

SYNOPSIS
This review summarizes data on translational studies to target the p53 pathway in cancer. It describes the functions of the p53 and Mdm-2 signaling pathways and discusses current therapeutic approaches to target p53 pathways, including reactivation of p53. In addition, direct interaction and colocalization of the p53 and focal adhesion kinase (FAK) proteins in cancer cells have been demonstrated, and different approaches to target this interaction are reviewed. In summary, this is a broad review of p53 function as it relates to the diagnosis and treatment of a broad range of cancers.
Keywords: p53, Mdm-2, FAK, survival, apoptosis
Introduction
P53 is a guardian of the genome and a tumor suppressor gene that controls genetic stability and prevents cancer. P53 is highly mutated in different types of tumors or is inactivated by different mechanisms such as interaction with Mdm-2, Mdmx, FAK and other proteins. In this review, we discuss different approaches to reactivate p53 pathway through either targeting its inhibiting protein interactions with Mdm-2 and FAK proteins, or through changing protein conformations with small molecule inhibitors, through introduction of wild type p53 or through other pathways, and we will focus on future directions and perspectives in translational research on developing p53-targeting therapeutics. There are several homologue proteins to p53, such as p63 and p73, with similar and unique functions that are not discussed in the present review.
p53 protein structure and function
The P53 gene is a tumor suppressor gene, whose genomic sequence contains 20 exons and spans 20 kilobases and is located at the short arm of the chromosome 17p13.1 region and encodes p53 protein (1). The p53 protein is a transcription factor that binds as a tetramer to the following DNA consensus sequence: 5’Pu-Pu-Pu-C-A/T-T/A-G-Py-Py-Py3’ (Pu-purine; Py-pyrimidine) in the promoters of the several genes, such as p21, Bax, Mdm-2, and PUMA and activates their transcription (2). It was also shown that p53 can repress transcription of several important genes such as survivin, Bcl-2, FAK and others (3). The promoter of p53 does not contain TATA box and consists of various binding sites for known transcription factors, such as NF-kappa B, Sp1 or c-Jun (4). The p53 gene encodes 393 amino-acid tumor suppressor protein, which contains several functional domains: two N-terminal transcriptional activating domains, TAD1 (1- 42 aa.) and TAD2 (43-65 aa); proline rich domain (64-92 aa); Central DNA-binding domain (102-292 aa); and C-terminal tetramerization domain (325-393 aa) and basic region (364-393 aa (5) (Fig. 1). The Nuclear localization signal is located after the DNA binding domain (305-322 aa) (Figure 1).
Figure 1. The p53 protein structure.
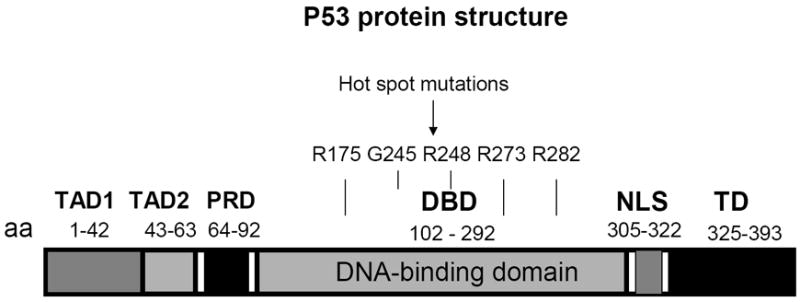
TAD-transactivating domain; DBD-DNA binding domain; NLS-nuclear localization sequence; TD-tetramerization domain.
[Tag: p53 protein; tumor suppressor protein]
N-terminal trans-activation domain of p53
The 17-28 aminoacids of the first N-terminal transactivation domain interact with Mdm-2 protein, double minute-2 homologue, which plays a major role in p53 degradation via the ubiquitin-proteasome pathway (1). P53 activates Mdm-2 transcription through binding to its promoter and up-regulates Mdm-2 expression. In turn, Mdm-2 binds to p53 and negatively regulates its stability and activity. Thus, Mdm-2 regulates p53 via an autoregulatory feedback loop in which both proteins control its cellular expression. The aminoacids 22-26 are involved in binding of histone acetyltransferase P300 (reviewed in (6). The second transactivation domain (43-63 aa) activates transcription of several p53 transcriptional targets
Proline-rich domain of p53
A proline-rich domain (64-92 aa) contains five PXXP motifs and is important for interaction with various proteins and regulates apoptosis (6). The PXXP motifs of this domain were shown to create a binding site for Src-homology SH3 domains (7). A mutant deleted for the proline-rich domain in human p53 (Δ62-91) was not able to cause apoptosis but was able to maintain cell cycle arrest (8). A polymorphism at the amino-acid 72 of either proline or arginine could cause differences in binding of transcription factors, in survival/apoptotic signaling and response to chemotherapy agents (9).
Central DNA-binding domain
The core central region of p53 is highly conserved, homologous to the regions of other p53 family members (p63 and p73). The DNA binding domain binds to promoter binding sites 5’Pu-Pu-Pu-C-A/T-T/A-G-Py-Py-Py3’ (1). The several hot-spot p53 mutation is located in this domain such as: Arg 175; Gly 245; Arg 248; Arg 273 and Arg 282 (Figure 1).
C-terminal tetramerization domain
This domain is involved in protein tetramerization and regulation of p53 activity. It contains tetramerization region (323-356 a.a.) and a negative regulatory region (363-393 a.a.) (reviewed in (6).
p53 Mutations in Cancer Cells
P53 is the most commonly mutated gene in tumors with up to 50% mutations in different types of cancers and 75% mutations in invasive cancers. The majority of mutations are missense mutations with four out of five mutations in the DNA-binding domain, only 1% are found in N-terminal domain and 4% in the C-terminal tetramerization domain (reviewed in (10). Most frequent “hot-spot” mutations are: 175 Arg→His (breaks bond between L2 and L3 loop); 248 Arg → Gln (Trp) (breaks contact with DNA in minor groove); 273 (Arg→His(Cys) (breaks contact with DNA in major groove) and 282 Arg →Trp (destabilizes H2 helix and DNA binding in the major groove and breaks contacts on the β-hairpin (reviewed in (10). Arginines 248 and 273 are involved in interaction of p53 with DNA and arginines 175 and 282 stabilize DNA-binding sequence of p53 (4). Wild type p53 binds to gene promoters with different affinity; for example, p53 activates p21 promoter with a higher affinity than the Bax promoter (4)
p53 function
P53 was discovered in 1979 and first was thought to play a role of oncogene and then revisited and found to be a tumor suppressor gene. The model of the main functions of p53 is shown in Figure 2 and is discussed in the recent review, summarizing the first 30 years of p53 research (11). It is known that p53 is mutated in almost 50% of all tumors (12). Inactivation of the p53 gene is a critical step in tumorigenesis (13). Following induction by a variety of cell stresses, such as DNA damage, hypoxia, or the presence of activated oncogenes, p53 up-regulates a set of genes that can promote either cell death, apoptosis, senescence, or growth arrest, such as p21, GADD45, cyclin G, and Bax, reviewed in (14). Recently, it was shown that p53 can repress promoter activities of a number of anti-apoptotic genes and cell-cycle genes [survivin (15), cyclin B1, cdc2, (16, 17), cdc25 c (18), stathmin(19), Map4(20), and bcl-2 (16)]. Overexpression of p53 induced apoptosis (21), (22), and p53 inactivation caused a decrease of radiation-induced apoptosis (23-25). Recently, new functions of p53, such as metabolism and anti-oxidant defense were reported and reviewed in (11) (Figure 2).
Figure 2. FAK, p53 and Mdm-2 interaction and down-stream signaling.
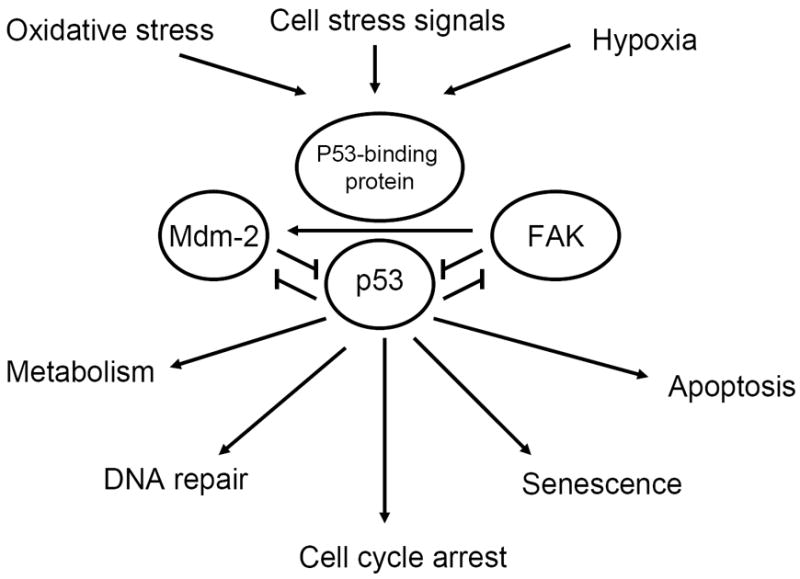
FAK binds to p53 and blocks its functions; FAK binds to Mdm-2 to facilitate p53 proteosomal degradation, as shown in [48]; Mdm-2 binds to p53 and causes its degradation. p53 activates Mdm-2 and inhibits FAK.
p53 binds Focal Adhesion Kinase (FAK) promoter and represses its activity
FAK (Focal adhesion Kinase) is a 125 non-receptor kinase which plays an important role in survival signaling, adhesion, motility, invasion, angiogenesis, and proliferation (26, 27). FAK was shown to be overexpressed in many types of tumors and proposed to be a therapeutic target (28),(29),(30).
The first report on the indirect link of FAK and p53 signaling in apoptosis was reported by Ilic et al. (31). The authors demonstrated that FAK suppressed a p53-regulated apoptotic pathway in anchorage-dependent cells such as fibroblasts and endothelial cells (31). The authors showed that in the absence of FAK function, p53-regulated apoptosis was activated by protein kinase C and phospholipase A2 and this process was inhibited by dominant-negative p53 and Bcl-2 (31). The authors did not study these processes in tumors and cancer cell lines (31), (32). In addition, immunohistochemical analysis of 115 endometrial carcinoma samples demonstrated a correlation between FAK and p53 overexpression (33).
Our group cloned the FAK promoter and found two p53 binding sites in this area (34). We have shown that p53 binds to the FAK promoter and inhibits its transcriptional activity (34). Moreover, mutant p53 with hot-spot mutations did not inhibit FAK promoter activity, while wild type did. The global analysis of p53 transcription factor binding sites demonstrated that induction of HCT116 colon cancer cells with 5-fluorouracil down-regulated FAK (35). Thus, the authors suggested that p53 can suppress metastasis through down-regulation of metastasis-linked genes, as FAK. Since p53 is often mutated in cancer, we performed the population-based study on 600 tumors from breast cancer patients and found that p53 mutations highly correlated with FAK overexpression (36). Recently, another group found high correlation between high FAK expression and low level p53 in oral squamous cell carcinoma (37) that can be explained by the absence of a repressive function on FAK promoter in p53-negative samples.
[Tag: FAK; Focal Adhesion Kinase, p125FAK]
Direct p53 and FAK protein binding and their feedback regulation
After finding that p53 regulates FAK promoter activity (38), we have demonstrated that the N-terminal transactivation domain of p53 is physically and directly associated with the N-terminal domain of FAK (39). There have been several reports on the localization and function of the N-terminal part of FAK in the nucleus (40), (41), (42), (40, 43). The N-terminal domain of FAK caused apoptosis in breast cancer cell lines (41) and its nuclear localization was regulated by caspase inhibitors in endothelial cells (43). p53 was also reported to be localized in the cytoplasm (44). P53 directly activated Bax and released pro-apoptotic molecules, activating multidomain protein complexes in the cytoplasm. This mechanism required the 62-91 proline-rich domain N-terminal domain of p53 (44). We also showed that FAK binds the 7 amino-acids (65-71 aa) in the proline region of p53 (45). In addition, we performed computer modeling and found that p53 peptides containing the 65-71 aa re-activated p53. We modeled this p53 peptide into the three dimensional structure of FAK and screened small molecules that docked into this site of interaction. We identified novel small molecules targeting the FAK and p53 interaction that were able to activate p53 (discussed below).
We have shown that FAK can suppress the transcriptional activity of p53 through its interaction, as p53-mediated activation of p53-targets - p21, Mdm-2, and Bax - was blocked by overexpression of FAK, but not by delta-FAK-NT with a deleted N-terminal domain that binds p53 (46). Thus, p53 regulates FAK, and in turn, FAK regulates p53. Thus FAK and p53 can be regulated through comprising a feedback mechanism (27, 47).
The binding of FAK and p53 was shown also by another group (48). The authors showed interaction of FAK with p53 and also with Mdm-2 proteins, where binding of FAK to Mdm-2 facilitated p53 ubiquitination and degradation (48). The link between FAK and p53 was supported by a report on the FAK-interacting protein, FIP200 (49). FIP200 overexpression resulted in increased p21 levels and caused growth arrest (49). The authors found that FIP200 interacted with FAK and p53 (49). The authors proposed a model of FAK-p53-FIP200 interaction, where FIP200/FAK and FIP200/p53 binding had competition and when FAK has low expression, FIP200 could bind p53 and increase its expression (50).
Recently, we showed that the stem cell marker and transcription factor Nanog, which maintains stem cell renewal and differentiation, was able to bind FAK promoter and up-regulate its expression (51) In addition, FAK phosphorylated Nanog (51). Interestingly, Nanog is repressed by p53 that induced differentiation of embryonic stem cells (52). FAK is also repressed by p53, while it is activated is by Nanog. The mechanism of Nanog and FAK regulation and its cross-talk with p53 in cancer and stem cells requires future studies. The novel mechanisms of FAK, p53 and Nanog survival function during carcinogenesis remain to be discovered.
p53 targeted therapy
In the next section, we will discuss therapeutic approaches to target p53 function. First, we will discuss p53-targeted therapy, and then we will focus on FAK-p53-targeted therapy based on data on FAK and p53 interaction.
Translational approaches that target p53 for cancer treatment include therapies targeting mutant p53 and therapies targeting wild type p53 (reviewed in (53)) and are shown in Figure 3.
Figure 3. p53-targeted therapies.
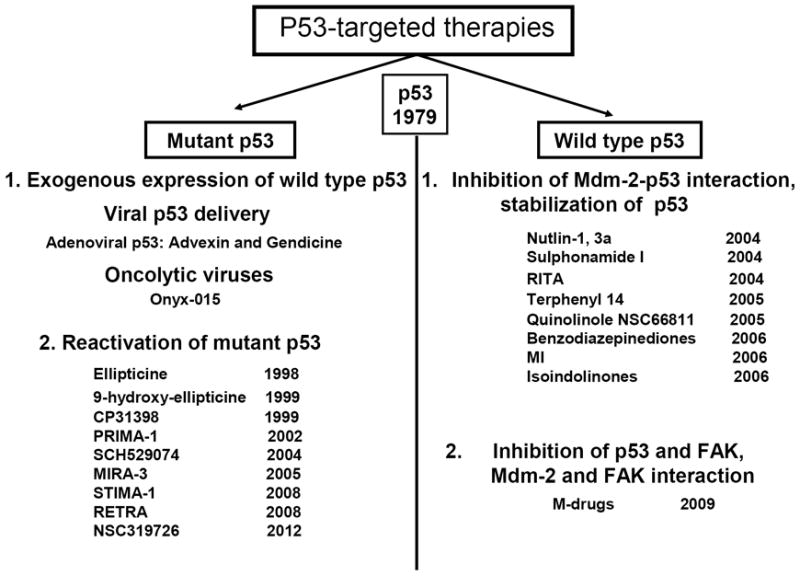
p53 therapies target either mutant p53 (left) or wild type p53 (right). Different therapeutic agents are shown with the year of their first report.
[Tag: p53-targeted therapy; therapy]
Mutant p53-targeted therapy
To target mutant type p53, the two therapeutic approaches were used: 1) to deliver wild type p53 with viruses (retroviruses or adenoviruses); 2) and to use oncolytic therapy to destroy tumor cells with deleted or mutated p53 by lysis.
Exogenous expression of wild type p53
Viral p53 delivery
Retroviruses are attractive agents for cancer gene therapy, as they integrate in the stable form into the genome and require cell division for transduction. The second strategy is to use adenoviruses, which are double stranded DNA viruses. In contrast to retroviruses, their effect is not limited to high proliferating cells and there is no risk of the insertional mutagenesis or tumorigenesis.
Advexin
One of the adenoviral drugs is Ad5CMV-p53 or Advexin developed by Introgen Therapeutics Inc.; Gendicine Shenzden SiBiono Gene Tch Co Ltd), which is a recombinant E1-deleted serotype 5 adenoviral vector encoding p53. In vitro and in vivo studies confirmed the therapeutic effect of Ad5CMVp53. This therapy approach was used effectively in clinical trials, Phase I and II, although some cases of tumor resistance were also demonstrated (reviewed in (4)). The drug was not approved by the FDA, while Gendicine (adenoviral p53) is used for the treatment of head and neck cancer in China (54) and is effective in the treatment of hepatocellular carcinoma (55).
Oncolytic viruses
The second viral approach is to apply oncolytic viruses to kill p53-defective tumor cells, including the use of E1B-defective adenoviruses, which is a major approach.
Onyx-015
The delta 1520 virus (dl1520; ONYX-015), developed by Onyx Phramaceuticals is an adenovirus that contains a deletion in the E1B region. ONYX-015 can be used as a selective drug against tumors with mutant p53. The problem of this and other adenoviral vectors is hepatotoxicity, and the FDA did not approve Onyx-15, which was used in numerous clinical trials.
Reactivation of mutant p53 with small molecules
To reactivate mutant p53, compounds were developed that targeted mutant p53 and reactivated wild p53 based on structural studies of p53 conformation, function, and regulation (Figure 3). The structures of all compounds are shown in review (53), and we present the compounds in chronological order, according to when the authors first reported them (shown in Figure 3).
Ellipticine (1998)
One of the first compounds that targeted mutant p53 was ellipticine, an alkaloid that had cytotoxic and anti-proliferative activity in a panel of cell lines with mutant p53 (56). Its derivative, 9-hydroxy ellipticine (9HE) was developed in 1999 (57). Later studies showed the off-target effects of ellipticine, such as targeting topoisomerase II, FOX, and other mechanisms.
CP31398 (1999)
In 1999, CP31398 (styrylquinazoline) compound, which stabilized the DNA binding domain of p53, was identified by high-thoughput screening of a library of >100,000 synthetic small molecule compounds by scientists from Pfizer Inc (58). The compound increased conformationally active p53 in cells with mutant p53, increased p53-transcriptional activity with p21 target in cells with mutant p53 and decreased tumor growth (58). CP31-398 compound caused apoptosis in different cancer cell lines and stabilized wild-type p53 protein independently from Mdm-2 (59, 60). However, different quantitative biophysical methods in a wide range of conditions did not detect the interaction of CP-31398 and p53 core domain, but detected intercalation of CP-31398 with DNA and destabilization of DNA-p53 complex (61), (62). The compound had several p53-independent functions, such as increase of apoptotic Bax protein (61). The CP31398 did not affect binding of wild type p53 with DNA and did not affect p63, p73 homologues binding to DNA, but increased the amount and affinity (K(d)) of mutant p53 to DNA (63). The CP31398 compound restored the functions of mutant p53 to suppress UVB-induced skin carcinogenesis in mice, which was associated with increased p53, p21, and BclXs (64, 65). CP31398 compound was shown to have a chemopreventive effect on intestinal adenoma development in an animal model of familial adenomatous polyposis (66). The compound suppressed development of tumors by 36% and 75% at low and high doses, respectively and increased expression of p53, p21, cleaved caapase-3 and PARP-1 (66). The chemopreventive effect of CP31398 was observed in combination with celecoxib on azoxymethane-induced aberrant crypt foci (ACF) and colon adenocarcinomas in F344 rats (67). The combination of CP31398 and celecoxib suppressed adenocarcinoma incidence by 78% and its multiplicity by 90% (67). The rats that were fed with a combination of these drugs increased p21 expression, and apoptosis and reduced tumor cell proliferation (67).
PRIMA-1 (2002)
Another p53 reactivating compound was found in 2002 and named PRIMA (p53 reactivation and induction of massive apoptosis) (68). PRIMA had anti-tumor activity in vivo in a mutant p53-dependent manner (68). It restored the p53-binding activity of R273H mutant and also other p53 mutants in the DNA_binding domain (68). A methylated form of PRIMA-1 was more active than the parent compound and synergized with cisplatin to induce tumor cell apoptosis, and induced apoptotic Bax and PUMA p53-targets (69). PRIMA-1MET had suppressive effects in different mutant p53-carrying mouse tumors: mouse sarcomas, mammary carcinoma, and chemically induced fibrosarcoma (70). PRIMA-1 entered into phase I clinical trials based on its high efficacy in the animal models.
SCH529074 (2004)
Another small molecule compound was discovered in 2004 and named SCH529074 (71). The compound was able to bind the DNA-binding domain of p53, restore the growth-suppressive functions of mutant p53, and interrupt the HDM2-regulated ubiquitination of wild type p53 (71)
MIRA-3 (2005)
The MIRA-1 derivative, MIRA-3, was also able to reactivate mutant p53 and to express anti-tumor activity in tumor xenografts with human mutant p53 in vivo (72).
STIMA-1 (2008)
STIMA-1 compound had structural similarities with CP-31398 and was able to reactivate mutant p53 (73).
RETRA (2008.)
Screening of a small molecule library to induce p53-dependent reporter activity in A431 cells with R273H mutation identified another compound, RETRA-1 (reactivation of transcriptional reporter activity) (74). RETRA-1 showed anti-tumor activity in mouse xenografts. It also targeted homologous p53 protein p73 (74). RETRA was suggested to sequester and release p73 from its inhibitory complex with mutant p53. Thus, targeting homologous to p53 proteins, such as p63 and p73 is a potential approach that can be used in therapy.
NSC319726 (2012)
In 2012 three new compounds NSC319725, NSC319726 and NSC328784 were found by in silico modeling screening of an NCI library of small molecule compounds in a panel of 60 cancer cell lines carrying p53 mutations and reported to selectively kill cancer cells while not affecting normal cells (75). After compound validation, NSC319726 compound had the best and specific killing effects on cancer cells, and it changed conformation of p53 and restored wild type function of p53R175 mutant. The compound killed R172Hp53 knock-in mice, caused apoptosis and activated p21 (p53 target) in mice xenograft tumors in a 175-allele-specific mutant p53-dependent manner(75). The activity of compound was dependent on the zinc chelating properties as well as redox changes. The authors conclude that the in silico method on a panel of cancer cell lines can be a future strategy to look for p53 reactivating compounds.
[Tag} Mutant p53-targeted therapy; mutant p53; small molecules targeting mutant p53
Wild type p53 targeted therapy
The small molecule compounds were described to mostly target the p53 and Mdm-2 interaction and will be described below (Figure 3). We will also describe the FAK-p53 pathway small molecule inhibitors developed in our group. We will analyze the structural data of these complexes and then describe small molecules targeting these complexes.
P53 and Mdm-2 interaction
The p53-Mdm-2 complex does not involve large protein areas, as many protein-protein interactions that make it suitable for targeted drug therapy developments. The six p53 amino acids from the p53 N-terminal transactivation domain one 18TFSDLW23 specifically bind to the pocket in the Mdm-2 protein (76). The co-crystallization of p53 peptide and Mdm-2 protein identified the 109 amino-acid region of amino-terminal domain of Mdm-2 bound to p53 peptide and detected that Phe 19, Tro23, and Leu 26 were deeply inserted inside the hydrophobic Mdm-2 cleft (77). Two hybrid genetic analyses identified amino-acids of Mdm-2 involved in interaction with p53 and included G58, D68, V75, and C77 amino acids of the N-terminal domain of Mdm-2 (78). The later study identified additional sites in the N-terminal transactivation domain of p53 - residues 40-45 and residues 49-54 - that are involved in binding with Mdm-2 (79). This binding is weaker than the main site of p53-Mdm-2 interaction, but can provide additional approaches for drug development. The first drugs that were developed to target p53-Mdm-2 interaction were peptides and then small molecule compounds due to their higher stability, bioavailability and other pharmacological and pharmacokinetic properties (described below).
Sulphonamide (2004)
Sulphonamide I was identified by computational screening using a 3D pharmacophore model and small molecules from the NCBI database, and this compound effectively inhibited p53-Mdm-2 interaction and activated p53 (80). In the binding assay IC50 was equal to 32 microM that was higher than p53 peptide, which caused 100% inhibition of p53-Mdm-2 binding at 100 μM. The functional efficacy was demonstrated in a reporter luciferase assay, where sulphonamide increased p53-transcriptuional activity in Mdm-2-overexpressing cell line.
Nutlins (2004)
The small molecule drugs that target p53 and Mdm-2 interaction were developed by La Roche company in Nutley, New Jersey and were named Nutlins. These compounds bind Mdm-2 in the p53-binding pocket with a nanomolar range of binding affinity. The first three compounds, Nutlin 1,2,3 share a common structure (81, 82). The binding of Nutlin with Mdm-2 was confirmed by co-crystallization studies. The compounds activated the p53 pathway in cancer cells, activated the p21 target, caused apoptosis, and led to inhibition of tumor xenograft growth. The Nutlin-3 anti-tumor activity was shown in different types of cancer, including acute myeloid leukemia, neuroblastoma, osteosarcoma, prostate cancer (83),(84), (85), (86), (87). The effect of Nutlins was p53-dependent. An oral Nutlin-3 entered clinical trials recently for treatment of patients with solid tumors and hematological cancers.
RITA (2004)
RITA (reactivation of p53 and induction of tumor apoptosis) was identified by screening a chemical library from the NCI set of compounds (88). RITA inhibited colon cancer cell growth HCT116 more efficiently than p53-/- cells and caused p53-dependent decrease of xenograft tumor growth in vivo. RITA prevented interaction of p53 and Mdm-2 in vitro due to p53 conformational change (88). RITA reactivated p53 and induced apoptosis in multiple myeloma cancer and leukemia cells (89, 90). RITA synergized with Nutlin to induce apoptosis in melanoma (91). RITA reactivated p53 and also activated the JNK-1 pathway in multiple myeloma cancer cells (92). RITA caused a decrease of cervical carcinoma xenograft growth in vivo, associated with reactivation with p53 transcriptional targets: Bax, Noxa-1, and PUMA-1 (93).
Terphenyl 14 (2005)
The computational screening of a small terphenyl library identified therphenyl 14, which was able to bind to the hydrophobic groove of Mdm-2 protein, to mimic the alpha-helical region of p53, and to disrupt p53-Mdm-2 protein interaction (94).
Benzodiazepinediones (2005)
High-throughput screening of chemical libraries using the assay that detected the effect of compounds on protein stability identified the benzodiazepinedione family of compounds, and the lead compound I stabilized p53 and induced p21 expression.(95). The optimization of compound I resulted in TDP521252 and TD665759, which both inhibited Mdm-2 (96). TDP521252 and TDP665759 inhibited the proliferation of wild type p53 cell lines with IC(50) of 14 and 0.7 microM, respectively, and disrupted the complex of Mdm-2 and p53 (96). Administration of TD665759 to mice increased p21 in liver samples. The compound synergized with doxorubicin in an A375 xenograft model and decreased tumor growth (96).
Quinolinole (NSC66811) (2006)
The computational screening of small molecules identified 354 Mdm-2 potential candidates to disrupt interaction with p53. The binding assay demonstrated that quinolinole (7-[anilino(phenyl)methyl]-2-methyl-8-quinolinol) ((NSC66811) is the best to inhibit Mdm-2 in nanomolar concentrations with Ki (120 nM), which was lower than p53 peptide (97). The compound mimicked three amino acids of p53 that were involved in interaction with Mdm-2 and disrupted interaction with Mdm-2.
MI (2006)
The MI compounds were generated by the University of Michigan and Ascenta and belong to the family of potent tryptophan-based inhibitors, which bind to Mdm-2 with high affinity, mimicking the binding of Trp 23 from the p53 peptide and disrupt the interaction of Mdm-2 with p53. At first oxindole was found to mimic the interaction of Trp23. The subsequent chemical modifications and optimizations resulted in a series of MI compounds (98). One of the most potent inhibitors was MI-63 with a Ki of 3 nM, and IC50 280 nM and induced p53 and p21 expression (98). The MI-43 was designed to target four amino-acids of p53 peptide: Phe19, Trp23, Leu22 and Leu26 (99). MI-43 inhibited cell growth and survival of colon cancer cells with wild type p53 (99). The MI-43 compound induced cell cycle arrest that required p53 and p21. The optimization of MI-63 resulted in MI-219, inhibiting tumor growth in vivo (99). The next derivative of MI-219 was MI319, which demonstrated efficient tumor growth suppression of lymphoma cells (100). MI-319 synergized with cisplatin and decreased pancreatic cancer cell growth (101).
Isoindolinones (2006)
Isoindolinones were identified as scaffold Mdm-2 inhibitors, blocking the pocket of interaction with p53 (102). The IC50 values of these inhibitors are 5-16 microM in growth inhibition assays and reporter assays and inhibit binding of Mdm-2 to p53. The most potent inhibitor was (+)-R-3-(4-Chlorophenyl)-3-(1-hydroxymethylcyclopropylmethoxy)-2-(4-nitrobenzyl)-2,3-dihydroisoindol-1-one (74a) or isoindolinone compound 74a, which effectively disrupted interaction of Mdm-2 and p53 and activated p53, p21 and Mdm-2 transcription (103). IC50 of 74a was 170 nM versus 61 nM of Nutlin in an Mdm-2 binding assay and was similar in viability assays (103). The cellular activity of 74a was p53-dependent.
[Tags: Wild type targeted therapy; wild type p53; small molecules targeting wild type p53; small molecules targeting p53 and Mdm-2 interaction]
Focal Adhesion Kinase as a therapeutic target
FAK inhibition with anti-sense oligonucleotides, siRNA and dominant-negative FAK
Recently, Focal Adhesion Kinase has been proposed to be a new therapeutic target (104). Several in vitro approaches were used to down-regulate FAK, such as adenoviral FAK-CD (dominant-negative FAK)(105), anti-sense oligonucleotides (106) and siRNA for FAK (107). We linked FAK expression to apoptosis by treating FAK positive tumor cell lines with different antisense oligonucleotides to FAK that specifically inhibited p125FAK expression (108). The cells treated with antisense oligonucleotides lost their attachment and underwent apoptosis (108). We also analyzed the FAK siRNA effect on tumor growth and found that silencing of FAK expression significantly decreased tumor growth in MCF-7 xenograft model (107). FAK siRNA directly affected important genes involved in survival/apoptotic, cell cycle pathways (107).
Small molecule inhibitors targeting FAK- kinase functions
Small molecules targeting FAK ATP-binding site
Several pharmaceutical companies such as Novartis, Pfizer, and Glaxo, developed several FAK inhibitors that targeted the ATP-binding site of FAK. The Novartis inhibitor, TAE-226, did not enter clinical trials due to toxicity and non-specifity. The Pfizer inhibitors: PF-573,228 did not enter clinical study (109), and PF-562271 did entered clinical study phase I, but has been reported to inhibit FAK homologue Pyk-1(110), and another Pfizer inhibitor, PF-04554878 was also not effective in the first clinical studies. Another group from Poniard developed a PND-1186 FAK inhibitor that also had non-specific effects by inhibiting different proteins at 1 microM, such as Aurora, Flt-3, Lck1, and others (111). Thus, these FAK inhibitors targeting the FAK ATP were non-specific due to the highly conservative nature of kinase domains with the ATP-binding site that limited their clinical use.
Allosteric FAK inhibitors
Another approach to FAK inhibition are the allosteric FAK inhibitors that targeted the FAK kinase-dependent and kinase independent (scaffolding) functions of FAK. The inhibitors targeting kinase function but not the active ATP-binding site (K454) were called allosteric inhibitors. They targeted other sites of FAK phosphorylation such as the main Y397 autophosphorylation site. Small-molecule drugs were found through computer modeling and database screening using three-dimensional target protein crystal structures and libraries of small molecule compounds. By using computer modeling and screening of >200,000 small molecules from the National Cancer Institute Database and docking compounds intro the structural functional pockets of FAK, we found 40 different potential FAK inhibitors. Using this method, we identified a FAK inhibitor targeting the Y397 site of FAK (112), called Y15 or FAK inhibitor 14, which blocked breast (112), pancreatic (113) and neuroblastoma tumor growth (114). The Y15 inhibitor also had anti-tumor activity in glioblastoma and colon cancer xenograft models. The similar allosteric FAK inhibitor, named Y11 also targeted Y397 FAK site and inhibited colon cancer tumor growth (115).
Small molecule inhibitors targeting FAK-scaffolding function
One of the approaches to inhibit FAK kinase-independent scaffolding function was targeting its protein-protein interaction with its binding partners, such VEGFR-3 or other p53 and Mdm-2. One of the lead FAK inhibitors, the FAK inhibitor C4, targeted the FAK-VEGFR-3 interaction and inhibited tumor growth in different types of tumor xenografts (116).
Small molecule drug inhibitors are effectively used to target p53 protein-protein interactions, particularly with Mdm-2 protein (117). The first potent inhibitors targeting p53-Mdm-2 interaction have been identified by high-throughput screening followed by structure-based optimization (117). The screening identified Nutlins that represent a class of cis-imidazole analogues that bind to the p53 pocket interacting with Mdm-2. The same strategy was used to target interaction with Mdm-2 and FAK (M-drugs) and p53 and FAK (P drugs). We also found the lead FAK and Mdm-2 targeting compound - M13 that reactivated p53 and inhibited tumor growth (118). In addition, we developed FAK-p53 targeting compounds (P-drugs) (Figure 3) that reactivated p53 and its transcriptional targets in p53+/+ HCT116 colon cancer cells but not in p53-/- cells. These drugs reactivated p53 transcriptional in vitro and in vivo and effectively decreased vuiability, clonogenicity and tumor growth.
[Tags: FAK, Focal Adhesion Kinase as a therapy target; FAK and p53 interaction therapy; small molecule inhibitors; kinase-dependent and kinase-independent function of FAK].
Perspectives
Thus, understanding FAK biology during tumorigenesis, the mechanisms of its up-regulation in different tumors, its role in stem cell biology, angiogenesis, motility, and especially mechanisms of its direct physical interaction with p53 protein and their downstream signaling pathways will be critical in developing targeted therapeutics. Studies with peptide inhibitors already have indicated that the blockade of specific protein–protein interactions has therapeutic promise for treating a variety of diseases, including cancer (119-123). Small molecule drugs are particularly attractive as inhibitors of intracellular protein–protein interactions due to the ability to modify their structures to achieve optimal target binding. As we further define the mechanisms of FAK signaling in cancer cells, we will identify the optimal sites for targeting this protein and disrupting its signaling to cause apoptosis in human tumors. Thus, targeting of the FAK and p53 interaction, as well as other binding partners, can be important therapeutic targets in cancer treatment programs.
The review of Levine and Oren, summarizing the first 30 years of p53 research (11) proposed the next decade of research will bring novel and important p53-targeted therapies. We hope that the p53 therapy approaches, discussed in this review will create new ideas and help in developing novel therapies targeting p53 signaling pathways in cancer cells.
Key Points.
The P53 gene is a tumor suppressor gene whose genomic sequence contains 20 exons and spans 20 kilobases.
P53 is the most commonly mutated gene in tumors with up to 50% mutations in different types of cancers and 75% mutations in invasive cancers.
Focal adhesion Kinase (FAK) can suppress the transcriptional activity of p53 through its interaction.
Understanding FAK biology during tumorigenesis, the mechanisms of its up-regulation in different tumors, its role in stem cell biology, angiogenesis, motility, and mechanisms of its direct physical interaction with p53 protein and their downstream signaling pathways will be critical in developing targeted therapeutics.
Acknowledgments
DISCLOSURES
Funding source:
NIH, NCI
Footnotes
Conflicts of interest:
Dr. Golubovskaya and Dr. Cance - Co-Founders and stock holders of CureFAKtor Pharmaceuticals, Buffalo, NY
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 2.Farmer G, Bargonetti J, Zhu H, et al. Wild-type p53 activates transcription in vitro. Nature. 1992;358:83–6. doi: 10.1038/358083a0. [DOI] [PubMed] [Google Scholar]
- 3.Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol. 2007;263:103–53. doi: 10.1016/S0074-7696(07)63003-4. [DOI] [PubMed] [Google Scholar]
- 4.Bouchet BP, de Fromentel CC, Puisieux A, et al. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol. 2006;58:190–207. doi: 10.1016/j.critrevonc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Khoury MP, Bourdon JC. The isoforms of the p53 protein. Cold Spring Harb Perspect Biol. 2:a000927. doi: 10.1101/cshperspect.a000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13:293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- 7.Yu H, Chen JK, Feng S, et al. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–45. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 8.Edwards SJ, Hananeia L, Eccles MR, et al. The proline-rich region of mouse p53 influences transactivation and apoptosis but is largely dispensable for these functions. Oncogene. 2003;22:4517–23. doi: 10.1038/sj.onc.1206726. [DOI] [PubMed] [Google Scholar]
- 9.Bergamaschi D, Gasco M, Hiller L, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell. 2003;3:387–402. doi: 10.1016/s1535-6108(03)00079-5. [DOI] [PubMed] [Google Scholar]
- 10.Stoklsa T, Golab J. Prospects for p53-based cancer therapy. Acta Biochim Pol. 2005;52:321–8. [PubMed] [Google Scholar]
- 11.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker SJ, Preisinger AC, Jessup JM, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–22. [PubMed] [Google Scholar]
- 13.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 14.Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12:2973–83. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 15.Hoffman WH, Biade S, Zilfou JT, et al. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Mehew JW, Heckman CA, et al. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001;20:240–51. doi: 10.1038/sj.onc.1204067. [DOI] [PubMed] [Google Scholar]
- 17.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–15. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 18.Krause K, Wasner M, Reinhard W, et al. The tumour suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res. 2000;28:4410–8. doi: 10.1093/nar/28.22.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnsen JI, Aurelio ON, Kwaja Z, et al. p53-mediated negative regulation of stathmin/Op18 expression is associated with G(2)/M cell-cycle arrest. Int J Cancer. 2000;88:685–91. doi: 10.1002/1097-0215(20001201)88:5<685::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 20.Murphy M, Hinman A, Levine AJ. Wild-type p53 negatively regulates the expression of a microtubule-associated protein. Genes Dev. 1996;10:2971–80. doi: 10.1101/gad.10.23.2971. [DOI] [PubMed] [Google Scholar]
- 21.Yonish-Rouach E, Resnitzky D, Lotem J, et al. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–7. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 22.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–42. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 23.Lowe SW, Ruley HE, Jacks T, et al. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 24.Lowe SW, Jacks T, Housman DE, et al. Abrogation of oncogene-associated apoptosis allows transformation of p53-deficient cells. Proc Natl Acad Sci U S A. 1994;91:2026–30. doi: 10.1073/pnas.91.6.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22:9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- 26.Owens LV, Xu L, Craven RJ, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Research. 1995;55:2752–5. [PubMed] [Google Scholar]
- 27.Cance WG, Golubovskaya VM. Focal adhesion kinase versus p53: apoptosis or survival? Sci Signal. 2008;1:pe22. doi: 10.1126/stke.120pe22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cance WG, Harris JE, Iacocca MV, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6:2417–23. [PubMed] [Google Scholar]
- 29.Owens LV, Xu L, Dent GA, et al. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Annals of Surgical Oncology. 1996;3:100–5. doi: 10.1007/BF02409059. [DOI] [PubMed] [Google Scholar]
- 30.Golubovskaya VM. Targeting focal adhesion kinase in cancer-part I. Anticancer Agents. Med Chem. 10:713. doi: 10.2174/187152010794728693. [DOI] [PubMed] [Google Scholar]
- 31.Ilic D, Almeida EA, Schlaepfer DD, et al. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–60. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Lu H, Dazin P, et al. Squamous cell carcinoma cell aggregates escape suspension-induced, p53-mediated anoikis: Fibronectin and integrin (alpha}v mediate survival signals through focal adhesion kinase. J Biol Chem. 2004 doi: 10.1074/jbc.M407953200. [DOI] [PubMed] [Google Scholar]
- 33.Livasy CA, Moore D, Cance WG, et al. Focal Adhesion Kinase Overexpression in Endometrial Neoplasia. Appl Immunohistochem Mol Morphol. 2004;12:342–5. doi: 10.1097/00129039-200412000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites*1. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 2004;1678:111–25. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 35.Wei CL, Wu Q, Vega VB, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 36.Golubovskaya VM, Conway-Dorsey K, Edmiston SN, et al. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int J Cancer. 2009;125:1735–8. doi: 10.1002/ijc.24486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosado P, Lequerica-Fernandez P, Pena I, et al. In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression. Virchows Arch. doi: 10.1007/s00428-012-1283-2. [DOI] [PubMed] [Google Scholar]
- 38.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–25. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Golubovskaya VM, Finch R, Cance WG. Direct Interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005 doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 40.Stewart A, Ham C, Zachary I. The focal adhesion kinase amino-terminal domain localises to nuclei and intercellular junctions in HEK 293 and MDCK cells independently of tyrosine 397 and the carboxy-terminal domain. Biochem Biophys Res Commun. 2002;299:62–73. doi: 10.1016/s0006-291x(02)02547-0. [DOI] [PubMed] [Google Scholar]
- 41.Beviglia L, Golubovskaya V, Xu L, et al. Focal adhesion kinase N-terminus in breast carcinoma cells induces rounding, detachment and apoptosis. Biochem J. 2003;373:201–10. doi: 10.1042/BJ20021846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones G, Stewart G. Nuclear import of N-terminal FAK by activation of the FcepsilonRI receptor in RBL-2H3 cells. Biochem Biophys Res Commun. 2004;314:39–45. doi: 10.1016/j.bbrc.2003.12.055. [DOI] [PubMed] [Google Scholar]
- 43.Lobo M, Zachary I. Nuclear localization and apoptotic regulation of an amino-terminal domain focal adhesion kinase fragment in endothelial cells. Biochem Biophys Res Commun. 2000;276:1068–74. doi: 10.1006/bbrc.2000.3547. [DOI] [PubMed] [Google Scholar]
- 44.Chipuk JE, Green DR. Cytoplasmic p53: Bax and Forward. Cell Cycle. 2004;3:429–31. [PubMed] [Google Scholar]
- 45.Golubovskaya VM, Finch R, Zheng M, et al. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem J. 2008;411:151–60. doi: 10.1042/BJ20071657. [DOI] [PubMed] [Google Scholar]
- 46.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 47.Golubovskaya VM, Cance W. Focal adhesion kinase and p53 signal transduction pathways in cancer. Front Biosci. 15:901–12. doi: 10.2741/3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim ST, Chen XL, Lim Y, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melkoumian ZK, Peng X, Gan B, et al. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 2005;65:6676–84. doi: 10.1158/0008-5472.CAN-04-4142. [DOI] [PubMed] [Google Scholar]
- 50.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006 doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 51.Ho B, Olson G, Figel S, et al. Nanog Increases Focal Adhesion Kinase (FAK) Promoter Activity and Expression and Directly Binds to FAK Protein to Be Phosphorylated. J Biol Chem. 287:18656–73. doi: 10.1074/jbc.M111.322883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–71. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 53.Essmann F, Schulze-Osthoff K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br J Pharmacol. 165:328–44. doi: 10.1111/j.1476-5381.2011.01570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi J, Zheng D. An update on gene therapy in China. Curr Opin Mol Ther. 2009;11:547–53. [PubMed] [Google Scholar]
- 55.Yang ZX, Wang D, Wang G, et al. Clinical study of recombinant adenovirus-p53 combined with fractionated stereotactic radiotherapy for hepatocellular carcinoma. J Cancer Res Clin Oncol. 136:625–30. doi: 10.1007/s00432-009-0701-6. [DOI] [PubMed] [Google Scholar]
- 56.Shi LM, Myers TG, Fan Y, et al. Mining the National Cancer Institute Anticancer Drug Discovery Database: cluster analysis of ellipticine analogs with p53-inverse and central nervous system-selective patterns of activity. Mol Pharmacol. 1998;53:241–51. doi: 10.1124/mol.53.2.241. [DOI] [PubMed] [Google Scholar]
- 57.Sugikawa E, Hosoi T, Yazaki N, et al. Mutant p53 mediated induction of cell cycle arrest and apoptosis at G1 phase by 9-hydroxyellipticine. Anticancer Res. 1999;19:3099–108. [PubMed] [Google Scholar]
- 58.Foster BA, Coffey HA, Morin MJ, et al. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–10. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- 59.Wang W, Takimoto R, Rastinejad F, et al. Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol Cell Biol. 2003;23:2171–81. doi: 10.1128/MCB.23.6.2171-2181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takimoto R, Wang W, Dicker DT, et al. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther. 2002;1:47–55. doi: 10.4161/cbt.1.1.41. [DOI] [PubMed] [Google Scholar]
- 61.Rippin TM, Bykov VJ, Freund SM, et al. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene. 2002;21:2119–29. doi: 10.1038/sj.onc.1205362. [DOI] [PubMed] [Google Scholar]
- 62.Rippin TM, Freund SM, Veprintsev DB, et al. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J Mol Biol. 2002;319:351–8. doi: 10.1016/S0022-2836(02)00326-1. [DOI] [PubMed] [Google Scholar]
- 63.Demma MJ, Wong S, Maxwell E, et al. CP-31398 restores DNA-binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. J Biol Chem. 2004;279:45887–96. doi: 10.1074/jbc.M401854200. [DOI] [PubMed] [Google Scholar]
- 64.Tang X, Zhu Y, Han L, et al. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J Clin Invest. 2007;117:3753–64. doi: 10.1172/JCI32481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El-Deiry WS. Targeting mutant p53 shows promise for sunscreens and skin cancer. J Clin Invest. 2007;117:3658–60. doi: 10.1172/JCI34251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rao CV, Swamy MV, Patlolla JM, et al. Suppression of familial adenomatous polyposis by CP-31398, a TP53 modulator, in APCmin/+ mice. Cancer Res. 2008;68:7670–5. doi: 10.1158/0008-5472.CAN-08-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rao CV, Steele VE, Swamy MV, et al. Inhibition of azoxymethane-induced colorectal cancer by CP-31398, a TP53 modulator, alone or in combination with low doses of celecoxib in male F344 rats. Cancer Res. 2009;69:8175–82. doi: 10.1158/0008-5472.CAN-09-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bykov VJ, Issaeva N, Selivanova G, et al. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 2002;23:2011–8. doi: 10.1093/carcin/23.12.2011. [DOI] [PubMed] [Google Scholar]
- 69.Bykov VJ, Zache N, Stridh H, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene. 2005;24:3484–91. doi: 10.1038/sj.onc.1208419. [DOI] [PubMed] [Google Scholar]
- 70.Zache N, Lambert JM, Wiman KG, et al. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol. 2008;30:411–8. doi: 10.3233/CLO-2008-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Demma M, Maxwell E, Ramos R, et al. SCH529074, a small molecule activator of mutant p53, which binds p53 DNA binding domain (DBD), restores growth-suppressive function to mutant p53 and interrupts HDM2-mediated ubiquitination of wild type p53. J Biol Chem. 285:10198–212. doi: 10.1074/jbc.M109.083469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bykov VJ, Issaeva N, Zache N, et al. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem. 2005;280:30384–91. doi: 10.1074/jbc.M501664200. [DOI] [PubMed] [Google Scholar]
- 73.Zache N, Lambert JM, Rokaeus N, et al. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol Oncol. 2008;2:70–80. doi: 10.1016/j.molonc.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kravchenko JE, Ilyinskaya GV, Komarov PG, et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci U S A. 2008;105:6302–7. doi: 10.1073/pnas.0802091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 21:614–25. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Picksley SM, Vojtesek B, Sparks A, et al. Immunochemical analysis of the interaction of p53 with MDM2;--fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene. 1994;9:2523–9. [PubMed] [Google Scholar]
- 77.Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 78.Freedman DA, Epstein CB, Roth JC, et al. A genetic approach to mapping the p53 binding site in the MDM2 protein. Mol Med. 1997;3:248–59. [PMC free article] [PubMed] [Google Scholar]
- 79.Chi SW, Lee SH, Kim DH, et al. Structural details on mdm2-p53 interaction. J Biol Chem. 2005;280:38795–802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- 80.Galatin PS, Abraham DJ. A nonpeptidic sulfonamide inhibits the p53-mdm2 interaction and activates p53-dependent transcription in mdm2-overexpressing cells. J Med Chem. 2004;47:4163–5. doi: 10.1021/jm034182u. [DOI] [PubMed] [Google Scholar]
- 81.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 82.Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–21. [PubMed] [Google Scholar]
- 83.Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tovar C, Higgins B, Kolinsky K, et al. MDM2 antagonists boost antitumor effect of androgen withdrawal: implications for therapy of prostate cancer. Mol Cancer. 10:49. doi: 10.1186/1476-4598-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barbarotto E, Corallini F, Rimondi E, et al. Differential effects of chemotherapeutic drugs versus the MDM-2 antagonist nutlin-3 on cell cycle progression and induction of apoptosis in SKW6.4 lymphoblastoid B-cells. J Cell Biochem. 2008;104:595–605. doi: 10.1002/jcb.21649. [DOI] [PubMed] [Google Scholar]
- 86.Secchiero P, Zerbinati C, Melloni E, et al. The MDM-2 antagonist nutlin-3 promotes the maturation of acute myeloid leukemic blasts. Neoplasia. 2007;9:853–61. doi: 10.1593/neo.07523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Maerken T, Ferdinande L, Taildeman J, et al. Antitumor activity of the selective MDM2 antagonist nutlin-3 against chemoresistant neuroblastoma with wild-type p53. J Natl Cancer Inst. 2009;101:1562–74. doi: 10.1093/jnci/djp355. [DOI] [PubMed] [Google Scholar]
- 88.Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 89.Saha MN, Jiang H, Mukai A, et al. RITA inhibits multiple myeloma cell growth through induction of p53-mediated caspase-dependent apoptosis and synergistically enhances nutlin-induced cytotoxic responses. Mol Cancer Ther. 9:3041–51. doi: 10.1158/1535-7163.MCT-10-0471. [DOI] [PubMed] [Google Scholar]
- 90.Kazemi A, Safa M, Shahbazi A. RITA enhances chemosensivity of pre-B ALL cells to doxorubicin by inducing p53-dependent apoptosis. Hematology. 16:225–31. doi: 10.1179/102453311X12953015767536. [DOI] [PubMed] [Google Scholar]
- 91.de Lange J, Ly LV, Lodder K, et al. Synergistic growth inhibition based on small-molecule p53 activation as treatment for intraocular melanoma. Oncogene. 31:1105–16. doi: 10.1038/onc.2011.309. [DOI] [PubMed] [Google Scholar]
- 92.Saha MN, Jiang H, Yang Y, et al. Targeting p53 via JNK pathway: a novel role of RITA for apoptotic signaling in multiple myeloma. PLoS One. 7:e30215. doi: 10.1371/journal.pone.0030215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao CY, Szekely L, Bao W, et al. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res. 70:3372–81. doi: 10.1158/0008-5472.CAN-09-2787. [DOI] [PubMed] [Google Scholar]
- 94.Yin H, Lee GI, Park HS, et al. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew Chem Int Ed Engl. 2005;44:2704–7. doi: 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- 95.Grasberger BL, Lu T, Schubert C, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48:909–12. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- 96.Koblish HK, Zhao S, Franks CF, et al. Benzodiazepinedione inhibitors of the Hdm2:p53 complex suppress human tumor cell proliferation in vitro and sensitize tumors to doxorubicin in vivo. Mol Cancer Ther. 2006;5:160–9. doi: 10.1158/1535-7163.MCT-05-0199. [DOI] [PubMed] [Google Scholar]
- 97.Lu Y, Nikolovska-Coleska Z, Fang X, et al. Discovery of a nanomolar inhibitor of the human murine double minute 2 (MDM2)-p53 interaction through an integrated, virtual database screening strategy. J Med Chem. 2006;49:3759–62. doi: 10.1021/jm060023+. [DOI] [PubMed] [Google Scholar]
- 98.Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–5. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 99.Shangary S, Ding K, Qiu S, et al. Reactivation of p53 by a specific MDM2 antagonist (MI-43) leads to p21-mediated cell cycle arrest and selective cell death in colon cancer. Mol Cancer Ther. 2008;7:1533–42. doi: 10.1158/1535-7163.MCT-08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mohammad RM, Wu J, Azmi AS, et al. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009;8:115. doi: 10.1186/1476-4598-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Azmi AS, Aboukameel A, Banerjee S, et al. MDM2 inhibitor MI-319 in combination with cisplatin is an effective treatment for pancreatic cancer independent of p53 function. Eur J Cancer. 46:1122–31. doi: 10.1016/j.ejca.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hardcastle IR, Ahmed SU, Atkins H, et al. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction based on an isoindolinone scaffold. J Med Chem. 2006;49:6209–21. doi: 10.1021/jm0601194. [DOI] [PubMed] [Google Scholar]
- 103.Hardcastle IR, Liu J, Valeur E, et al. Isoindolinone inhibitors of the murine double minute 2 (MDM2)-p53 protein-protein interaction: structure-activity studies leading to improved potency. J Med Chem. 54:1233–43. doi: 10.1021/jm1011929. [DOI] [PubMed] [Google Scholar]
- 104.McLean GW, Carragher NO, Avizienyte E, et al. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 105.Xu LH, Yang X, Craven RJ, et al. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998;9:999–1005. [PubMed] [Google Scholar]
- 106.Smith CS, Golubovskaya VM, Peck E, et al. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005;15:357–62. doi: 10.1097/00008390-200510000-00003. [DOI] [PubMed] [Google Scholar]
- 107.Golubovskaya VM, Zheng M, Zhang L, et al. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer. 2009;9:280. doi: 10.1186/1471-2407-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu LH, Owens LV, Sturge GC, et al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–8. [PubMed] [Google Scholar]
- 109.Slack-Davis JK, Martin KH, Tilghman RW, et al. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 110.Roberts WG, Ung E, Whalen P, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 111.Tanjoni I, Walsh C, Uryu S, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 9:764–77. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Golubovskaya VM, Nyberg C, Zheng M, et al. A Small Molecule Inhibitor, 1,2,4,5-Benzenetetraamine Tetrahydrochloride, Targeting the Y397 Site of Focal Adhesion Kinase Decreases Tumor Growth. J Med Chem. 2008;51:7405–16. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hochwald SN, Nyberg C, Zheng M, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8:2435–43. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beierle EA, Ma X, Stewart J, et al. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 9:1005–15. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Golubovskaya VM, Figel S, Ho BT, et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis. 33:1004–13. doi: 10.1093/carcin/bgs120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kurenova EV, Hunt DL, He D, et al. Small Molecule Chloropyramine Hydrochloride (C4) Targets the Binding Site of Focal Adhesion Kinase and Vascular Endothelial Growth Factor Receptor 3 and Suppresses Breast Cancer Growth in Vivo. J Med Chem. 2009;52 doi: 10.1021/jm900159g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vassilev LT. p53 Activation by small molecules: application in oncology. J Med Chem. 2005;48:4491–9. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]
- 118.Golubovskaya V, Palma NL, Zheng M, et al. A Small-Molecule Inhibitor, 5’-O-Tritylthymidine, Targets FAK And Mdm-2 Interaction, And Blocks Breast And Colon Tumorigenesis In Vivo. Anticancer Agents Med Chem. 2012 doi: 10.2174/1871520611313040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akhter SA, Luttrell LM, Rockman HA, et al. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–7. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 120.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–33. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 121.May MJ, D’Acquisto F, Madge LA, et al. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–4. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 122.van Rooij E, Doevendans PA, de Theije CC, et al. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. 2002;277:48617–26. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]