

Abstract
Following transient forebrain ischemia, astrocytes play a key role in determining whether or not neurons in the hippocampal CA1 sector go on to die in a delayed fashion. MicroRNAs (miRNAs) are a novel class of RNAs that control gene expression at the post-transcriptional level and the miR-29 family is highly expressed in astrocytes. In this study we assessed levels of miR-29 in hippocampus following forebrain ischemia and found that after transient forebrain ischemia and short periods of reperfusion, miR-29a significantly increased in the resistant dentate gyrus, but decreased in the vulnerable CA1 region of the hippocampus. We demonstrate that miR-29a targets BH3-only pro-apoptotic BCL2 family member PUMA by luciferase reporter assay and by Western blot. Comparing primary neuron and astrocyte cultures, and postnatal brain, we verified the strongly astrocytic expression of miR-29a. We further found that miR-29a mimic protects and miR-29a inhibitor aggravates cell injury and mitochondrial function after ischemia-like stresses in vitro. Lastly, by overexpressing and reducing miR-29a we demonstrate the protective effect of miR-29a on CA1 delayed neuronal death after forebrain ischemia. Our data suggest that by targeting a pro-apoptotic BCL2 family member, increasing levels of miR-29a might emerge as a strategy for protection against ischemia-reperfusion injury.
Keywords: microRNA, PUMA, astrocyte
Introduction
Cerebral injury resulting from cardiac arrest and resuscitation (rodent models of global/forebrain ischemia) leads to death and neurological impairment, and has only been effectively treated in the clinical setting with hypothermia (Bernard et al., 2002; THACAS, 2002). Lack of consideration of the role of astrocytes is thought to be a factor in the failure of potential neuroprotective strategies (Nedergaard and Dirnagl, 2005). Our previous study showed that selective dysfunction of hippocampal CA1 astrocytes occurs at early reperfusion times, hours to days before the death of CA1 neurons following forebrain ischemia (Ouyang et al., 1997). We further demonstrated that targeting protective proteins to hippocampal astrocytes improves the survival of CA1 neurons (Xu et al., 2010).
MicroRNAs (miRNAs) are a novel and abundant class of ~22-nucleotide (nt) RNAs that control gene expression at the post-transcriptional level by binding mRNAs. Numerous miRNAs are expressed in a cell-specific manner and miR-29 was found to be enriched in astrocytes (Smirnova et al., 2005). The miR-29 family consists of three members (a, b, and c) and map to two distinct genomic loci in clusters (Fig. 1A): miR-29 a/b-1 on chromosome six and miR-29c/b-2 on chromosome one in mouse. Loss of the miRNA cluster miR-29a/b-1 was reported in Alzheimer’s disease (Hébert et al., 2008; Shioya et al., 2010). Studies profiling changes in miRNAs in cerebral ischemia have reported different effects on miR-29. After focal ischemia the miR-29 family was reported to be downregulated in cortex (Dharap et al., 2009; Jeyaseelan et al., 2008) but after forebrain ischemia the miR-29 family was upregulated in hippocampus (Yuan et al., 2010).
Fig. 1.

A. Schematic representation of the genomic organization of mouse miR-29. The two clusters are on chromosomes 1 and 6. B. Sequences of mature wild type (WT) and seed mutated (SM) miR-29a, miR-29bb and miR-29c. SMs are used as negative controls. C. Vector MWX-PGK-IRES-GFP to express pri-miR-29 and its SM. D. Sequence and alignment of the miR-29-binding sites in the 3′UTR of PUMA. BBC3 has two predicted target sites (from TargetScan): the first (316–322) is less broadly conservative than the second one (401–407). E. Two wild type (BBC3-1 and BBC3-2) and seed mutant (BBC3-1-SM and BBC3-2-SM) in the 3′UTR of PUMA. F. Renilla luciferase reporter vector phRL-TK to express 3′UTRs of PUMA.
In this study, in a forebrain ischemia model in rats, we found that the expression levels of miR-29a increased in the ischemia resistant hippocampal dentate gyrus (DG) area and decreased in the vulnerable CA1 area. We validated that miR-29a targets the BH3-only pro-apoptotic protein PUMA (p53 upregulated modulator of apoptosis). By comparing primary neurons, astrocytes, and postnatal brain we verified the dominant astrocytic expression of miR-29a. By overexpressing and knocking down miR-29a we further confirm the importance of miR-29 for outcome in cerebral ischemia.
Materials and Methods
miRNA, pri-miRNA, antagomir/inhibitor, 3′UTRs and controls
miRNA biogenesis starts with a long primary miRNA transcript (pri-miRNA), which is processed to a ~70-nt precursor miRNA (pre-miRNA), and then to a mature ~22nd miRNA (see Fig. 1 in (Ouyang et al., 2013)). DNA fragments containing the pri-miR-29ab or pri-miR-29c hairpin and ~250–300 nt flanking sequence on each side were cloned downstream of the PGK promoter in MWX-PGK-IRES-GFP to create plasmids for pri-miR-29 overexpression, as described previously (Ouyang et al., 2012a; Ouyang et al., 2012b). The seed sequence (5–7 nt long) in the miRNA determines the specificity of binding to the mRNA. Since pri-miR-29a and pri-miR-29b-1 are very close (Fig. 1A), we made pri-miR-29a/b-1 constructs first and then made different seed mutations (miR-29ab-SM is the double mutant negative control, a-SM contains the native pri-miR-29b sequence and b-SM contains the native pri-miR-29a) to distinguish a and b functions. Wild-type mature miR-29a-c and their seed mutant sequences are listed in Fig. 1B. MWX-PGK-IRES-GFP vector (Fig. 1C) was a kind gift from Dr. Chang-Zheng Chen at Stanford University. miRNA mimics are small, chemically modified, double stranded RNA molecules that load the active strand into the RNA induced silencing complex (RISC) which then binds the target mRNA to induce translational silencing. miRNA inhibitors and antagomirs (which differ in their chemical modifications and intended use in vivo) are modified single-stranded antisense oligonucleotides harboring the full or partial complementary sequence to the mature miRNA, to reduce endogenous levels of the miRNA. Fluorescent-tagged miRNA transfection control, negative control, miR-29a mimic, and inhibitor/antagomir were purchased from Thermo Scientific via Dharmacon (Chicago, IL); catalogue numbers are in Table 1.
Table 1.
Essential materials bought from companies and their catalog numbers
| Name | Catalog # | Company |
|---|---|---|
| miR-29a Primer | 002112 | Applied Biosystems (Foster City, CA, USA) |
| miR-29b Primer | 000413 | |
| miR-29c Primer | 000587 | |
| U6 snRNA Primer | 001973 | |
| miR-29a Mimic | C-310521-07-0005 | Thermo Scientific via Dharmacon (Chicago, IL, USA) |
| miR-29a Inhibitor | IH-310521-08-0005 | |
| miR-29a Antagomir | IH-120642-00-30 | |
| Positive Control | CP-004500-01-05 | |
| Negative Control | CN-001000-01-05 |
Sequence and alignment of the miR-29-binding sites in the 3′UTR of BBC3 are shown in Fig. 1D. The primer sets (5′ to 3′) used to generate specific 3′UTR fragments of BBC3 are: forward: ACTTTTTCTGCACCATGTAGC and reverse: TGTCCTTACAGGTAGTGCCAG. Both wild-type and seed mutant inserts were confirmed by sequencing. The mouse 3′UTRs of BBC3 (Fig. 1E) were cloned into the Renilla luciferase reporter vector phRL-TK (Promega) (Fig. 1F).
Reverse Transcription Quantitative real-time Polymerase Chain Reaction (RT-qPCR) for miRNA quantitation
All materials used for RT-qPCR were from Applied Biosystems (Foster City, CA). Total RNA was isolated with TRIzol®. Reverse transcription was performed using the TaqMan MicroRNA Reverse Transcription Kit. Equal amounts of total RNA (200 ng) were reverse-transcribed with 1.3 mM dNTPs (with dTTP), 50 U reverse transcriptase, 10 U RNase inhibitor, and specific miRNA reverse transcriptase primers (Applied Biosystems) at 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min. PCR reactions were then conducted using the TaqMan® MicroRNA Assay Kit at 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 min. Each reaction contained 0.75 μl of the RT reaction product, 5 μl TaqMan 2×Universal PCR Master Mix in a total volume of 10 μl using the 7900HT. Predesigned primer/probes for miRNAs and mouse U6 were from Applied Biosystems. The expression of miR-29a/b/c was normalized using U6 as the internal control. Measurements were normalized to U6 (ΔCt) and comparisons calculated as the inverse log of the ΔΔCT to give the relative fold change for all miRNA levels (Livak and Schmittgen, 2001). Liu et al have validated U6 as not changing in cerebral ischemia (Liu et al., 2010). The PCR experiments were repeated 3 times, each using separate sets of samples.
Luciferase reporter assay
The luciferase reporter assay was performed as described (Ouyang et al., 2012a; Ouyang et al., 2012b). BOSC23 cells were plated at a density of 1.2–1.5 × 104 cells/well in 96-well plates one day before transfection. Cells were co-transfected with 0.25 ng firefly luciferase control reporter plasmid, 0.05ng Renilla luciferase target reporter, and 40 ng miRNA expression vector using Fugene (Roche) according to the manufacturer’s instructions. At 24 h post-transfection, 100 μl of culture medium was added to each well. Cells were harvested 48 h post-transfection and assayed using the Dual-Luciferase system (E1960, Promega, Sunnyvale, CA). Results were expressed as relative luciferase activity by first normalizing to the firefly luciferase transfection control, then to the Renilla/firefly value of the empty control vector and finally to the corresponding seed mutant reporter control.
Stereotactic infusion and forebrain ischemia
All experimental protocols using animals were performed according to protocols approved by the Stanford University Animal Care and Use Committee and in accordance with the NIH guide for the care and use of laboratory animals. Stereotactic infusion just outside CA1 of the hippocampus was performed in Sprague-Dawley rats as described previously (Sun et al., 2006; Xu et al., 2010). One or two days later depending on whether plasmid pri-miR-29 (2 days) or antagomir (1 day) was used, rats were either sacrificed to assess expression, or subjected to forebrain ischemia.
Forebrain ischemia was induced with the two-vessel occlusion plus hypotension to <40 mm Hg as performed before (Ouyang et al., 2007; Xu et al., 2010). After 10 min bilateral carotid occlusion, recirculation was induced by reinfusing the shed blood and releasing the carotid clamps. After various durations of reperfusion rats were sacrificed and brains perfused with saline, then ice-cold 4% phosphate-buffered paraformaldehyde for histological analysis. Coronal vibratome sections (35 μm) were used for immunohistochemistry or cresyl violet staining to assess injury (Ouyang et al., 2007). For biochemical assays or isolation of RNA or protein, brains were rapidly removed after cold saline perfusion.
Cell cultures and transfection
Primary astrocyte cultures were prepared from postnatal day 1 Swiss Webster mice as described previously (Ouyang et al., 2011). Briefly, neocortices were dissected, treated with trypsin, and plated as a single-cell suspension. BOSC 23 was purchased from American Type Culture Collection (ATCC, Manassas, VA) and grown in a Dulbecco’s Modified Eagle Medium supplemented with 10% FBS and 100 μg/ml penicillin/streptomycin. Primary cultures in 24-well plates were transfected on day 5 in vitro with pri-miR-29 plasmids, mimic or inhibitor of miR-29a, or controls using Lipofectamine from Invitrogen (Foster City, CA) according to the manufacturer’s instructions. Cells were then used in injury experiments on day 6 and day 10 in vitro. For comparison of miR-29 levels, primary neuronal cultures were made as we described before from E16 mouse embryos (Xu et al., 2004). Overexpression or downregulation of miR-29a was confirmed by RT-qPCR.
Injury paradigms and assessment of cell injury
Glucose deprivation (GD) or oxygen glucose deprivation (OGD) was performed as described previously (Ouyang et al., 2011; Ouyang et al., 2006). Identification of damaged cells based on nuclear staining was performed after labeling with Hoechst 33258 dye at 5 mg/ml followed by propidium iodide at 5 mg/ml for 10 minutes at room temperature (Xu et al., 2004). In some experiments, LDH activity was measured to quantitate cell injury as previously described (Ouyang et al., 2006). Medium was sampled at the end of the experiments. Cells were frozen/thawed to provide maximum LDH release values. The percent death (% of LDH release) was calculated by dividing the experimental time point by the maximum values × 100.
Immunoblotting
Immunoblotting of 100 μg protein for astrocyte cultures and 60 μg protein of hippocampal tissue was performed as previously (Ouyang et al., 2007; Ouyang et al., 2011) with PUMA polyclonal antibody (# PA1313 from Insight Genomics, Falls Church, VA, 1:200 dilution) or GFAP monoclonal antibody (# 2555 from Cell Signaling, Danvers, MA, 1:200 dilution). Immunoreactive bands were visualized using the LICOR Odyssey infrared imaging system (Licor Biosciences, Lincoln, NE). Densitometric analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD) and band intensities were normalized to β-actin.
Live cell imaging
Mitochondrial membrane potential (MMP) and reactive oxygen species (ROS) were monitored using tetramethylrhodamine methyl ester (TMRE) and hydroethidine (HEt) respectively, as previously described (Ouyang et al., 2011) using a Zeiss Axiovert 200M fluorescence microscope. Fluorescence changes were quantified (Ouyang et al., 2006) by selecting a cytoplasmic region of each cell that was fluorescent at baseline and normalizing subsequent fluorescence measurements to the basal fluorescence for each cell at the start of the experiment.
Statistics
All data reported represent at least 3 independent experiments for n=3–6 cultures in each experiment; numbers of animals are indicated in figure legends. Data reported are means ± SD. Statistical difference was determined using T-test for comparison of two groups or ANOVA followed by Newman Keuls post test for experiments with >2 groups. P < 0.05 was considered significant.
Results
Forebrain ischemia induces changes of miR-29 levels in hippocampus
To evaluate a possible role for miR-29 in ischemic forebrain injury we first assessed levels of miR-29 in hippocampus following forebrain ischemia, comparing levels in the CA1 and DG regions (Fig. 2A–C). While initial levels were similar in CA1 and DG, there was a significant increase of miR-29a in the DG, while there was a significant decrease in the CA1 region immediately after 10 min forebrain ischemia and these opposite changes increased through 5 h of reperfusion (Fig. 2A). Thus increased miR-29a is associated with survival of transient forebrain ischemia in DG, while decreased levels are associated with delayed neuronal death in CA1 (Ouyang et al., 2007). miR-29b and c did not show this pattern of change, with differences between the regions first apparent at 5 h reperfusion. miR-29b decreased initially in both CA1 and DG, and returned at 5 h reperfusion with significantly higher levels in DG (Fig. 2B). There was no significant change in miR-29c level in CA1 at all reperfusion times tested compared to sham control (Fig. 2C). At 5 h reperfusion miR-29c level decreased in DG significantly compared to CA1 or sham control.
Fig. 2. Levels of miR-29 and distribution of PUMA in hippocampus after 10 min forebrain ischemia.

By RT-qPCR levels of miR-29a (A), miR-29b (B) and miR-29c (C) were assessed in hippocampal DG and CA1 area after 10 min forebrain ischemia and 0 to 5 h reperfusion. Hippo: hippocampus. N=4–6 animals in each group. *P<0.05 compared to DG group at the same time point. D. Sections from rat brain fixed 1 h after sham or 10 min forebrain ischemia, and double labeled for PUMA (green) and the astrocyte marker GFAP (red). GFAP co-labeled for PUMA shows yellow/orange color in merged panel. The CA1 pyramidal neuron cell layer is indicated by CA1- on the left side of the micrographs.
Forebrain ischemia induces changes of PUMA levels in CA1 astrocytes
From computational miRNA target prediction algorithms, as detailed at TargetScan (http://targetscan.org), we found that miR-29 could potentially target the 3′UTR of the mRNA for BBC3 (protein name PUMA) in an evolutionarily conserved way with two predicted target sites (Fig. 1D): the first is less broadly conserved than the second one. We therefore checked PUMA levels in the CA1 region after forebrain ischemia. Brain sections collected 1 h after 10 min forebrain ischemia or sham were double immunostained using antibodies to PUMA and GFAP, an astrocyte marker. Little PUMA (green) was detected in hippocampus from sham treated animals (Fig. 2D), but by 1 h reperfusion, PUMA increased and colocalized with GFAP (red)- the merged image shows yellow/orange astrocytes (Fig. 2D). At 1 h reperfusion the CA1 neuronal cell body layer (indicated as CA1- in the figure) also shows increased PUMA expression compared to sham control. This is consistent with prior reports of PUMA increase in neurons after global ischemia (Niizuma et al., 2009).
miR-29 directly targets the BH3-only proapoptotic protein PUMA
The luciferase assay was used to directly test whether miR-29 recognizes the 3′UTR of PUMA. We cotransfected cells with luciferase control reporter, luciferase target reporter containing the wild type 3′UTR of BBC3 (PUMA) and miRNA (pri-miR-29 or pri-miR-29 mutant). Pri-miR-29ab (WT) represses expression from these 3′UTRs ~ 60% compared to vector controls (Fig. 3A). Seed mutation of both sites in pri-miR-29ab (ab-SM) and miR-29a (a-SM) but not miR-29b (b-SM), de-repressed the targets indicating that within pri-miR-29ab, miR-29a is the effective miRNA for BBC3. Compared to vector controls, pri-miR-29c (c-WT) is also able to repress these 3′UTRs significantly. However, seed mutation of pri-miR-29c did not de-repress the target indicating that the repression is not specific.
Fig. 3.
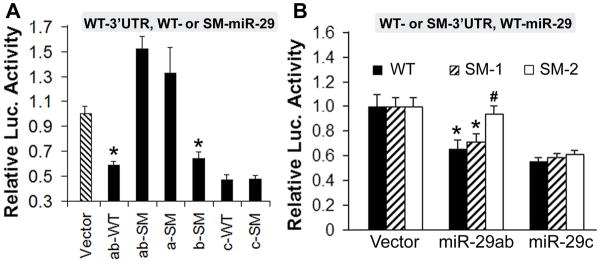
miR-29 targets PUMA. A. Dual luciferase activity assays using co-transfection with wild type 3′UTR of BBC3 and pri-miR-29 (WT) or pri-miR-29 mutant (SM). B. Dual luciferase activity assays using co-transfection with miR-29 and the wild type 3′UTR (WT) of BBC3, and its first seed mutant (SM-1) or second seed mutant (SM-2) as well as wild type pri-miR-29. Assays were performed 3 times in triplicate. P<0.01 compared to the *vector control group or to the #3′UTR-WT group.
To further exclude off-target effects and distinguish which site in the BBC3 3′UTR is effective we performed the complementary experiment of mutating the seed sequences within the 3′UTR of BBC3 as shown in Fig. 1D, E. We generated mutant 3′UTRs at each of the two potential sites with 6 base substitutions, shown underlined in Fig. 1E, to distinguish site 1 and site 2 functions. This mutation abrogated repression of luciferase expression by pri-miR-29ab and pri-miR-29c. This time we cotransfected cells with luciferase control reporter, luciferase target reporter containing the wild type 3′UTR of BBC3 (WT) or its first seed mutant (SM-1) or second seed mutant (SM-2), and wild type pri-miR-29. While pri-miR-29ab reduced luciferase activity from the construct containing both WT sequences and SM-1 compared to vector control group, SM-2 blocked this reduction showing that the second site is the active binding site (Fig. 3B). Pri-miR-29c reduced luciferase activity in 3′UTR-WT, -SM1 and -SM2 groups, with no difference between the 3 groups, thus BBC3 is not a genuine target of miR-29c (Fig. 3B). In summary, within pri-miR-29ab, miR-29a is the effective miRNA for BBC3 and of the two potential target sites within the BBC3 3′UTR only the second site is the target for miR-29a.
miR-29a is dominant in hippocampus
We quantitated levels of miR-29a, b, and c in hippocampus (Fig. 4A). miR-29 is strongly expressed, with miR-29a expressed at levels 10–50 times higher than miR-29c or b. We thus focused on miR-29a for the rest of this study for three reasons: 1) of the 3 miR-29 family members it is most highly expressed in hippocampus, 2) changes in miR-29a correlated best with survival or death, that is opposite changes in DG compared to CA1 and 3) miR-29a effectively targets the 3′UTR of PUMA, which increased when miR-29a decreased in CA1 following transient forebrain ischemia.
Fig. 4.
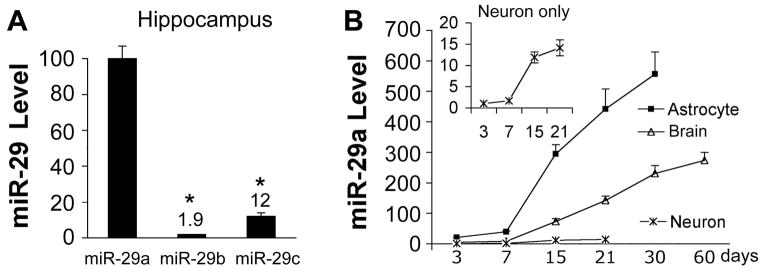
miR-29a is dominant in hippocampus and enriched in astrocytes. A. Relative levels of miR-29a, b, and c in normal rat hippocampus. N=4–6/group. B. Relative miR-29a levels in primary cultures of cortical neurons and astrocytes from 3 to 30 days in vitro, and postnatal day 3 to 60 brain cortex. All experiments were performed 3 times in triplicate. All values were normalized to neuronal miR-29a level at 3 days.
miR-29a is highly expressed in astrocytes
To investigate the cell type specificity of miR-29a expression, we prepared RNA from primary cultures of neurons or astrocytes after 3, 7, 15, 21 and 30 days in vitro, and from brains of 3, 7, 15, 21, 30 and 60 day old mice. While miR-29a increased in brain, astrocytes, and neurons with development, at each time point levels of miR-29 in cultured astrocytes were 20–40 times higher than in cultured neurons, and levels in brain tissue were about 1/2 that seen in cultured astrocytes (Fig. 4B). This suggests that the increase of miR-29a in the brain with development may largely reflect increases in astrocyte miR-29a.
miR-29a protects astrocytes from ischemia-like injury in vitro
In our preliminary experiments we transfected primary cortical astrocytes with pri-miR-29a plasmid or controls (vector or seed mutant). After 24 h we observed 50–80% transfection efficiency, and a 2–3 fold increase in miR-29a level (data not shown). Since transfection with plasmid was more variable than with mimic we repeated the experiments using miR-29a mimic and inhibitor. Transfection with high amounts of miRNA (50 pmol mimic or inhibitor) was toxic to astrocytes, so we established the dose-response to miR-29a mimic and inhibitor and found that 30 pmol mimic or inhibitor significantly increased or decreased miR-29a without overt toxicity (Fig. 5A and B); this amount was therefore used for subsequent experiments.
Fig. 5. Effect of miR-29a on in vitro ischemia in astrocytes.

A, B. Dose-response of miR-29a levels to transfection with increasing amounts of miR-29a mimic (A) or inhibitor (B) in primary cultures of astrocytes, relative to control (Ctrl=1). Arrows indicate the dose we used for the following experiments. C. Representative immunoblots show PUMA protein levels in primary astrocytes without stress or after 6 h GD after transfection with control, mimic, or inhibitor. D. The graph shows the quantification of the Western blots. E. Effect of miR-29a mimic or inhibitor on astrocyte injury induced by 24 h glucose deprivation (GD). F. Effect of miR-29a mimic or inhibitor on cell injury induced by 7.5 h oxygen glucose deprivation (OGD) in primary astrocyte cultures. G. Effect of miR-29a mimic and inhibitor on mitochondrial membrane depolarization in astrocytes subjected to 3 h GD. Depolarization is indicated by decreased TMRE fluorescence. H. Effect of miR-29a mimic or inhibitor on ROS in astrocytes subjected to 3 h GD. Increasing HEt fluorescence indicates increasing ROS. Fluorescence values are normalized to the starting fluorescence = 1.0. Ctrl: transfection control. All experiments were performed 3 times in triplicate. *P<0.01 compared to Ctrl.
To test if miR-29a targets PUMA in astrocytes we measured protein levels of PUMA by Western blot after transfection of primary astrocytes with either mimic or inhibitor. PUMA protein is very low in non-stressed control astrocytes (Fig. 5C, non-stress), so we stressed the cells with 6 h glucose deprivation (GD). PUMA protein increased significantly in control astrocytes after 6 h GD (Fig. 5C, 6 h GD), and miR-29a mimic decreased and miR-29a inhibitor increased PUMA protein levels significantly when compared to the corresponding control (Fig. 5D).
To see if altering miR-29a levels influences astrocyte injury, levels of miR-29a were increased or decreased in primary astrocytes by transfection with mimic or inhibitor. Increasing miR-29a in cultured astrocytes decreased GD cell injury, while inhibitor increased the injury (Fig. 5E). To confirm and extend these observations we used a second injury paradigm combined oxygen glucose deprivation (OGD) followed by restoration of oxygen and glucose to better mimic ischemia and reperfusion. Astrocytes exposed to 7.5 h OGD suffered about 70% cell death when assayed at 24 h; mimic decreased and inhibitor increased injury significantly (Fig. 5F).
miR-29 protects mitochondrial function in astrocytes
To determine how miR-29a functions to inhibit apoptosis we tested whether miR-29 could influence mitochondrial membrane potential (MMP) and reactive oxygen species (ROS) generation in response to stress. We previously described the time course of MMP decrease and ROS increase in astrocytes subjected to GD (Ouyang et al., 2002; Ouyang et al., 2007; Ouyang et al., 2006). Primary astrocytes were transfected with miR-29a mimic or inhibitor and subjected to GD. There were no apparent changes in MMP or ROS when astrocytes were transfected with miR-29a mimic, inhibitor, or SM controls under normal growth conditions. At 3 h GD astrocyte MMP was < 60% of starting values in the GD control group; miR-29a mimic reduced this decrease while inhibitor led to greater reduction of MMP (Fig. 5G). Upon exposure to GD control astrocytes showed an increase in ROS accumulation, reaching approximately 8 times the initial fluorescence level at 3 h of GD; miR-29a mimic reduced while inhibitor led to a greater increase in ROS with GD (Fig. 5H).
miR-29a attenuates hippocampal CA1 injury after forebrain ischemia
To further confirm the biological role of miR-29a we stereotactically injected either pri-miR-29a plasmid or miR-29a antagomir outside the right hippocampus in rat. Two days after administration of pri-miR-29a, we observed about a 2.5 fold increase in miR-29a level compared to the seed mutant control (Fig. 6A). One day after administration of the antagomir miR-29a levels were reduced to 70% of the control (Fig. 6B) in the hippocampus. As shown above in vitro, we also found reciprocal changes in expression of miR-29a and PUMA in hippocampus after pri-miR-29ab or antagomir injection (Fig. 6C–D). While levels of PUMA were very low in non-stressed control astrocytes, normal control hippocampus showed readily detectable PUMA protein levels (Fig. 6C–D). This may be because astrocytes in culture differ from astrocytes in vivo (Fig. 4B) but may also reflect expression of PUMA in other cell types in the hippocampus. The 2.5 fold increase in miR-29a reduced PUMA levels about 40% (Fig. 6A, C). This is consistent with the extent of reduction of protein often seen after increasing a miRNA.
Fig. 6.

Effect of miR-29a on CA1 delayed neuronal damage after transient forebrain ischemia. A, B. Hippocampal levels of miR-29a in rats treated with pri-miR-29a plasmid (A) or miR-29a antagomir (B). N=6/group, *P<0.01 compared to seed mutant (SM) or Ctrl group. C, D. Western blots show PUMA protein levels in the hippocampus of rats pretreated with pri-miR-29a plasmid (C), or miR-29a antagomir (D). Representative immunoblots are shown above the graphs. N=6 rats in each group. *P<0.01 compared to SM or Ctrl group. E. Representative cresyl violet-stained coronal sections demonstrate selective loss of CA1 hippocampal neurons (between white arrows) at 6 days reperfusion after 10 min forebrain ischemia. F. Loss of CA1 neurons was quantified by cresyl violet staining density. N=12/group, P<0.01 compared to sham (*) or to ischemic control/antagomir group (#).
The effect of altering miR-29a levels on delayed neuronal death was assessed 6 days after 10 min forebrain ischemia. Photomicrographs in Fig. 6E show that selective loss of CA1 neurons was markedly reduced in the pri-miR-29a injected brain, while antagomir resulted in extension of the injury to include CA2-4. Quantitation of CA1 survival in Fig. 6F demonstrates that overexpression of miR-29a significantly attenuated hippocampal CA1 injury. Antagomir injected brain showed a trend to even further reduced CA1 survival compared to ischemic control, but this did not reach statistical significance. However, as noted above, neuronal loss extended to include CA2-4 with antagomir (Fig. 6E).
Discussion
PUMA (Han et al., 2001; Nakano and Vousden, 2001; Yu et al., 2001) is one of the most potent killers among the BCL-2 homology 3 (BH3)-only subgroup of BCL-2 family members. PUMA mainly localizes to mitochondria and is a key mediator of p53-dependent and p53-independent apoptosis (for a recent review, see Hikisz and Kiliańska (2012)). It binds and antagonizes all known anti-apoptotic BCL-2 family members and activates two key multidomain pro-apoptotic BCL-2 family proteins, BAX and BAK. This leads to permeabilization of the mitochondrial membrane, leading to mitochondrial dysfunction and caspase activation (Yu and Zhang, 2008). PUMA also drives apoptosis induced by p53-independent signals, such as growth factor deprivation or exposure to glucocorticoids or phorbol ester (Jeffers et al., 2003). Selective CA1 injury, which is mainly apoptotic, was strongly reduced in PUMA knockout mice (Bonner et al., 2010; Tsuchiya et al., 2011). PUMA was previously shown to be upregulated in CA1 neurons after transient global cerebral ischemia and inhibiting PUMA upregulation protected CA1 neurons from delayed ischemic death (Niizuma et al., 2009). The study by Niizuma and colleagues focused on the role of PUMA in neurons following global ischemia, but did not look at astrocytes. This is the first observation of changes in PUMA in astrocytes after forebrain ischemia (Fig. 2D).
Little is known about the post-transcriptional and post-translational regulation of the pro-apoptotic activity of PUMA. One group (Fricker et al., 2010) reported that this protein is subject to phosphorylation. We observed that miR-29a targets PUMA mRNA in cultured astrocytes in vitro and hippocampal tissues in vivo. A recent study published while this work was in progress found that miR-29b is activated during neuronal maturation and targets several BH3-only genes including PUMA to reduce apoptosis in maturing sympathetic neurons (Kole et al., 2011). Kole and colleagues (Kole et al., 2011) used mature miR-29b mimic in their luciferase assays, while we used a primary miR-29ab construct, which has to be processed by the cell to mature miR-29a and b. This may account for the difference in results we obtained for miR-29b. We observed that within pri-miR-29ab, miR-29a is the effective endogenous component for targeting PUMA. We further demonstrated that miR-29a protects mitochondrial function and reduces cell injury in astrocytes, in concert with reducing levels of PUMA. Our study showed that miR-29c does not target PUMA (Fig. 3B). A recent study showed that treatment with miR-29c, which was down-regulated after focal ischemia and after OGD in PC12 cells, reduced infarct volume and neuronal death by targeting DNA methyltransferase 3a (Pandi et al., 2013). Interestingly, increasing miR-29b had the effect of promoting neuronal cell death in focal ischemia by inhibiting BCL2L2 (protein BCL-w), an anti-apoptotic member of the BCL2 protein family (Shi et al., 2012). The role of different miR-29 family members in different tissues and under different stress conditions deserves further study.
Loss of the miRNA cluster miR-29a/b-1 was reported in sporadic Alzheimer’s disease (Hébert et al., 2008; Shioya et al., 2010), and this work identified β site APP-cleaving enzyme 1 (BACE1) as a target of miR-29. Our results suggest that decreased levels of miR-29a expression may leave neurons more vulnerable to neurodegeneration, and may affect the ability of astrocytes to protect neurons, emphasizing the importance of miR-29 for neuronal survival by at least two mechanisms.
In this study we also confirm higher levels of miR-29 expression in astrocytes. Prior work demonstrated that miR-29a/b-1 is developmentally regulated in mouse brain with the highest expression observed in adults (Hébert et al., 2008; Kole et al., 2011), and higher in astrocytes compared to neurons (Smirnova et al., 2005).
Astrocytes are the most abundant glial cells within the mammalian brain. They support neurons by providing antioxidant protection, and are metabolically coupled to neurons, as well as influencing their synaptic function; they also release “gliotransmitters” and participate in intercellular communication (Giaume et al., 2007; Volterra and Meldolesi, 2005). Inhibition of astrocyte mitochondrial function (Voloboueva et al., 2007) or astrocyte glutamate uptake (Dugan et al., 1995; Rosenberg and Aizenman, 1989) impairs neuronal survival after excitotoxic injury. We previously reported that selective dysfunction of hippocampal CA1 astrocytes occurs at early reperfusion times, long before CA1 neurons die (Ouyang et al., 2007) and that targeting protective proteins to hippocampal astrocytes markedly improves the survival of CA1 neurons (Xu et al., 2010). In our in vivo study whether the overexpressed miR-29a protects only astrocytes and then indirectly neurons, or protects both astrocytes and neurons directly needs to be further studied. Since increased PUMA leads to mitochondrial impairment and increased oxidative stress, this leads to the likelihood that reduced PUMA due to increasing miR-29a could lower oxidative stress and thereby preserve astrocyte glutamate transporter 1 (GLT1) leading to better survival of neurons. This could also indirectly lead to reduced PUMA levels in the neurons if the astrocytes are better able to protect them.
The major finding of this report is that a miRNA (miR-29) that is highly expressed in astrocytes and targets the BH3-only protein PUMA regulates outcome from ischemic injury in vitro and in vivo. This work suggests that miRNAs constitute a potential new approach to reduce ischemic brain injury.
Acknowledgments
Supported by NIH grants GM049831, NS053898, and NS080177 to RGG. The authors would like to thank William Magruder for help preparing the manuscript.
Footnotes
The authors have no conflicting financial interests.
References
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Bonner HP, Concannon CG, Bonner C, Woods I, Ward MW, Prehn JHM. Differential expression patterns of Puma and Hsp70 following proteasomal stress in the hippocampus are key determinants of neuronal vulnerability. J Neurochem. 2010;114:606–616. doi: 10.1111/j.1471-4159.2010.06790.x. [DOI] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29:675–87. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan L, Bruno V, Amagasu S, Giffard R. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, O’Prey J, Tolkovsky AM, Ryan KM. Phosphorylation of Puma modulates its apoptotic function by regulating protein stability. Cell Death Dis. 2010;1:e59. doi: 10.1038/cddis.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Kirchhoff F, Matute C, Reichenbach A, Verkhratsky A. Glia: the fulcrum of brain diseases. Cell Death Differ. 2007;14:1324–1335. doi: 10.1038/sj.cdd.4402144. [DOI] [PubMed] [Google Scholar]
- Han J-w, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L, Chittenden T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Nat Acad Sci. 2001;98:11318–11323. doi: 10.1073/pnas.201208798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/β-secretase expression. Proc Nat Acad Sci. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikisz P, Kiliańska Z. Puma, a critical mediator of cell death — one decade on from its discovery. Cell Mol Biol Lett. 2012;17:646–669. doi: 10.2478/s11658-012-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jeyaseelan K, Lim KY, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke. 2008;39:959–66. doi: 10.1161/STROKEAHA.107.500736. [DOI] [PubMed] [Google Scholar]
- Kole AJ, Swahari V, Hammond SM, Deshmukh M. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev. 2011;25:125–130. doi: 10.1101/gad.1975411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DZ, Tian Y, Ander BP, Xu H, Stamova BS, Zhan X, Turner RJ, Jickling G, Sharp FR. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30:92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–286. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential Role of PUMA in Delayed Death of Hippocampal CA1 Neurons After Transient Global Cerebral Ischemia. Stroke. 2009;40:618–625. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Carriedo SG, Giffard RG. Effect of Bcl-XL overexpression on reactive oxygen species, intracellular calcium, and mitochondrial membrane potential following injury in astrocytes. Free Radical Biol Med. 2002;33:544–551. doi: 10.1016/s0891-5849(02)00912-7. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Kuroda S, Kristián T, Siesjö BK. Release of mitochondrial aspartate aminotransferase (mAST) following transient focal cerebral ischemia suggests the opening of a mitochondrial permeability transition pore. Neurosci Res Comm. 1997;20:167–173. [Google Scholar]
- Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE, Sun X, Giffard RG. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis. 2012a;45:555–563. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Stary C, Yang GY, Giffard R. microRNAs: innovative targets for cerebral ischemia and stroke. Current Drug Targets. 2013;14:90–101. doi: 10.2174/138945013804806424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012b;12:213–219. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–60. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. Overexpressing GRP78 influences Ca(2+) handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. 2011;11:279–86. doi: 10.1016/j.mito.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Sun YJ, Giffard RG. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chap. 2006;11:180–6. doi: 10.1379/CSC-182R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandi G, Nakka VP, Dharap A, Roopra A, Vemuganti R. MicroRNA miR-29c Down-Regulation Leading to De-Repression of Its Target DNA Methyltransferase 3a Promotes Ischemic Brain Damage. PLoS ONE. 2013;8:e58039. doi: 10.1371/journal.pone.0058039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Shi G, Liu Y, Liu T, Yan W, Liu X, Wang Y, Shi J, Jia L. Upregulated miR-29b promotes neuronal cell death by inhibiting Bcl2L2 after ischemic brain injury. Experimental Brain Research. 2012;216:225–230. doi: 10.1007/s00221-011-2925-3. [DOI] [PubMed] [Google Scholar]
- Shioya M, Obayashi S, Tabunoki H, Arima K, Saito Y, Ishida T, Satoh J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol Appl Neurobiol. 2010;36:320–30. doi: 10.1111/j.1365-2990.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- Smirnova L, Gräfe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005;21:1469–1477. doi: 10.1111/j.1460-9568.2005.03978.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ouyang Y-B, Xu L, Chow AM-Y, Anderson R, Hecker JG, Giffard RG. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2006;26:937–950. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- THACAS. Mild Therapeutic Hypothermia to Improve the Neurologic Outcome after Cardiac Arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Bonner HP, Engel T, Woods I, Matsushima S, Ward MW, Taki W, Henshall DC, Concannon CG, Prehn JHM. Bcl-2 homology domain 3-only proteins Puma and Bim mediate the vulnerability of CA1 hippocampal neurons to proteasome inhibition in vivo. Eur J Neurosci. 2011;33:401–408. doi: 10.1111/j.1460-9568.2010.07538.x. [DOI] [PubMed] [Google Scholar]
- Voloboueva L, Suh S, Swanson R, Giffard R. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Xu L, Chock V, Yang EY, Giffard RG. Susceptibility to apoptosis varies with time in culture for murine neurons and astrocytes: changes in gene expression and activity. Neurol Res. 2004;26:632–643. doi: 10.1179/016164104225017587. [DOI] [PubMed] [Google Scholar]
- Xu L, Emery JF, Ouyang YB, Voloboueva LA, Giffard RG. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia. 2010;58:1042–1049. doi: 10.1002/glia.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27:S71–S83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA Induces the Rapid Apoptosis of Colorectal Cancer Cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Wang JY, Xu LY, Cai R, Chen Z, Luo BY. MicroRNA expression changes in the hippocampi of rats subjected to global ischemia. J Clin Neurosci. 2010;17:774–8. doi: 10.1016/j.jocn.2009.10.009. [DOI] [PubMed] [Google Scholar]