

Abstract
Differential ion mobility spectrometry (FAIMS) separates ions in gases based on the difference between their mobilities in strong and weak electric fields, captured directly employing a periodic waveform with dissimilar profiles in opposite polarities. As that difference is not tightly correlated with the ion size or mass, FAIMS separations are generally quite orthogonal to both conventional IMS (based on the absolute ion mobility that reflects the physical ion size) and mass spectrometry (based on mass). Until a few years ago, that advantage was largely offset by poor FAIMS resolving power (∼10–20), an order of magnitude below that achieved with conventional (drift-tube) IMS. This article summarizes the major recent technical developments that have raised FAIMS resolving power up to ∼500. These include use of higher and more stable voltages provided by new waveform generators, novel buffer gas compositions comprising high helium or hydrogen fractions, and extended filtering times up to ∼1 s. These advances have enabled previously unthinkable analyses such as broad baseline separations of peptide sequence inversions, localization variants (post-translationally modified peptides with differing PTM attachment sites) even for the larger “middle-down” peptides and smallest PTMs, and lipid regioisomers.
Keywords: ion mobility spectrometry, differential IMS, FAIMS, proteomics, post-translational modifications, lipidomics
Fast gas-phase separations using ion mobility spectrometry (IMS) prior to mass spectrometry (MS) are becoming ubiquitous, moving beyond research laboratories to practical everyday analyses thanks to the recent introduction of IMS/MS systems by major vendors including Waters (Synapt),1) AB Sciex (Selexion),2) and Thermo Scientific.3) While the IMS peak capacity in present commercial platforms is short of the need for most complex samples, it allows drastically reducing the separation power required of upfront condensed-phase stages such as liquid chromatography (LC) and thus raising the throughput and/or dynamic range of analyses.4)
As the single-step drift-tube (DT) IMS technology developed since 1960s becomes routine,5) research and instrumentation development are moving to novel approaches beyond simple separations by low-field ion mobility. Sciences studying perturbations in media (such as optics, acoustics, and fluid dynamics) have at some point shifted from the linear to nonlinear paradigm, where a perturbation propagates depending on its magnitude and driving force.6) While the technology within linear description remains industrially important (for example, eyewear and architectural glass for optics), frontline research has moved to nonlinear phenomena. IMS is undergoing that transition now with the rise of differential or field asymmetric waveform IMS (FAIMS) based on the evolution of mobility (K) as a function of electric field intensity E, rather than the absolute K in conventional IMS.7) We expect that process to continue as the understanding of foundations improves, new modalities and applications emerge, and vendors introduce further instruments.
The K(E) function is neither constant for any ion in any gas nor identical for two different species other than enantiomers. Therefore, in principle, different ions can always be distinguished by either conventional IMS or FAIMS. The key advantage of FAIMS (and, generally, nonlinear IMS methods) is a greater orthogonality to MS. The mobility itself is strongly correlated to the ion size and thus mass (at least, for a given charge state), which severely compresses the useful IMS/MS separation space. The correlation is much weaker for the mobility increment between two E values.8) That aspect commonly lets FAIMS to distinguish both targeted isomers and biomolecular classes better than conventional IMS at equal formal resolving power (customarily defined as the position of a peak divided by its width at half maximum, fwhm).9) It also means that FAIMS is substantially orthogonal to conventional IMS, and 2-D FAIMS/DTIMS platforms can resolve more features than either stage alone.10)
However, the utility of FAIMS for over two decades since its invention7) in 1982 was severely restricted by low resolving power, typically11,12) R ∼10 versus >100 achieved for DTIMS by late 1990s.13) That deficiency has muted the orthogonality benefit of FAIMS when coupled to MS. Hence, we have strived to improve the R metric and associated resolution of FAIMS and map the new application classes opened as the result. The nonlinear ion motion is far more sensitive to the gas composition, temperature, and magnitude of E than the K value,7) while the resolution further depends on the electrode geometry and separation time. This wide multidimensional space of instrumental parameters creates exceptional opportunities for flexible development of analytical methods.
While the early commercial FAIMS units featured coaxial cylindrical electrodes forming a curved gap,3) modeling has revealed that high resolution requires homogeneous electric fields possible only in straight gaps between planar electrodes.14) Indeed, new planar devices uniformly provided much better resolution than cylindrical analogs with otherwise equal gap dimensions, asymmetric waveform amplitude (“dispersion voltage,” DV), and separation time (t).14) With a gap of 1.9 mm width and ∼50 mm length, DV of 4 kV, and t=0.2 s, we obtained R ∼40 using the nitrogen gas and R ∼70 with 1 : 1 helium/N2 mixtures (for multiply-charged peptides).12,14)
While the resolving power scales roughly as (DV)3 and rapidly improves upon He addition because of increasing ion mobility in gases of lighter molecules,15) the feasible combinations of DV and He fraction are limited by electrical (avalanche) breakdown across the gap. However, the breakdown threshold is materially greater for high-frequency rf than dc voltages, and a 1.9-mm gap can hold16,17) DV=4 kV with up to 75% He (rest N2) or up to 5.4 kV at 50% He. Operation in these regimes increases R (for peptides) up to ∼200, which compares with the metrics for the most advanced existing DTIMS systems.18)
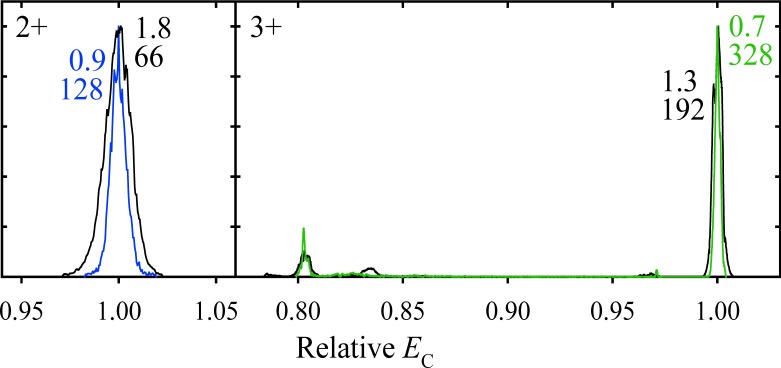
The resolving power of separations in media, including FAIMS, is normally proportional15,19) to t1/2 because the positions of peaks scale as t while the widths, governed by diffusion, scale as t1/2. The ion filtering time in FAIMS is proportional to the inverse gas flow velocity and thus can be set by adjusting the volume flow rate, Q. For our planar-gap device, going from the previously standard Q=2 L/min to 0.5 L/min extends t from 0.2 to 0.8 s.19) As theory predicted, that has increased R up to twofold (to >300 for peptides) at the cost of sensitivity19)Fig. 1
Fig. 1. Normalized spectra for the 2+ and 3+ ions of the Syntide 2 peptide, measured using the “standard” t=0.2 s (black lines) and “extended” t=0.5 s (colored lines) with DV=5.4 kV and 1 : 1 He/N2 gas.19) The peak widths (fwhm, V/cm) are marked with the resolving power values underneath.
Whereas fundamentally the use of He is an effective path to elevated FAIMS resolution, the approach is severely constrained by He being about the easiest gas to break down.20) The only gas lighter than He, thus supporting yet higher ion mobilities that lead to better resolution,15) is hydrogen. Fortunately, H2 has much higher breakdown threshold than He,20) permitting up to 90% H2 (vs. 50% for He) even at our maximum DV=5.4 kV.21) As anticipated from simulations, separations in He/N2 and H2/N2 with equal N2 fractions are similar, with slightly narrower peaks and thus higher resolution for the latter.21) The main attraction, however, is stable operation at up to 85% H2, with the resolving power for larger 1+ ions improving by up to threefold as absolute compensation voltages or CVs (the FAIMS separation parameter) increase while peaks narrow further (Fig. 2). One can trade a higher R for faster analyses, e.g., accelerating the separation by up to fourfold at same resolution.21) The gains for multiply-charged peptides with already very high R are less, but still significant.21)
Fig. 2. Resolving power of FAIMS measured for the reserpine 1+ benchmark using DV=5.4 kV. Solid line for He/N2 mixtures and solid circles for H2/N2 reflect the “standard” t=0.2 s. The data at extended t are represented by blank circles for H2/N2, the dashed line shows the maximum R with He/N2 compositions.21).

As analytical methods mature, the resolution usually becomes limited more by the instrument parameter instabilities than physical factors. For example, the resolving power in modern time-of-flight MS is controlled mostly by the field uniformity, pusher voltage stability, and detector jitter rather than ion–ion Coulomb repulsion or other physical effects.22) Likewise, the peak widths in FAIMS are affected by the fluctuations of waveform incorporating CV, gas pressure and temperature, and variations of the gap width due to imperfect electrode surface or alignment.23) With the peak positions now employed to identify analytes, the reproducibility of measured CVs also becomes crucial.24) While internal calibration helps, its utility is limited by the inevitably unequal K(E) dependences for different species.24) Starting to address these issues, we have constructed a new CV supply with a more precise and less noisy output.23) This electronics has narrowed the features at highest resolving power by ∼40 mV or ∼1/3, pushing the maximum R (for multiply-charged peptides) into the ∼400–500 range. The temporal drift and jitter of peaks have noticeably decreased, tightening the error margins for assignments based on measured CVs. The resulting combination of high resolving power and CV precision enables high-definition ion mobility spectrometry.
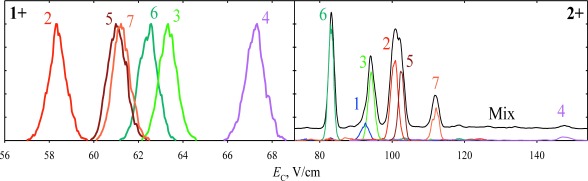
This unprecedented performance opens major new applications to biological analyses, including proteomics and metabolomics. In (bottom-up or top-down) proteomics, one ubiquitously encounters peptide or protein isomers of several types. A major one is sequence inversions, where two or more residues on the peptide backbone are permuted. These, as exemplified for seven isomers possible for a model nitrated tryptic octapeptide nYAAAAAAK, can be broadly resolved baseline by high-resolution FAIMS21,25) (Fig. 3). Separations are independent of the residue shift distance (i.e., shorter nY moves along the backbone are uncorrelated with lower peak resolution), so FAIMS remains effective for isomers with adjacent alternatively modified sites that challenge MS/MS most (as only one unique fragment may be observed).25) Separations for different charge states (1+ and 2+) are about orthogonal, hence the peak capacities for individual charge states are nearly additive.25) This would be more important for larger peptides that exhibit ions of three or more charge states.
Fig. 3. Spectra for the 1+ (normalized) and 2+ nitropeptides of (nY)AAAAAAK stoichiometry, the number indicates the nY position on the backbone (e.g., 4 stands for AAAnYAAAK).25) The result for an equimolar mixture of all seven isomers is added for 2+. The data were acquired using DV=5.4 kV, the He/N2 mixtures with 20% He (1+) and 40% He (2+), and t of 0.7 s (1+) and 0.2 s (2+).
Another common isomer type are localization variants, which differ in the attachment site of one or more post-translational modifications (PTM). Separating such variants by LC is often difficult and lengthy,26) while MS/MS is challenging or even fundamentally impossible. In particular, MS/MS can tell apart only two of three or more localization isomers because all fragments of others (with either collision-induced or electron capture/transfer dissociation) are isobaric to those from a mixture of the first two.24,27) Consequently, PTMs are often assigned to a protein region rather than a residue,28) although variants with PTMs at nearby sites may have different and even opposite activities in vivo.29)
High-resolution FAIMS appears able to generally separate the localization variants for all PTMs baseline, as demonstrated for phosphorylation24,30) (80 Da, Fig. 4), O-linked glycosylation (by N-acetyl-galactosamine, 221 Da),31) acetylation (42 Da),32) and the smallest additive PTM–methylation (14 Da).29) The capability extends to larger “middle-down” (∼3–4 kDa) peptides such as histone tails that are richly modified and allow thousands of localization variants29,32) (Fig. 5). Counterintuitively, the resolution does not necessarily worsen for larger peptides or smaller PTMs, e.g., some methylated variants are separated better than their acetylated analogs of same size and similar PTM shift distance.29) Again, separation is unrelated to the PTM shift involved and even the peptides with adjacent alternatively modified sites (which challenge LC and MS/MS methods most) are resolved well. Separations are equally good for peptides with one and more PTMs, and PTMs on different sites (e.g., serine vs. threonine).30) Separations for different charge states are independent as for sequence inversions, and the variants co-eluting for one are often resolved for another.29,30,32) The peak capacity is thus multiplied by the number of observed states, which is increasingly consequential for larger peptides as explained above.30)
Fig. 4. Normalized spectra for the 2+ and 3+ ions of 226–240 segment of human tau protein (VAVVRT1PPKS2PS3S4AK) singly phosphorylated at one of the residues (1–4) as marked, obtained using DV=4 kV, 7 : 3 He/N2 mixture, and t=0.2 s.30).
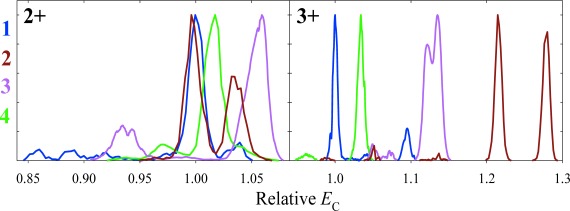
Fig. 5. Normalized spectra for the 5+ and 6+ ions of the 1–25 H4 histone tail (SGRGK5GGK8GLGK12GGAK16RHRKVLRDN) with the biotinylated terminal GSGSK linker, singly acetylated on one of the lysine resides as marked, obtained using DV=5.4 kV, 46 : 54 He/N2 mixture, and t=0.2 s.32).

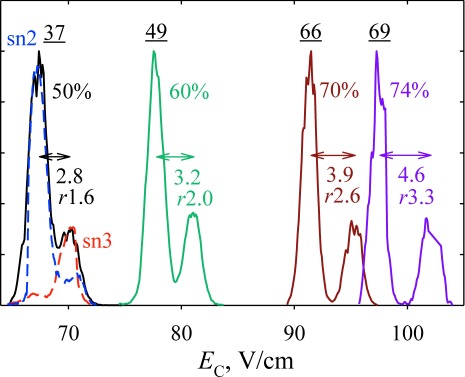
Another area where separation and identification of isomers is critical, yet existing LC and MS/MS tools are insufficient, is lipidomics.33) In a brief survey, FAIMS has separated various lipid classes (such as phospholipids, lysophospholipids, monoacylglycerols, and diacylglycerols), saturated and unsaturated lipids, and even subclasses with different head groups by trend lines (domains) in the FAIMS/MS space markedly better than conventional IMS, again manifesting that FAIMS is correlated with MS less than IMS is9) (Fig. 6). While here a significant correlation remains (unlike for peptides), a perfect orthogonality would preclude domains in the FAIMS/MS palettes and thus classification. Specific isomers, such as the regioisomers of diacylglycerols with sn2 versus sn3 site for fatty acid attachment, were also resolved baseline9) (Fig. 7).
Fig. 6. Trend lines for various lipid classes in the FAIMS/MS space, measured9) using DV=4 kV and 1 : 1 He/N2. The abbreviations are: P for phospholipids, LP for lysophospholipids, and MG, DG, and TG for mono-, di-, and tri-acylglycerols. The number after abbreviation gives the number of unsaturated bonds.
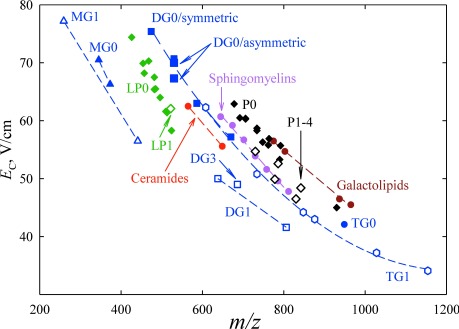
Fig. 7. Separation of 1+ ions for the 1 : 1 mixture of 16 : 0 (sn1)/12 : 0 (sn2) and 16 : 0 (sn1)/12 : 0 (sn3) regioisomers of diacylglycerol, using DV=4 kV, He/N2 mixtures with He fractions as marked (solid lines), and t=0.2 s.9) The resolving power values for the major peak are underlined. Values below are the distances between two peaks with their resolution (r, the peak distance divided by the average fwhm peak width) underneath. Spectra for each isomer at 50% He are shown in dashed lines.
Concluding, the FAIMS resolving power has been raised by over an order of magnitude via thorough theoretical and experimental optimization of the device geometry, buffer gas composition, waveform parameters, and separation time. This has enabled numerous new applications to biological analyses focused on either smaller molecules found in metabolomics or larger ones in bottom-up or middle-down proteomics. Preliminary data suggest that the major gains of resolving power and resolution extend to whole proteins such as ubiquitin, which would enhance top-down and intact protein analyses and perhaps allow resolving certain sequence inversions or PTM localization variants on the protein level. With that, the resolving power of FAIMS has matched or exceeded that of the best known conventional IMS platforms, allowing the orthogonality advantage of FAIMS and MS dimensions to truly shine.
Acknowledgments
This work was supported by the US National Institutes of Health and carried out in EMSL, a US DoE national scientific user facility at PNNL.
References
- 1) S. D. Pringle, K. Giles, J. L. Wildgoose, J. P. Williams, S. E. Slade, K. Thalassinos, R. H. Bateman, M. T. Bowers, J. H. Scrivens. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int. J. Mass Spectrom. 261: 1–12, 2007. [Google Scholar]
- 2) B. B. Schneider, T. R. Covey, S. L. Coy, E. V. Krylov, E. G. Nazarov. Chemical effects in the separation process of a differential mobility/mass spectrometer system. Anal. Chem. 82: 1867–1880, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3) D. A. Barnett, M. Belford, J. J. Dunyach, R. W. Purves. Characterization of a temperature-controlled FAIMS system. J. Am. Soc. Mass Spectrom. 18: 1653–1663, 2007 [DOI] [PubMed] [Google Scholar]
- 4) E. S. Baker, E. A. Livesay, D. J. Orton, R. J. Moore, W. F. Danielson 3rd, D. C. Prior, Y. M. Ibrahim, B. L. LaMarche, A. M. Mayampurath, A. A. Schepmoes, D. F. Hopkins, K. Tang, R. D. Smith, M. E. Belov. An LC-IMS-MS platform providing increased dynamic range for high-throughput proteomic studies. J. Proteome Res. 9: 997–1006, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5) G. A. Eiceman, Z. Karpas. Ion Mobility Spectrometry, CRC Press, Boca Raton, FL, 2004
- 6) E. Wolf (ed.). Progress in Optics (vol. 44), North-Holland, 2003 [Google Scholar]
- 7) A. A. Shvartsburg. Differential Ion Mobility Spectrometry: Nonlinear Ion Transport and Fundamentals of FAIMS, CRC Press, Boca Raton, FL, 2008
- 8) A. A. Shvartsburg, S. V. Mashkevich, R. D. Smith. Feasibility of higher-order differential ion mobility separations using new asymmetric waveforms. J. Phys. Chem. A 110: 2663–2673, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9) A. A. Shvartsburg, G. Isaac, N. Leveque, R. D. Smith, T. O. Metz. Separation and classification of lipids using differential ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 22: 1146–1155, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10) K. Tang, F. Li, A. A. Shvartsburg, E. F. Strittmatter, R. D. Smith. Two-dimensional gas-phase separations coupled to mass spectrometry for analysis of complex mixtures. Anal. Chem. 77: 6381–6388, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11) R. Guevremont, G. Thekkadath, C. K. Hilton. Compensation voltage (CV) peak shapes using a domed FAIMS with the inner electrode translated to various longitudinal positions. J. Am. Soc. Mass Spectrom. 16: 948–956, 2005 [DOI] [PubMed] [Google Scholar]
- 12) A. A. Shvartsburg, K. Tang, R. D. Smith. Differential ion mobility separations of peptides with resolving power exceeding 50. Anal. Chem. 82: 32–35, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13) P. Dugourd, R. R. Hudgins, D. E. Clemmer, M. F. Jarrold. High-resolution ion mobility measurements. Rev. Sci. Instrum. 68: 1122–1129, 1997 [Google Scholar]
- 14) A. A. Shvartsburg, F. Li, K. Tang, R. D. Smith. High-resolution field asymmetric waveform ion mobility spectrometry using new planar geometry analyzers. Anal. Chem. 78: 3706–3714, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15) A. A. Shvartsburg, R. D. Smith. Scaling of the resolving power and sensitivity for planar FAIMS and mobility-based discrimination in flow- and field-driven analyzers. J. Am. Soc. Mass Spectrom. 18: 1672–1681, 2007 [DOI] [PubMed] [Google Scholar]
- 16) A. A. Shvartsburg, W. F. Danielson, R. D. Smith. High-resolution differential ion mobility separations using helium-rich gases. Anal. Chem. 82: 2456–2462, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17) A. A. Shvartsburg, D. C. Prior, K. Tang, R. D. Smith. High-resolution differential ion mobility separations using planar analyzers at elevated dispersion fields. Anal. Chem. 82: 7649–7655, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18) C. A. Srebalus, J. Li, W. S. Marshall, D. E. Clemmer. Gas-phase separations of electrosprayed peptide libraries. Anal. Chem. 71: 3918–3927, 1999 [DOI] [PubMed] [Google Scholar]
- 19) A. A. Shvartsburg, R. D. Smith. Ultrahigh-resolution differential ion mobility spectrometry using extended separation times. Anal. Chem. 83: 23–29, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20) J. M. Meek, J. D. Craggs. Electrical Breakdown of Gases, Wiley, NY, 1978
- 21) A. A. Shvartsburg, R. D. Smith. Accelerated high-resolution differential ion mobility separations using hydrogen. Anal. Chem. 83: 9159–9166, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22) A. F. Dodonov, V. I. Kozlovski, I. V. Soulimenkov, V. V. Raznikov, A. V. Loboda, Z. Zhou, T. Horwath, H. Wollnik. High-resolution electrospray ionization orthogonal-injection time-of-flight mass spectrometer. Eur. J. Mass Spectrom. (Chichester, Eng.) 6: 481–490, 2000. [Google Scholar]
- 23) A. A. Shvartsburg, T. A. Seim, W. F. Danielson, R. Norheim, R. J. Moore, G. A. Anderson, R. D. Smith. High-definition differential ion mobility spectrometry with resolving power up to 500. J. Am. Soc. Mass Spectrom. 24: 109–114, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24) A. A. Shvartsburg, A. J. Creese, R. D. Smith, H. J. Cooper. Separation of peptide isomers with variant modified sites by high-resolution differential ion mobility spectrometry. Anal. Chem. 82: 8327–8334, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25) A. A. Shvartsburg, A. J. Creese, R. D. Smith, H. J. Cooper. Separation of a set of peptide sequence isomers using differential ion mobility spectrometry. Anal. Chem. 83: 6918–6923, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26) D. Singer, J. Kuhlmann, M. Muschket, R. Hoffmann. Separation of multiphosphorylated peptide isomers by hydrophilic interaction chromatography on an aminopropyl phase. Anal. Chem. 82: 6409–6414, 2010 [DOI] [PubMed] [Google Scholar]
- 27) Y. Xuan, A. J. Creese, J. A. Horner, H. J. Cooper. High-field asymmetric waveform ion mobility spectrometry (FAIMS) coupled with high-resolution electron transfer dissociation mass spectrometry for the analysis of isobaric phosphopeptides. Rapid Commun. Mass Spectrom. 23: 1963–1969, 2009 [DOI] [PubMed] [Google Scholar]
- 28) P. J. Boersema, S. Mohammed, A. J. R. Heck. Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 44: 861–878, 2009 [DOI] [PubMed] [Google Scholar]
- 29) A. A. Shvartsburg, Y. Zheng, R. D. Smith, N. L. Kelleher. Separation of variant methylated histone tails by differential ion mobility. Anal. Chem. 84: 6317–6320, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30) A. A. Shvartsburg, D. Singer, R. D. Smith, R. Hoffmann. Ion mobility separation of isomeric phosphopeptides from a protein with variant modification of adjacent residues. Anal. Chem. 83: 5078–5085, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31) A. J. Creese, H. J. Cooper. Separation and identification of isomeric glycopeptides by high field asymmetric waveform ion mobility spectrometry. Anal. Chem. 84: 2597–2601, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32) A. A. Shvartsburg, Y. Zheng, R. D. Smith, N. L. Kelleher. Ion mobility separation of variant histone tails extending to the “middle-down” range. Anal. Chem. 84: 4271–4276, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33) T. W. Mitchell, H. Pham, M. C. Thomas, S. J. J. Blanksby. Identification of double bond position in lipids: From GC to OzID. J. Chromatogr. B 877: 2722–2735, 2009 [DOI] [PubMed] [Google Scholar]