, Verhoeven NM, et al. Carnitine-acylcarnitine translocase deficiency: phenotype, residual enzyme activity and outcome. Eur J Pediatr. 2001;160:101–4. doi: 10.1007/s004310000644. [DOI] [PubMed] [Google Scholar]
- 30.He M, Rutledge SL, Kelly DR, et al. A New Genetic Disorder in Mitochondrial Fatty Acid β-Oxidation: ACAD9 Deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schara U, von Kleist-Retzow JC, Lainka E, et al. Acute liver failure with subsequent cirrhosis as the primary manifestation of TRMU mutations. J Inherit Metab Dis. 2011;34:197–201. doi: 10.1007/s10545-010-9250-z. [DOI] [PubMed] [Google Scholar]
- 32.Zeharia A, Shaag A, Pappo O, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85:401–7. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Coster R, Smet J, George E, et al. Blue native polyacrylamide gel electrophoresis: a powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr Res. 2001;50:658–65. doi: 10.1203/00006450-200111000-00020. [DOI] [PubMed] [Google Scholar]
- 34.Ye F, Hoppel CL. Measuring oxidative phosphorylation in human skin fibroblasts. Anal Biochem. 2013;437:52–8. doi: 10.1016/j.ab.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 35.Kotarsky H, Karikoski R, Morgelin M, et al. Characterization of complex III deficiency and liver dysfuntion in GRACILE syndryome caused by a BCS1L mutation. Mitochondrion. 2010;10:497–509. doi: 10.1016/j.mito.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Valnot I, Osmond S, Gigarel N, et al. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet. 2000;67:1104–9. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vedrenne V, Galmiche L, Chretien D, et al. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J Hepatol. 2012;56:294–7. doi: 10.1016/j.jhep.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Lee NC, Dimmock D, Hwu WL, et al. Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation. Arch Dis Child. 2009;94:55–8. doi: 10.1136/adc.2008.139584. [DOI] [PubMed] [Google Scholar]
- 39.Smet J, Seneca S, De Paepe B, et al. Subcomplexes of mitochondrial complex V reveal mutations in mitochondrial DNA. Electrophoresis. 2009;30:3565–72. doi: 10.1002/elps.200900213. [DOI] [PubMed] [Google Scholar]
- 40.Teitelbaum JE, Berde CB, Nurko S, et al. Diagnosis and Management of MNGIE Syndrome in Children: Case Report and Review of the Literature. J Pediatr Gastroenterol Nutr. 2002;35:377–83. doi: 10.1097/00005176-200209000-00029. [DOI] [PubMed] [Google Scholar]
- 41.Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet. 2012;21:4521–29. doi: 10.1093/hmg/dds294. [DOI] [PubMed] [Google Scholar]
- 42.Hirano M, Garone C, Quinzii CM. CoQ(10) deficiencies and MNGIE: two treatable mitochondrial disorders. Biochim Biophys Acta. 2012;1820:625–31. doi: 10.1016/j.bbagen.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dimmock DP, Dunn JK, Feigenbaum A, et al. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transpl. 2008;14:1480–85. doi: 10.1002/lt.21556. [DOI] [PubMed] [Google Scholar]
- 44.Thomson M, McKiernan P, Buckels J, et al. Generalised Mitochondrial Cytopathy Is an Absolute Contraindication to Orthotopic Liver Transplant in Childhood. J Pediatr Gastroenterol Nutr. 1998;26:478–81. doi: 10.1097/00005176-199804000-00024. [DOI] [PubMed] [Google Scholar]
- 45.Dubern B, Broue P, Dubuisson C, et al. Orthotopic Liver Transplantation for Mitochondrial Respiratory Chain Disorders: A Study of 5 Children1. Transplantation. 2001;71:633–37. doi: 10.1097/00007890-200103150-00009. [DOI] [PubMed] [Google Scholar]
- 46.Sokal EM, Sokol R, Cormier V, et al. Liver transplantation in mitochondrial respiratory chain disorders. Eur J Pediatr. 1999;158:S81–S84. doi: 10.1007/pl00014328. [DOI] [PubMed] [Google Scholar]
- 47.Cohen BH. Pharmacologic effects on mitochondrial function. Dev Disabil Res Rev. 2010;16:189–99. doi: 10.1002/ddrr.106. [DOI] [PubMed] [Google Scholar]
- 48.Kam PCA, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62:690–701. doi: 10.1111/j.1365-2044.2007.05055.x. [DOI] [PubMed] [Google Scholar]
- 49.Parikh S, Saneto R, Falk MJ, et al. A modern approach to the treatment of mitochondrial disease. Current treatment options in neurology. 2009;11:414–30. doi: 10.1007/s11940-009-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cormier-Daire V, Bonnefont J-P, Rustin P, et al. Mitochondrial DNA rearrangements with onset as chronic diarrhea with villous atrophy. J Pediatr. 1994;124:63–70. doi: 10.1016/s0022-3476(94)70255-1. [DOI] [PubMed] [Google Scholar]