

Abstract
Intein-mediated expressed protein ligation (EPL) permits the site-specific chemical customization of proteins. While traditional techniques have used purified, soluble proteins, we have extended these methods to release and modify intein fusion proteins expressed on the yeast surface, thereby eliminating the need for soluble protein expression and purification. To this end, we sought to simultaneously release yeast surface-displayed proteins and selectively conjugate with chemical functionalities compatible with EPL and click chemistry. Single-chain antibodies (scFv) and green fluorescent protein (GFP) were displayed on the yeast surface as fusions to the N-terminus of the Mxe GyrA intein. ScFv and GFP were released from the yeast surface with either a sulfur nucleophile (MESNA) or a nitrogen nucleophile (hydrazine) linked to an azido group. The hydrazine azide permitted the simultaneous release and azido functionalization of displayed proteins, but nonspecific reactions with other yeast proteins were detected, and cleavage efficiency was limited. In contrast, MESNA released significantly more protein from the yeast surface while also generating a unique thioester at the carboxy-terminus of the released protein. These protein thioesters were subsequently reacted with a cysteine alkyne in an EPL reaction and then employed in an azide–alkyne cycloaddition to immobilize the scFv and GFP on an azide-decorated surface with >90% site-specificity. Importantly, the immobilized proteins retained their activity. Since yeast surface display is also a protein engineering platform, these approaches provide a particularly powerful tool for the rapid assessment of engineered proteins.
INTRODUCTION
The capability to append unique chemical functionalities to proteins is valuable for a variety of applications, including protein immobilization,1–6 therapeutic drug delivery,7–9 and imaging.10,11 Inserting these chemical functionalities into proteins in a site-specific manner is often preferred over nonspecific modifications to provide uniformly modified protein populations while minimizing potential deleterious effects on native protein activity.4,7,12–14 One approach that has been employed for site-specific protein modification is the reaction of a non-self cleaving intein fusion protein with nucleophiles possessing desired chemical properties.2,5,6,15–17 Non self-cleaving inteins have been engineered to block native protein splicing activity, enabling an exogenous nucleophile to catalyze protein release. Typically, a sulfur nucleophile is used to cleave the target protein from the intein moiety, forming a thioester intermediate at the C-terminus of the target protein.18,19 The thioester can subsequently be reacted with an N-terminal cysteine or cysteine derivative to form a native amide bond.20 Using this method of expressed protein ligation (EPL), functional groups such as tyrosine analogs,21 biotin,5,6,20 azides, and alkynes6 have been installed at the C-terminus of proteins. This traditional two-step approach was reduced to a single reaction step by employing a nitrogen nucleophile bearing an azido group, hydrazine azide, that could cleave the intein while installing a C-terminal azide on the protein, obviating the need to form the thioester intermediate.2,17
Such intein-mediated chemical functionalization strategies are typically performed at a preparative scale with soluble, purified proteins.2,17,18,20–24 It would, however, be desirable to integrate these modification strategies into a protein engineering platform, such as yeast surface display, where multiple clones could be evaluated rapidly on a small scale and in a high-throughput manner. In this way, intein-mediated release of engineered proteins from display platforms would permit the downstream analysis of many clones as soluble proteins without tedious preparation steps such as subcloning, expression, and purification. Furthermore, the ability to simultaneously append unique chemical functionalities to the C-terminus of the protein could allow for rapid integration into a variety of protein-based assays such as immobilization, imaging, and drug conjugation.
In this study, we have extended intein-mediated chemical functionalization approaches to proteins displayed on the surface of yeast. Intein fusion proteins displayed as active proteins on the yeast surface were released and subsequently chemically functionalized to be compatible with EPL and click chemistry. The released and functionalized proteins were used directly for immobilization on surfaces without any protein purification steps, and immobilized antibodies demonstrated specific capture of their target antigens.
MATERIALS AND METHODS
Yeast Strains and plasmids
Yeast surface display was performed using Saccharomyces cerevisiae strain EBY100 25 (MATa AGA1::GAL1-AGA1::URA3 ura3–52 trp1 leu2Δ1 his3 200 pep4::HIS3 prb1Δ1.6R can1 GAL). The pCT4Re yeast surface display vector was created by insertion of the constructs shown in Figure 1a into the pCT-302 yeast surface display vector25 between the GAL1-10 promoter and the alpha factor terminator sequences using the restriction sites EcoRI and XhoI. The Mxe GyrA intein sequence was subcloned from the pTXB1 vector (New England Biolabs). The anti-fluorescein single-chain antibody (scFv) (4-4-20) was subcloned from the pCT-302 vector,25 creating pCT4Re-4420, and the yeast enhanced green fluorescent protein (GFP) was subcloned from the pCT-GFP plasmid,26 creating pCT4Re-GFP. The anti-epidermal growth factor receptor (EGFR) scFv, scFv2, was subcloned from the pCDNA3.1-scFv2 plasmid generously donated by Dr. Winfried Wels27 to create pCT4Re-scFv2. An N-linked glycosylation site that appears in the scFv2 amino acid sequence (N-x-T) was altered to prevent N-linked glycosylation by changing the asparagine residue to a serine (S-x-T) with the Quikchange II Site-Directed Mutagenesis Kit (Agilent). Yeast were transformed using the LiAc/ssDNA/PEG method28 and transformants were selected on tryptophan and uracil deficient SD-CAA agar plates (20.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids, 10.19 g/L Na2HPO4.7H2O, 8.56 g/L NaH2HPO4.H2O, 15 g/L agar).
Figure 1.

Expression and activity of yeast surface-displayed intein-fusion proteins. (a) Yeast surface display construct pCT4Re anchors the C-terminus of the Aga2p-protein fusions (shown here as Aga2p-scFv fusions) to the surface via disulfide bonds. A FLAG epitope tag is expressed on the N-terminus of the construct to represent full-length expression. In the intein-containing construct, the Mxe GyrA intein is expressed on the C-teminus of the target protein. The non-intein construct is identical except it contains no intein. (b) Using flow cytometry, expression of target proteins in the pCT4Re construct (black line in the histograms) and the pCT4Re construct with intein (solid gray in the histograms) was detected with a FLAG-tag antibody (shown for scFv2). These expression levels corresponded to ~24,000 4-4-20-intein constructs, ~48,000 scFv2-intein constructs, and ~86,000 GFP-intein constructs per yeast. Activity of the proteins in the pcT4Re (black line in the histograms) and the pcT4Re with intein (solid gray in the histograms) constructs was also evaluated by detecting binding to the scFv antigens at saturating concentrations or by measuring GFP fluorescence (shown for scFv2). (c) The geometric mean fluorescence of the FLAG-positive yeast populations was quantified to determine relative expression levels, and these values were normalized to the pCT4Re construct without intein for each protein. Activity per molecule was determined by ratio of the geometric means for activity (binding or fluorescence) to FLAG expression levels, and normalized to the pCT4Re construct without intein. Plotted are the mean ± SD for yeast display results from three independent yeast transformants.
Yeast growth and surface display induction
Yeast cells were grown in 50 mL SD-CAA medium (20.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids, 10.19 g/L Na2HPO4.7H2O, 8.56 g/L NaH2PO4.H2O) overnight at 30°C, 260 rpm. The following day, cultures were reset to an optical density at 600 nm (OD600) of 0.3 and grown for ~4 h in SD-CAA until a culture density OD600=1.0 was reached. Surface display was induced by replacing the media with 50 mL SG-CAA (20.0 g/L galactose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids, 10.19 g/L Na2HPO4.7H2O, 8.56 g/L NaH2PO4.H2O) for 20 h at 20°C, 260 rpm. Following induction, an OD600 of ~2–3 was typically reached, corresponding to a total culture of ~1×109–1.5×109 yeast. The medium was removed, and the yeast were washed in PBS containing 0.1% w/v bovine serum albumin (BSA) (PBS-BSA).
Flow cytometry
The following immunolabeling steps for flow cytometry analysis were carried out at 4°C using 2×106 yeast per labeling experiment. Surface display expression levels were evaluated by incubating induced yeast with an anti-FLAG rabbit polyclonal antibody (Sigma–Aldrich, diluted 1:500 in PBS-BSA) for 30 min, washing once with PBS-BSA, and secondary labeling with anti-rabbit allophycocyanin (APC) (Invitrogen, diluted 1:500 in PBS-BSA) for 30 min followed by a final wash with PBS-BSA. To evaluate the binding of 4-4-20 to fluorescein, yeast were incubated with 10 μM fluorescein isothiocyanate-functionalized dextran in PBS-BSA (FITC-dextran, Sigma–Aldrich) for 30 min and washed once with PBS-BSA to remove non-specifically bound antigen. To evaluate the binding of scFv2 to EGFR, yeast were incubated with purified human EGFR (4 μg/mL in PBS-BSA, purified human EGFR was generously donated by Greg Wiepz, Biomolecular Chemistry, University of Wisconsin-Madison) for 1 h, followed by a PBS-BSA wash to remove unbound protein. Yeast were subsequently incubated with anti-EGFR mouse antibody cocktail Ab-12 (Lab Vision Corporation, diluted 1:200 in PBS-BSA) for 30 min, washed once with PBS-BSA, and labeled with anti-mouse PE (Sigma–Aldrich, diluted 1:40 in PBS-BSA) for 30 minutes followed by a final wash with PBS-BSA. GFP activity was assessed by measuring the GFP fluorescence of the yeast at 488 nm excitation. The fluorescence of the immunolabeled yeast cells was measured using a FACSCalibur flow cytometer (Becton Dickinson), and the geometric mean fluorescence intensities of the protein expressing populations were quantified with the FlowJo software package to determine relative display levels and activity.
For absolute quantitation of surface expression levels, Quantum Simply Cellular™ anti-mouse IgG microbeads (Bangs Laboratories) were used. Yeast displaying the scFv-intein and GFP-intein constructs were incubated with mouse anti-FLAG antibody (Invitrogen, diluted 1:200 in PBS-BSA) for 30 min and washed once with PBS-BSA. The microbeads were also incubated and washed under the same conditions. The yeast and microbeads were subsequently incubated with anti-mouse Alexa 647 (Invitrogen, 1:500 dilution in PBS-BSA) for 30 min, washed once with PBS-BSA, and analyzed via flow cytometry.
SDS-PAGE and Western blotting of reacted proteins
Protein samples were resolved on 12.5% w/v SDS-PAGE gels. Samples were boiled for 10 min prior to resolution on the SDS-PAGE gels for non-reducing conditions. For reducing SDS-PAGE, 1mM 2-mercaptoethanol was added to the sample buffer and the samples were boiled for 10 min prior to resolution on the gels. Gels were either stained with Coomassie blue, or the proteins were transferred to a nitrocellulose membrane for Western blot analysis. To detect the FLAG tag, membranes were probed with anti-FLAG M2 mouse monoclonal antibody (Sigma–Aldrich, diluted 1:3000), followed by anti-mouse HRP conjugate (Sigma–Aldrich, diluted 1:2000). For detection of biotinylated proteins, membranes were probed with anti-biotin mouse monoclonal antibody Ab-2 clone BTN.4 (Lab Vision Corporation, diluted 1:500), followed by anti-mouse HRP conjugate. Membranes were subsequently developed with ECL reagents and exposed to Hyperfilm (GE Healthcare). For quantitative Western blotting, the band intensities were measured using the NIH ImageJ program, and the slopes of the unsaturated band intensities versus exposure time were compared to determine the relative amounts of protein.
Hydrazine release of yeast surface-displayed proteins
For protein release with hydrazine, induced yeast (~1×109 cells containing an estimated total of ~40–140 pmole scFv or GFP) were suspended in 500 μl MOPS-NaOH (0.5 M) buffer at pH 8.0 containing NaCl (0.50 M), EDTA (0.10 mM), 0.1% w/v BSA (added as carrier to prevent non-specific adsorption losses after protein release), and 500 mM of either azide-terminated hydrazine (synthesized as described previously17), or a simple hydrazine, ethyl hydrazinoacetate hydrochloride (Sigma–Aldrich). The reaction was carried out for 3 days at room temperature with gentle rotation. Subsequently, the yeast were removed by centrifugation, and the supernatant containing the released proteins recovered. The samples were dialyzed against PBS using a Slide-A-Lyzer dialysis cassette (Pierce) to remove unreacted hydrazine. To purify the hydrazine-released proteins, the dialyzed solution was added to 25 μl of anti-FLAG M2 resin (Sigma–Aldrich) and incubated for 2 h at room temperature with gentle rotation. The FLAG resin was subsequently washed three times with 500 μl PBS to remove unbound proteins. The proteins were eluted by incubating with 50 μl of 100 μg/mL 3x FLAG peptide (Sigma–Aldrich) in PBS for 30 min, followed by 3 serial incubations for 10 min each in the same buffer.
To perform the CuAAC reactions, the following was added to 100 μl solution of hydrazine-released proteins in PBS: 5 μl of 5 mM alkyne biotin (Invitrogen) in DMSO, 2.5 μl of 4 mM CuSO4 (Thermo-Fisher) in water, 5 μl of 1.6 mM TBTA ligand (Sigma–Aldrich) in 80% v/v tert-butanol, and 5 μl of 100 mM sodium ascorbate (Sigma–Aldrich) in water. The reactions were carried out for 1 h at room temperature.
MESNA release and EPL of yeast surface-displayed proteins
Release of the scFvs with the sulfur nucleophile was conducted by suspending induced yeast cells (~1×109 cells containing an estimated total of ~40–140 pmole scFv or GFP) in 500 μl HEPES (50 mM pH 7.2) containing 0.1% w/v BSA and 50 mM 2-mercapthoethanesulfonic acid (MESNA, Sigma–Aldrich). Initially, this reaction was carried out for 20 h at room temperature followed by yeast removal by centrifugation and recovery of the supernatant containing the released proteins. To analyze the mechanism of release via MESNA, yeast expressing pcT4Re-GFP constructs were reacted with MESNA for 45 min at room temperature prior to yeast removal. The proteins were either frozen at −20°C to prevent further reaction, or the proteins were allowed to react in the MESNA solution in the absence of yeast for an additional 20 h at room temperature. Where indicated, the N-linked glycans were removed from the samples with EndoH (New England Biolabs) for 1 h at 16°C. Since the 45 minute protocol led to nearly complete removal of all surface displayed fusions, all further MESNA experiments were carried out using the 45 minute, 20 h approach. When necessary, a cysteine reagent was added during the 20 h step to perform EPL, as described below.
Three cysteine reagents were used in this study to perform the EPL reactions, cysteine azide (Anaspec), cysteine alkyne (Anaspec), and Bio-P1, an N-terminal cysteine peptide containing a biotin (synthesized by the University of Wisconsin Biotechnology Center based upon the Bio-P1 peptide from New England Biolabs. Sequence: NH2-CDPEK(Bt)DS-CONH2). Proteins were released from yeast displaying scFv-intein or GFP-intein fusion proteins with either simple hydrazine (3 days) or MESNA (45 min), and the supernatants containing released proteins were separated from the yeast by centrifugation. For biotinylation, 15 μl of 15 mM Bio-P1 in PBS was added to 100 μl of the released protein products and allowed to react for 20 h at room temperature. To generate azide and alkyne proteins, either 3 μl of 800 mM cysteine azide in PBS or 3 μl of 800 mM cysteine alkyne in PBS was added directly to 500 μl of the released protein products and the reactions were allowed to proceed for 20 h at room temperature. The reactions were subsequently dialyzed against PBS with a Slide-A-Lyzer dialysis cassette to remove unreacted cysteines.
The azide and alkynyl functionalization of the EPL proteins was evaluated via CuAAC reaction. To 100 μl of azide protein, alkyne protein, or protein thioester, the following was added: either 5 μl of 5 mM alkyne biotin in DMSO or 5 μl of 5 mM azide biotin (Invitrogen) in DMSO, 2.5 μl of 4 mM CuSO4 in water, 5 μl of 1.6 mM TBTA ligand in 80% v/v tert-butanol, and 5 μl of 100 mM sodium ascorbate in water. The reactions were carried out for 1 h at room temperature.
Immobilization of alkynyl proteins on agarose beads
Azide-functionalized surfaces were generated using NHS-activated agarose beads (Thermo-Fisher). The agarose beads (250 μl) were washed three times with PBS to remove the storage buffer. The beads were resuspended in 1 mL PBS, and a PEG-azide containing a primary amine, O-(2-aminoethyl)-O′-(2-azidoethyl)nonaethylene glycol (Sigma–Aldrich), was added to the suspension to a final concentration of 100 mM. The mixture was incubated for 2 h at room temperature followed by an overnight incubation at 4°C with gentle rotation. The beads were washed three times with PBS to remove the PEG-azide reagent, and unreacted NHS groups were quenched by incubating the agarose beads in 1 M Tris buffer (pH 7) for 2 h at room temperature. The beads were subsequently washed 3 times in PBS-BSA and resuspended in PBS-BSA.
Alkynyl proteins (100 μl) were added to 5 μl of azide-functionalized beads along with 2.5 μl of 4 mM CuSO4 in water, 5 μl of 1.6 mM TBTA ligand in 80% v/v tert-butanol, and 5 μl of 100 mM sodium ascorbate in water. The reaction was incubated for 16 h at room temperature with gentle rotation. The reacted beads were washed three times with 500 μl of PBS-BSA. To assay for immobilized proteins bearing a FLAG tag, the agarose beads were incubated with anti-FLAG rabbit polyclonal antibody (1:2000 dilution in PBS-BSA) for 1 h, washed three times in PBS with 0.1% w/v Tween (PBST), incubated with anti-mouse Alexa 594 (1:500 dilution in PBS-BSA) (Invitrogen) for 30 min, and washed three times in PBST. To assay 4-4-20 activity, protein-loaded agarose beads were incubated with 10 μM FITC-dextran in PBS-BSA for 30 min and washed three times in PBST. To assay scFv2 activity, protein-loaded agarose beads were incubated with purified human EGFR (4 μg/mL in PBS-BSA) for 2 h, washed three times with PBST, immunolabeled with anti-EGFR antibody cocktail Ab-12 (1:200 dilution in PBS-BSA) for 1 h, washed three times with PBST, immunolabeled with secondary anti-mouse Alexa 488 antibody (1:500 dilution in PBS-BSA) for 30 min, and washed three times with PBST. The agarose beads were subsequently imaged with an Olympus IX70 fluorescence microscope, and bead-associated fluorescence was quantified with a Tecan Infinite M1000 fluorescent microplate reader.
RESULTS
Surface display of intein fusion proteins
The scFv, GFP, scFv-intein and GFP-intein fusions were expressed as fusion partners to Aga2p, as in standard yeast surface display25 (Figure 1a). However, while most yeast display systems employ fusion to the C-terminus of the Aga2p, the display constructs used here are fused to the N-terminus of Aga2p to allow for intein-mediated protein release from the yeast surface (Figure 2b). The resultant constructs enable display of unmodified scFv or GFP or display of the same proteins fused to the N-terminus of the Mxe GyrA intein, a genetically modified intein that undergoes nucleophile-induced protein splicing at its N-terminus29 (Figure 1a). Three different target proteins were evaluated: GFP,26 anti-fluorescein scFv 4-4-20,25 and anti-epidermal growth factor receptor (EGFR) scFv2.27 First, the effects of intein fusion on protein display and activity were evaluated. Each of the proteins was successfully displayed on the surface (Figure 1b and 1c), with the two scFvs, 4-4-20 and scFv2, having ~40% reduced expression when displayed as a fusion to intein, and GFP expression being unaffected by intein fusion (Figure 1c). The activity of the displayed proteins was determined by evaluating scFv binding to cognate antigen (4-4-20, fluorescein and scFv2, EGFR), or by measuring the GFP fluorescence. While expression was attenuated for 4-4-20 or scFv2, the proteins displayed as intein fusions exhibited nearly the same activity per molecule as the proteins displayed without intein (Figure 1c), indicating that the presence of the intein did not negatively impact the function of the displayed protein.
Figure 2.

Intein-mediated protein release. (a) Two nucleophiles were used to release the proteins, 2-mercapthoethanesulfonic acid (MESNA), and a hydrazine azide. (b) For intein-mediated protein release, the intein facilitates an N- to S-acyl shift to generate a thioester. Nucleophilic attack results in protein release from the display construct while a functional group is simultaneously appended to the C-terminus of the protein. When MESNA is used to release the protein, a thioester is installed on the protein of interest; when the hydrazine azide is used, an azido group is installed. (c) Western blots of the supernatants obtained after incubating protein-displaying yeast with the hydrazine azide for 3 days or MESNA for 20 h, probed with an anti-FLAG antibody to detect the presence of released target protein. Prior to Western blotting, proteins were resolved by SDS-PAGE using nonreducing conditions to prevent unwanted intein-mediated protein release by the beta-mercaptoethanol nucleophile present in reducing SDS-PAGE sample buffer. Released scFv or GFP at their unfused ~30-kDa size were only detected when intein was present. (d) GFP-displaying yeast with and without intein were reacted with MESNA for 45 min and the resulting supernatant removed from the yeast and incubated for an additional 20 h where noted. Anti-FLAG Western blots performed under nonreducing conditions to compare the MESNA-released GFP products after reaction for 45 min and 20 h. The released proteins were first deglycosylated with EndoH revealing Aga2p-GFP (~55 kDa) fusion release in the absence of intein (compared to 2c -intein without deglycosylation). Release of both Aga2p-intein-GFP (~75 kDa) and unfused GFP (~30 kDa) is observed when MESNA reacts with the yeast for 45 min, but upon allowing the reaction to proceed an additional 20 h, GFP is nearly quantitatively released from the intein. (e) Quantitative Western blot analysis was used to determine the relative amount of proteins released, and revealed that the 20-h reaction with MESNA releases 4 times more protein than does the 3-day reaction with the hydrazine azide. Plotted are means and standard deviations for three independent reactions originating from three independent yeast surface display transformants.
Intein-mediated release and C-terminal functionalization of surface-displayed proteins
Key to intein-mediated release and functionalization, a spontaneous, reversible N- to S- acyl shift occurs at the amino-terminal cysteine of the Mxe GyrA intein backbone, forming a thioester that is susceptible to nucleophilic attack. Reaction with the nucleophile releases the protein from the intein, and hence, from the yeast surface display construct (Figures 2a and 2b). The ability of the thiol and hydrazine nucleophiles to promote intein-mediated release of target proteins from the yeast surface was evaluated. Two different nucleophiles were compared in this study, a nitrogen nucleophile containing an azido group, hydrazine azide,17 and a sulfur nucleophile, 2-mercapthoethanesulfonic acid (MESNA)18 (Figure 2a). After treating yeast displaying either scFv or GFP with the nucleophiles, Western blotting with an anti-FLAG antibody demonstrated that both nucleophiles release the yeast-displayed protein from the intein in a 3-day reaction for the hydrazine azide or a 20-h reaction for MESNA (Figure 2c).
Since MESNA is also a reducing agent and potentially capable of reducing the disulfide bonds between Aga1p and Aga2p (Figure 1a), the contribution of intein-mediated release versus disulfide-bond reduction was assessed. Intein-release typically requires an overnight reaction for completion,29 and thus, an abbreviated MESNA reaction was examined to determine if GFP-intein-Aga2p fusions are released via disulfide reduction. Yeast displaying GFP-Aga2p or GFP-intein-Aga2p fusions were reacted with MESNA for 45 min, and the supernatant containing released proteins was removed from the yeast. Given that glycosylation can cause Aga2p fusion proteins to have diffuse bands at high molecular weights on Western blots that are difficult to detect26 (Figure 2c), samples were treated with glycosidase prior to Western blot analysis. An anti-FLAG Western blot revealed that with or without intein, MESNA released GFP from the yeast surface within 45 min, consistent with disulfide bond reduction (Figure 2d). When the released proteins were allowed to further react in the MESNA solution for 20 h after yeast removal as performed in Figure 2c, GFP was nearly completely released from the intein-Aga2p fusion. As expected, the increased reaction time had no effect on the non-intein construct, as MESNA did not release GFP from Aga2p in the absence of intein (Figure 2d). These results indicate that MESNA first releases the displayed proteins from the yeast surface by reducing the disulfide bonds between Aga1p and Aga2p and/or by intein-mediated reaction, and subsequently further reacts with intein to completely release the scFvs or GFP from the rest of the display construct. Likely, because of this dual release mechanism, the treatment with MESNA removed nearly 90% of the fusions from the yeast surface, while treatment with hydrazine azide only removed 20% of the fusions from the yeast surface, as determined by flow cytometry. This resulted in the MESNA reaction yielding ~4 times more released protein than the hydrazine azide reaction, as determined by a quantitative Western blot (Figure 2e).
Following intein-mediated release of the yeast surface displayed proteins, the carboxy-terminal functionalization of the proteins was confirmed by further reacting the released proteins with biotinylated reagents (Figure 3a). After 3 days, the hydrazine-released proteins were separated from the yeast by centrifugation and subsequently reacted with a biotin alkyne in a copper-catalyzed azide–alkyne cycloaddition (CuAAC) (Figure 3b). Anti-biotin Western blotting demonstrates that when the displayed proteins were released with a non-azido hydrazine, there was no reaction with the biotin alkyne, as expected (Figure 3c). However, when performing the CuAAC reaction with the hydrazine azide-released proteins, multiple proteins reacted with the biotin alkyne reagent (Figure 3c). Thus, although the target protein was released by nucleophile treatment (Figure 2c), other yeast proteins are also non-specifically released (Supplemental Figure 1), a subset of which were functionalized by the hydrazine azide (Figure 3c). This issue could be resolved by purifying the hydrazine-azide released proteins with anti-FLAG resin prior to reaction with biotin alkyne, as was demonstrated for GFP, resulting in azide-dependent biotinylation of the released protein (Figure 3d).
Figure 3.

Site-specific biotinylation of released proteins. (a) Structures of the biotinylation reagents, biotin alkyne, biotin azide and cysteine-terminated biotinylated peptide. (b) The biotin alkyne reacts with the azide-functionalized proteins obtained via cleavage reaction with the hydrazine azide in a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. The protein thioester obtained by the cleavage reaction with MESNA reacts with a N-terminal cysteine peptide in an expressed protein ligation (EPL) reaction. (c) A Western blot of the CuAAC reaction probed with an anti-biotin antibody reveals that multiple proteins react with the biotin alkyne when the hydrazine azide is used to release the proteins from the yeast surface. When a non-azido hydrazine is used to cleave the proteins, no reaction with the biotin alkyne is detected. (d) The hydrazine-released products were subjected to FLAG tag purification before CuAAC reaction with biotin alkyne. A Western blot of the reaction probed an anti-biotin antibody reveals biotinylation corresponds to the hydrazine-azide released proteins (shown for GFP only). (e) An anti-biotin Western blot of the EPL reaction between the protein thioesters and biotinylated cysteine peptide shows specific biotinylation of the released protein. Hydrazine-released proteins were reacted with the biotinylated cysteine peptide as a non-thioester negative control to demonstrate that the reaction is specific to the thioester.
To examine thioester functionality of the MESNA-released proteins, these proteins were reacted with a biotinylated peptide having an N-terminal cysteine (Figure 3a) in an EPL reaction (Figure 3b). Following the initial 45-min reaction with MESNA, released proteins were separated from the yeast by centrifugation. The intein-release reaction via MESNA was allowed to proceed for an additional 20 h in the presence of the biotinylated cysteine peptide to complete the EPL reaction. Anti-biotin Western blotting demonstrates specific biotinylation of the thioester scFvs and GFP but no biotinylation of proteins released with hydrazine that lack a thioester moiety (Figure 3e). In contrast to the results achieved with the hydrazine azide, the main biotinylated product of the EPL reaction corresponded to the target protein. Although MESNA will release other yeast proteins because of its reducing character (Figure 2d and Supplemental Figure 1), the thioester functionality was selectively added to the target protein via the intein-mediated reaction. These results further demonstrate that although MESNA does initially release the yeast-displayed constructs via disulfide bond reduction (Figure 2d), the subsequent MESNA-mediated release of the scFv or GFP from the remainder of the display construct produces specific carboxy-terminal functionalization of the target protein. Thus, protein release with MESNA eliminates the need for protein purification prior to use in downstream applications. Although the hydrazine azide proved useful to release and chemically functionalize purified intein fusion proteins,2,17 its use with yeast surface displayed proteins is less advantageous because subsequent protein purification steps are necessary (Figure 3d). This disadvantage, along with the greater absolute efficiency of the MESNA release, led us to move forward with the MESNA-EPL approach.
Azide and alkyne functionalization of thioester proteins
While EPL with the thioester functionalized proteins in solution can yield specific modification of a protein, it would be desirable to adapt these methods for site-specific immobilization of proteins on surfaces. The protein thioester formed is not ideally suited for protein immobilization applications via EPL since millimolar concentrations of the reactants are typically required for efficient conjugation.30,31 This has previously resulted in inefficient thioester surface immobilization reactions because these concentrations are difficult to obtain on surfaces.15 As an alternative that is compatible with the MESNA-mediated release approaches, it is possible to employ EPL reactions to append azido and alkynyl groups onto the MESNA-released proteins. Subsequently the azide and alkyne functionalities can be used to immobilize proteins on surfaces since CuAAC reactions require lower concentrations of substrates than does EPL.6,15,31,32 Thus, by performing the EPL reaction with cysteine azide or cysteine alkyne in solution, high concentrations of the derivatized cysteine can be used to drive efficient functionalization of the proteins.
To generate these CuAAC-compatible proteins, proteins released after the initial 45-min MESNA reaction were separated from the yeast by centrifugation, and MESNA-mediated intein release was allowed to proceed for an additional 20 h in the presence of either cysteine azide or cysteine alkyne (Figure 4a and 4b). Resultant protein functionalization was detected by reacting the azido proteins with biotin alkyne (Figure 3a) and the alkynyl proteins with biotin azide (Figure 3a) in a CuAAC reaction, followed by anti-biotin Western blotting analysis of the reaction. The CuAAC reaction resulted in biotinylation of the target proteins that were functionalized with an azide or alkyne (Figure 4c); whereas, MESNA-released proteins that had not been reacted with the cysteine reagents and used as non-alkyne and non-azido negative controls were not functionalized. Although other yeast surface proteins are released in the MESNA reaction and are present in the subsequent reaction mixtures (Supplemental Figure 1), the primary products of the EPL and CuAAC reactions correspond to biotinylated target protein. However, more non-specific biotinylation of yeast proteins was observed when EPL was performed with the cysteine azide followed by CuAAC with biotin alkyne than with the opposite chemical orientation (Figure 4c). Thus, by functionalizing the proteins with an alkyne via EPL followed by CuAAC with an azide target, protein functionalization is achieved with high specificity and no purification or post-processing after release from the yeast surface.
Figure 4.

Azide and alkyne functionalization of thioester proteins via EPL. (a) Cysteine azide and cysteine alkyne were used in the EPL reaction. Biotin alkyne and biotin azide (shown in Figure 3a) were used to detect the chemical functionalization. (b) Thioester proteins obtained though the MESNA-release reaction react with cysteine azide or cysteine alkyne to install an azide and alkyne, respectively, on the C-terminus of the protein. The azide-functionalized protein is subsequently reacted with the biotin alkyne in the presence of Cu(I) via CuAAC to yield biotinylated protein. Similarly, the alkyne-functionalized protein reacts with biotin azide to yield biotinylated protein. (c) Western blots of the CuAAC reactions probed with an anti-biotin antibody demonstrate biotinylation only when the azide or alkyne is present in the protein. Proteins containing only the thioester modification were used as negative controls lacking azide and alkyne functionalization.
Immobilization of alkyne-functionalized proteins
Previous studies have demonstrated a greater reaction efficiency by immobilizing alkynyl proteins on azido surfaces than reacting azido proteins on alkynyl surfaces.6 Given this result and the higher specificity of the CuAAC reaction in solution with the alkynyl protein, protein immobilization was performed with the alkynyl proteins (Figure 4c). Azido-functionalized agarose beads were reacted with either the alkynyl protein or the control, thioester proteins, under CuAAC conditions. Substantial protein immobilization in terms of FLAG epitope tag detection was observed only when the reaction was performed with a protein containing a carboxy-terminal alkynyl group (Figure 5a). Quantification revealed that 10–13 fold more scFv or GFP was present on the beads when the reaction was performed with scFv alkyne or GFP alkyne compared to scFv or GFP without the alkyne (Figure 5a), indicating that >90% of the alkynyl protein immobilization is site-specific and mediated via the alkyne.
Figure 5.

Immobilization and activity of alkynyl proteins. (a) Proteins released and functionalized with alkyne as described are further reacted with the azide functionalized agarose beads via CuAAC. Microscopic images indicate the presence of immobilized proteins by immunofluorescent labeling of the FLAG tag. Protein immobilization is quantified by measuring the total bead fluorescence. The fluorescence reading was independently normalized to the alkyne-linked protein for each of the three proteins, and represents the mean ± SD values of the immobilization reaction performed with proteins released from 6 independent yeast transformants, thereby representing the full measure of method variability. (b) Fluorescent microscope images demonstrate the activity of immobilized proteins on agarose beads. Top panel: Beads reacted with alkyne 4-4-20 exhibit binding to the 4-4-20 ligand, FITC-dextran, while beads reacted with non-alkyne 4-4-20 or alkyne scFv2 did not bind fluorescein. Middle panel: Binding of EGFR to immobilized proteins is detected with an anti-EGFR antibody. Immobilized alkyne scFv2 was shown to bind its ligand, EGFR, while beads reacted with alkyne 4-4-20 or non-alkyne scFv2 do not demonstrate binding to EGFR. Bottom panel: Beads exhibit GFP fluorescence only when reacted with alkyne GFP. Protein activity is quantified by measuring the resultant fluorescence intensity of the beads and the fluorescence values are independently normalized based upon the signal for alkynyl protein binding its respective antigen (or GFP fluorescence). Plotted are the mean ± SD of activity measurements from three independent bead immobilization reactions.
Following confirmation of site-specific protein immobilization, the activity of the proteins conjugated to the beads was evaluated to ensure that the protein release and modification strategies did not have deleterious effects upon protein activity. In particular, scFvs contain two intrachain disulfide bonds that are critical for their stability and proper folding,33 and so the exposure to thiol-based reagents such as MESNA and cysteine alkyne could in principle reduce these disulfide bonds, causing the protein to unfold and become inactive. Using the various combinations of activity (GFP) and antigens (4-4-20, FITC and scFv2, EGFR), it was demonstrated that proteins indeed retained activity and antigen specificity after immobilization on the beads. Azido beads loaded with alkyne-conjugated 4-4-20 specifically bound fluorescein, while beads loaded with alkyne-conjugated scFv2 instead specifically bound EGFR (Figure 5b). GFP fluorescence was also detected on beads loaded with alkyne-conjugated GFP (Figure 5b). Although a small amount of non-specific protein immobilization was detected with anti-FLAG immunofluorescence labeling (Figure 5a), the adsorbed proteins appeared to exhibit little to no antigen binding or GFP fluorescence, indicating the importance of site-specific immobilization for the activity of these proteins (Figure 5b). Antigen binding selectivity was greater than 30:1 for the alkyne-immobilized scFvs compared to beads loaded with negative control scFv or non-alkyne containing scFvs (Figure 5b). Similarly, site-specific GFP immobilization yielded 100-fold more fluorescence than did non-alkyne containing GFP (Figure 5b). These results suggest that the requisite MESNA release, cysteine alkyne EPL, and CuAAC reactions are collectively compatible to yield functional scFv and GFP
DISCUSSION
Intein-mediated protein functionalization strategies permit the addition of unique chemical functionalities to the C-terminus of proteins. In this study, we have demonstrated that target proteins displayed as intein fusion partners on the surface of yeast can be released by appropriate nucleophiles and subsequently functionalized to be compatible with EPL and CuAAC chemistries. The optimized release and chemical modification strategy illustrated in Figure 6 enabled facile and rapid functionalization of the C-terminus of our target proteins with alkynes while obviating the need for purification steps for downstream applications. To this end, we obtained >90% CuAAC-mediated immobilization of the alkynyl proteins on surfaces and demonstrated that the functionalization and immobilization strategies produced active antibodies that were capable of binding antigen.
Figure 6.
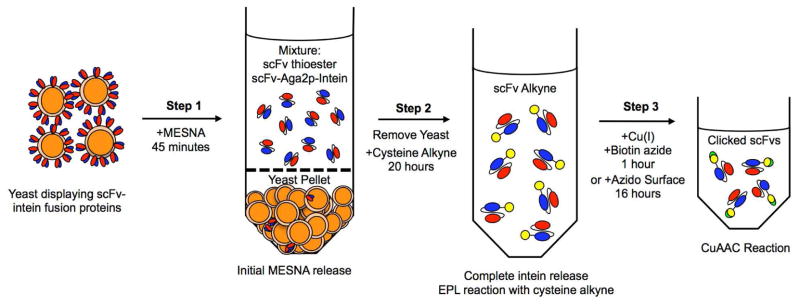
The overall intein-based release and functionalization method is performed in three steps, as shown in this example for an scFv. Step 1: Yeast displaying scFv-intein fusion proteins are reacted with MESNA for 45 min. Step 2: Released proteins in the MESNA solution (a mixture of scFv-thioesters and scFv-intein-Aga2p fusions) are removed from the yeast and cysteine alkyne is added. This reaction proceeds for 20 h to complete release of the scFv from intein and to conjugate the alkyne via EPL. Step 3: The alkyne-conjugated scFvs are reacted in a CuAAC reaction with biotin alkyne for 1 h or with an azido surface for 16 h.
Typical intein-mediated functionalization procedures are performed using soluble, purified proteins.2,5,15–17,22–24,32 The techniques described in this study provide a facile alternative for obtaining small amounts of modified protein by directly releasing and reacting intein fusion proteins that are displayed on the surface of yeast. Whereas proteins that contain disulfide bonds such as scFvs are often expressed as insoluble inclusion bodies in prokaryotic organisms like E. coli,34 we displayed the intein fusions as folded, active proteins on the yeast surface. This eliminated protein solubilization and refolding procedures that not only require multiple steps to obtain soluble protein, but that can also result in inactive intein fusion proteins incapable of nucleophile-mediated release.23,35 However, in contrast to nucleophile-mediated release with soluble proteins, performing the reactions directly on the yeast surface generates the possibility that the nucleophile will react with other proteins present on the yeast surface. These undesirable side reactions were observed when the hydrazine azide was used to release the target proteins from the yeast surface. Although the hydrazine azide is known to react chemoselectively with purified protein thioesters,2,17 the lack of chemoselectivity apparent in Figure 3c is likely due to the formation of hydrazones with carbonyl groups on glycosylated proteins that are secreted by yeast cells during the 3 day reaction or that reside on their surface. This competitive reaction would not confound the use of the hydrazine azide in the release of proteins displayed on the surface of E. coli or other cells that do not glycosylate their proteins. In contrast, yeast displayed protein released with MESNA contained a carboxy-terminal thioester capable of undergoing an EPL reaction, which led to uniquely functionalized scFv or GFP, thus obviating the need to perform any protein purification. Therefore, by directly releasing and modifying surface displayed proteins, we have eliminated intermediate protein preparation steps including inclusion body solubilization, protein refolding, and protein purification to provide a simplified protein functionalization method.
Yeast surface display is a powerful protein engineering technique that can be used to perform high-throughput selections of scFv clones from large libraries in order to identify novel antibodies from nonimmune libraries36,37 or to fine-tune antibody properties such as affinity, stability, and specificity.38–40 By releasing and functionalizing proteins displayed on the yeast surface, we have integrated these intein-mediated protein modification strategies into a protein engineering platform, potentially enabling downstream analysis of engineered clones as modified, soluble proteins without time-consuming intermediate steps. This platform could prove particularly useful to analyze panels of engineered clones where protein subcloning, soluble expression, and purification become limiting factors for clonal fitness assessment. Furthermore, standard enzymatic approaches for protein release from the yeast surface25,41 and functionalization methods such as biotinylation42 would yield proteins that are immobilized or conjugated in a noncovalent fashion. In contrast, our strategy results in protein release and insertion of CuAAC-compatible groups that instead can enable stable, covalent conjugation of released proteins to many different linkers, proteins, surfaces and nanoparticles15,31,43–45. In addition, the general approaches employing EPL chemistries described here could also be used for covalent introduction of numerous other useful chemical functionalities.20,46
The methods described in this study would be especially well suited for applications where small amounts of protein are sufficient for downstream analysis. Based upon the surface expression of our proteins (Figure 1b, 24,000–86,000 fusions per cell), it is possible to obtain between ~14 μg and ~58 μg of protein using a 1 L yeast culture and a 20-h surface display induction time. As one example, typical microarrays require antibody spotting at concentrations ranging from 25 to 400 μg/mL47–49 and spotting volumes between 50 and 350 pL,49–51 and the amount of released protein would permit, at a minimum, 100,000 array spots. Thus, it is conceivable that by using the intein-linked yeast surface display method, a large selection of novel scFv clones could be expressed, released, functionalized, and immobilized in parallel to rapidly generate an antibody microarray. In conclusion, a combination of yeast surface display with intein-based tools provide a facile method for direct chemical functionalization of proteins, likely enabling a variety of downstream applications.
Supplementary Material
Acknowledgments
Dr. Greg Wiepz at the University of Wisconsin Department of Biomolecular Chemistry provided the soluble EGFR. The anti-EGFR scFv, scFv2, was donated by Winfried Wels, Institute for Biomedical Research Georg-Speyer-Haus. This work was funded by National Institutes of Health grant R01 CA108467. Additional support was received from the Materials Research Science and Engineering Center at the University of Wisconsin–Madison (NSF DMR-0520527) and National Institutes of Health grants R01 GM044783 and R01 NS052649.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Supporting information includes a Coomassie-stained SDS-PAGE gel showing overall protein release from the yeast surface in the hydrazine azide and MESNA reactions. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.de Araújo AD, Palomo JM, Cramer J, Köhn M, Schröder H, Wacker R, Niemeyer C, Alexandrov K, Waldmann H. Diels–Alder Ligation and Surface Immobilization of Proteins. Angew Chem, Int Ed. 2006;45:296–301. doi: 10.1002/anie.200502266. [DOI] [PubMed] [Google Scholar]
- 2.Kalia J, Abbott NL, Raines RT. General Method for Site-Specific Protein Immobilization by Staudinger Ligation. Bioconjugate Chem. 2007;18:1064–1069. doi: 10.1021/bc0603034. [DOI] [PubMed] [Google Scholar]
- 3.Khan F, He M, Taussig MJ. Double-Hexahistidine Tag with High-Affinity Binding for Protein Immobilization, Purification, and Detection on Ni-Nitrilotriacetic Acid Surfaces. Anal Chem. 2006;78:3072–3079. doi: 10.1021/ac060184l. [DOI] [PubMed] [Google Scholar]
- 4.Soellner MB, Dickson KA, Nilsson BL, Raines RT. Site-Specific Protein Immobilization by Staudinger Ligation. J Am Chem Soc. 2003;125:11790–11791. doi: 10.1021/ja036712h. [DOI] [PubMed] [Google Scholar]
- 5.Lesaicherre ML, Lue RYP, Chen GYJ, Zhu Q, Yao SQ. Intein-Mediated Biotinylation of Proteins and Its Application in a Protein Microarray. J Am Chem Soc. 2002;124:8768–8769. doi: 10.1021/ja0265963. [DOI] [PubMed] [Google Scholar]
- 6.Lin PC, Ueng SH, Tseng MC, Ko JL, Huang KT, Yu SC, Adak AK, Chen YJ, Lin CC. Site-Specific Protein Modification through CuI-Catalyzed 1,2,3-Triazole Formation and Its Implementation in Protein Microarray Fabrication. Angew Chem. 2006;118:4392–4396. doi: 10.1002/anie.200600756. [DOI] [PubMed] [Google Scholar]
- 7.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 8.Deiters A, Cropp TA, Summerer D, Mukherji M, Schultz PG. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg Med Chem Lett. 2004;14:5743–5745. doi: 10.1016/j.bmcl.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 9.Liu XM, Thakur A, Wang D. Efficient synthesis of linear multifunctional poly(ethylene glycol) by copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition. Biomacromolecules. 2007;8:2653–2658. doi: 10.1021/bm070430i. [DOI] [PubMed] [Google Scholar]
- 10.Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 11.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franco EJ, Hofstetter H, Hofstetter O. A comparative evaluation of random and site-specific immobilization techniques for the preparation of antibody-based chiral stationary phases. J Sep Sci. 2006;29:1458–1469. doi: 10.1002/jssc.200600062. [DOI] [PubMed] [Google Scholar]
- 13.Gaertner HF, Offord RE. Site-Specific Attachment of Functionalized Poly(ethylene glycol) to the Amino Terminus of Proteins. Bioconjugate Chem. 1996;7:38–44. doi: 10.1021/bc950074d. [DOI] [PubMed] [Google Scholar]
- 14.Huang W, Wang J, Bhattacharyya D, Bachas LG. Improving the Activity of Immobilized Subtilisin by Site-Specific Attachment to Surfaces. Anal Chem. 1997;69:4601–4607. doi: 10.1021/ac970390g. [DOI] [PubMed] [Google Scholar]
- 15.Elias DR, Cheng Z, Tsourkas A. An Intein-Mediated Site-Specific Click Conjugation Strategy for Improved Tumor Targeting of Nanoparticle Systems. Small. 2010;6:2460–2468. doi: 10.1002/smll.201001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohlmann SB, Peter, Simone Greven, Axel Harrenga. Site-specific modification of ED-B-targeting antibody using intein-fusion technology. BMC Biotechnol. 2011;11:76–85. doi: 10.1186/1472-6750-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalia J, Raines RT. Reactivity of Intein Thioesters: Appending a Functional Group to a Protein. ChemBioChem. 2006;7:1375–1383. doi: 10.1002/cbic.200600150. [DOI] [PubMed] [Google Scholar]
- 18.Evans TC, Benner J, Xu M-Q. Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci. 1998;7:2256–2264. doi: 10.1002/pro.5560071103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fong BA, Wu W-Y, Wood DW. The potential role of self-cleaving purification tags in commercial-scale processes. Trends Biotechnol. 2010;28:272–279. doi: 10.1016/j.tibtech.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Tolbert TJ, Wong CH. Intein-Mediated Synthesis of Proteins Containing Carbohydrates and Other Molecular Probes. J Am Chem Soc. 2000;122:5421–5428. [Google Scholar]
- 21.Wang D, Cole PA. Protein Tyrosine Kinase Csk-Catalyzed Phosphorylation of Src Containing Unnatural Tyrosine Analogues. J Am Chem Soc. 2001;123:8883–8886. doi: 10.1021/ja010540b. [DOI] [PubMed] [Google Scholar]
- 22.Guo C, Li Z, Shi Y, Xu M, Wise JG, Trommer WE, Yuan J. Intein-mediated fusion expression, high efficient refolding, and one-step purification of gelonin toxin. Protein Expression Purif. 2004;37:361–367. doi: 10.1016/j.pep.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 23.Sydor JR, Mariano M, Sideris S, Nock S. Establishment of Intein-Mediated Protein Ligation under Denaturing Conditions: C-Terminal Labeling of a Single-Chain Antibody for Biochip Screening. Bioconjugate Chem. 2002;13:707–712. doi: 10.1021/bc025534z. [DOI] [PubMed] [Google Scholar]
- 24.Wood RJ, Pascoe DD, Brown ZK, Medlicott EM, Kriek M, Neylon C, Roach PL. Optimized Conjugation of a Fluorescent Label to Proteins via Intein-Mediated Activation and Ligation. Bioconjugate Chem. 2004;15:366–372. doi: 10.1021/bc0341728. [DOI] [PubMed] [Google Scholar]
- 25.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 26.Huang D, Shusta EV. Secretion and surface display of green fluorescent protein using the yeast Saccharomyces cerevisiae. Biotechnol Prog. 2005;21:349–357. doi: 10.1021/bp0497482. [DOI] [PubMed] [Google Scholar]
- 27.Hyland S, Beerli RR, Barbas CF, Hynes NE, Wels W. Generation and functional characterization of intracellular antibodies interacting with the kinase domain of human EGF receptor. Oncogene. 2003;22:1557–1567. doi: 10.1038/sj.onc.1206299. [DOI] [PubMed] [Google Scholar]
- 28.Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 29.Telenti A, Southworth M, Alcaide F, Daugelat S, Jacobs WR, Perler FB. The Mycobacterium xenopi GyrA protein splicing element: characterization of a minimal intein. J Bacteriol. 1997;179:6378–6382. doi: 10.1128/jb.179.20.6378-6382.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muralidharan V, Muir TW. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
- 31.Gupta SS, Kuzelka J, Singh P, Lewis WG, Manchester M, Finn M. Accelerated bioorthogonal conjugation: a practical method for the ligation of diverse functional molecules to a polyvalent virus scaffold. Bioconjugate Chem. 2005;16:1572–1579. doi: 10.1021/bc050147l. [DOI] [PubMed] [Google Scholar]
- 32.Steinhagen M, Holland-Nell K, Meldal M, Beck-Sickinger AG. Simultaneous “One Pot” Expressed Protein Ligation and CuI-Catalyzed Azide/Alkyne Cycloaddition for Protein Immobilization. ChemBioChem. 2011;12:2426–2430. doi: 10.1002/cbic.201100434. [DOI] [PubMed] [Google Scholar]
- 33.Wörn A, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 34.Verma R, Boleti E, George AJT. Antibody engineering: Comparison of bacterial, yeast, insect and mammalian expression systems. J Immunol Methods. 1998;216:165–181. doi: 10.1016/s0022-1759(98)00077-5. [DOI] [PubMed] [Google Scholar]
- 35.Bastings MMC, Van Baal I, Meijer E, Merkx M. One-step refolding and purification of disulfide-containing proteins with a C-terminal MESNA thioester. BMC Biotechnol. 2008;8:76. doi: 10.1186/1472-6750-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21:163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 37.Wang XX, Cho YK, Shusta EV. Mining a yeast library for brain endothelial cell-binding antibodies. Nat Methods. 2007;4:143–145. doi: 10.1038/nmeth993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shusta EV, Holler PD, Kieke MC, Kranz DM, Wittrup KD. Directed evolution of a stable scaffold for T-cell receptor engineering. Nat Biotechnol. 2000;18:754–759. doi: 10.1038/77325. [DOI] [PubMed] [Google Scholar]
- 39.Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc Natl Acad Sci U S A. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kato M, Maeda H, Kawakami M, Shiraga S, Ueda M. Construction of a selective cleavage system for a protein displayed on the cell surface of yeast. Appl Microbiol Biotechnol. 2005;69:423–427. doi: 10.1007/s00253-005-0006-x. [DOI] [PubMed] [Google Scholar]
- 42.Parthasarathy R, Bajaj J, Boder ET. An Immobilized Biotin Ligase: Surface Display of Escherichia coli BirA on Saccharomycescerevisiae. Biotechnol Prog. 2005;21:1627–1631. doi: 10.1021/bp050279t. [DOI] [PubMed] [Google Scholar]
- 43.Bundy BC, Swartz JR. Site-Specific Incorporation of p- Propargyloxyphenylalanine in a Cell-Free Environment for Direct Protein-Protein Click Conjugation. Bioconjugate Chem. 2010;21:255–263. doi: 10.1021/bc9002844. [DOI] [PubMed] [Google Scholar]
- 44.Li M, De P, Gondi SR, Sumerlin BS. Responsive polymer-protein bioconjugates prepared by RAFT polymerization and copper-catalyzed azide-alkyne click chemistry. Macromol Rapid Commun. 2008;29:1172–1176. [Google Scholar]
- 45.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn M. Bioconjugation by copper (I)-catalyzed azide-alkyne [3+ 2] cycloaddition. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 46.Chaisemartin L, Chinestra P, Favre G, Blonski C, Faye JC. Synthesis and Application of a N-1′ Fluorescent Biotinyl Derivative Inducing the Specific Carboxy-Terminal Dual Labeling of a Novel RhoB-Selective scFv. Bioconjugate Chem. 2009;20:847–855. doi: 10.1021/bc800272r. [DOI] [PubMed] [Google Scholar]
- 47.Angenendt P, Glökler J, Sobek J, Lehrach H, Cahill DJ. Next generation of protein microarray support materials: Evaluation for protein and antibody microarray applications. J Chromatogr A. 2003;1009:97–104. doi: 10.1016/s0021-9673(03)00769-6. [DOI] [PubMed] [Google Scholar]
- 48.Olle EW, Sreekumar A, Warner RL, McClintock SD, Chinnaiyan AM, Bleavins MR, Anderson TD, Johnson KJ. Development of an internally controlled antibody microarray. Mol Cell Proteomics. 2005;4:1664–1672. doi: 10.1074/mcp.M500052-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Olle EW, Messamore J, Deogracias MP, McClintock SD, Anderson TD, Johnson KJ. Comparison of antibody array substrates and the use of glycerol to normalize spot morphology. Exp Mol Pathol. 2005;79:206–209. doi: 10.1016/j.yexmp.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Kristensen T, Mikkelsen JD, Knox JP. Sugar-coated microarrays: a novel slide surface for the high-throughput analysis of glycans. Proteomics. 2002;2:1666–1671. doi: 10.1002/1615-9861(200212)2:12<1666::AID-PROT1666>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 51.Silzel JW, Cercek B, Dodson C, Tsay T, Obremski RJ. Mass-sensing, multianalyte microarray immunoassay with imaging detection. Clin Chem. 1998;44:2036–2043. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.