

Abstract
The optimization and truncation of our lead peptide-derived ligand TY005 possessing eight amino-acid residues was performed. Among the synthesized derivatives, NP30 (Tyr1-DAla2-Gly3-Phe4-Gly5-Trp6-O-[3′,5′-Bzl(CF3)2]) showed balanced and potent opioid agonist as well as substance P antagonist activities in isolated tissue-based assays, together with significant antinociceptive and antiallodynic activities in vivo.
Keywords: bifunctional compounds, opioid receptor agonists, neutokinin-1 receptor antagonists, Truncation of peptide sequence, NMR structure
The clinical treatment of pain, especially prolonged and neuropathic pain is still a major challenge. Current analgesic drugs, such as opioid drugs are widely used following major surgery and controlling the pain of terminal diseases such as cancer, but their use is limited by several undesired side effects, including the development of tolerance and physical dependence. The mechanisms for these side effects are still largely unclear, but it is clear that prolonged pain as well as sustained opioid administration develops neuroplastic changes in the central nerve system (CNS) in which pain-enhancing neurotransmitters, such as substance P, and their corresponding receptors are up-regulated to lead to more pain and tolerance1-3. Current treatment of prolonged and/or neuropathic pain generally can only modulate pain, and cannot counteract against these induced neuroplastic changes. Thus, it is not surprising that current analgesic drugs do not work well in these pathological conditions.1
In order to address these problems, we are working at a new approach in which the opioid agonist and neurokinin 1 (NK1) antagonist activities were combined into one ligand, to neutralize the induced neuroplastic changes. The desirable pharmacological activities of our ligand would include potent analgesic affects in both acute pain and in neuropathic pain states without the development of tolerance. In fact, our lead compound TY005 (1: Tyr1-DAla2-Gly3-Phe4-Met5-Pro6-Leu7-Trp8-O-[3′,5′-Bzl(CF3)2]) has been shown to reverse neuropathic pain in a rodent model, no sign of opioid-induce tolerance, and no development of reward liability, validating our hypothesis that a single compound possessing opioid agonist/NK1 antagonist activities could be an effective treatment against neuropathic pain.2a The designed multivalent chimeric molecules also have simple metabolic and pharmacokinetic properties compared to a cocktail of individual drugs for easy administration, a simple ADME property and no drug-drug interaction.
As previously reported, our drug-design strategy is based on the overlapping pharmacophores concept, in which the opioid agonist pharmacophore is incorporated at the N-terminus and the NK1 antagonist pharmacophore locates at the C-terminus of a single peptide-derived molecule (Fig 1). 3 The opioid pharmacophore of these chimeric peptides were designed based on the sequence of biphalin and DADLE,3,4 while the structures from 3′,5′-(bistrifluoromethyl)-benzyl ester of N-acylated tryptophans was modified into the NK1 antagonist pharmacophore5. These two pharmacophores could be divided into “address” and “message” regions. Based on the previous structure-activity relationship (SAR) studies in our bifunctional ligands,3 opioid agonist pharmacophore works as a message region for NK1 antagonist activity, and vise versa, implying that 1 has four-residues (Met-Pro-Leu-Trp-O-[3′,5′-Bzl(CF3)2]) message region for opioid activity and seven-residues (Tyr-DAla-Gly-Phe-Met-Pro-Leu) message sequence for NK1 pharmacophore with Met5-Pro6-Leu7 overlapping for both of them (Fig. 1).
Figure 1.

Sequences of bifunctional ligands 1-10.
In this article, the contraction of the peptide sequence of 1 in its address regions was examined. The changes in their activities and selectivities were observed and discussed, and analgesic activity of a contracted peptide derivative was confirmed in vivo. It should be stressed that the contracted peptides have several advantages over longer peptides for easier synthesis, lower preparative cost, and being a better template to be orally-available as small molecule peptide mimetics.3e,6
We initiated this research with the optimization of 1 in the Met5-Pro6-Leu7 sequence, which is the address region for both pharmacophores. Since the importance of Met5 and Leu7 were already validated especially for opioid activities,3 the Pro6 was modified into Ala (2) (Table 1). The ligand 2 showed reduced affinities at both human delta opioid receptor (DOR) and rat mu opioid receptor (MOR), and its functional activities as a mu opioid agonist were also reduced compared to those of 1 (EC50 in GTPγS binding assay, 72 nM; IC50 in GPI assay, 1200 nM). The binding affinity of 2 at the rat NK1 receptor (rNK1) was also reduced. 3, 4 and 5, possessing CLeu, Aib and D Pro at the sixth position, respectively, have the same trend in the binding affinities and functional activities: lower binding affinities at DOR, MOR and rNK1 receptors, and decreased functional activity in MVD and GPI assays, compared to those in 1. These results combined with our previous SAR clearly suggested that the Met5-Pro6-Leu7 is a crucial sequence for both opioid and NK1 activities in 1. In fact, ligand 6, which possesses a seven aminoacid sequence with only a missing Met5 compared to the sequence of 1, displayed reduced binding affinities for DOR and MOR, as well as for all the functional activities in the GTPγS binding and the isolated tissue-based assays.
Table 1. Binding affinities and functional activities of bifunctional peptide derivatives at δ/μ opioid receptors and NK1 receptors.
| Opioid agonist activities | NK1 antagonist activities | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| radioligand binding assays | [35S] GTPγS | binding assays | MVD (δ) | GPI (μ) | radioligand binding assays | GPI | |||||||
|
| |||||||||||||
| hDORa,b | rMORa,c | Ki(μ) /Ki(δ) |
hDORa | rMORa | Opioid agonist | hNK1d,e | rNK1d,f | Ki(hNK1) /Ki(rNK1) |
Substance P antagonist | ||||
|
|
|
|
|||||||||||
| no | Ki (nM)g |
Ki (nM)g |
EC50 (nM)h |
Emax (%)i |
EC50 (nM)h |
Emax (%)i |
IC50 (nM)j |
IC50 (nM)j |
Ki (nM)g |
Ki (nM)g |
Ke (nM)k |
||
| 1l | 2.8 | 36 | 13 | 2.9 | 45 | 32 | 42 | 22 | 360 | 0.082 | 0.29 | 3.5 | 25 |
| 2 | 25 | 150 | 6.0 | 2.5 | 26 | 72 | 51 | 12 | 1200 | 0.034 | 1.1 | 32 | 380 |
| 3 | 5.0 | 110 | 22 | 0.26 | 41 | 54 | 52 | 5.1 | 6.8%@1μM | 0.066 | 7.8 | 118 | 2.0 |
| 4 | 3.1 | 63 | 20 | 5.5 | 28 | 100 | 27 | 6.9 | 42%@1μM | 0.12 | 9.8 | 82 | 12 |
| 5 | 13 | 76 | 5.8 | 190 | 25 | 120 | 30 | 8.5 | 25%@1μM | 0.88 | 3.6 | 4.1 | 29 |
| 6m | 50 | 180 | 3.6 | 35 | 16 | 140 | 26 | 400 | 520 | 0.0023 | 1.6 | 696 | 3.6 |
| 7 | 2.9 | 23 | 7.9 | 4.8 | 60 | 14 | 54 | 26 | 45 | 0.09 | 0.29 | 3.2 | Not tested |
| 8 | 0.6 | 33 | 55 | 1.1 | 36 | 0.80 | 51 | 9.1 | 390 | 0.0090 | 0.089 | 9.9 | 2.0 |
| 9 | 5.7 | 15 | 2.6 | 76 | 56 | 37 | 47 | 85 | 260 | 0.097 | 1.6 | 16 | 1.8 |
| 10 | 4.7 | 0.29 | 0.062 | 27 | 87 | 10 | 36 | 21 | 26 | 0.0057 | 4.2 | 737 | 59 |
| Biphalinn | 2.6 | 1.4 | 0.54 | 1.1 | 83 | 2.7 | 8.8 | ||||||
| DAMGO | 37 | 150 | |||||||||||
| L-732,138 | 0.73 | 130 | 180 | 250 | |||||||||
Competition analyses were carried out using membrane preparations from transfected HN9.10 cells that constitutively expressed the DOR and MOR. See reference 3 for detailed procedures.
[3H]DPDPE; Kd = 0.45 ± 0.1 nM.
[3H]DAMGO; Kd = 0.50 ± 0.1 nM.
Competition analyses were carried out using membrane preparations from transfected CHO cells that constitutively expressed rat or human NK1 receptors. See reference 3 for detailed procedures.
[3H]Substance P; Kd = 0.16 ± 0.03 nM.
[3H] Substance P; Kd = 0.40 ± 0.17 nM.
The data were collected from at least two independent experiments performed in duplicate. The Ki values are calculated using the Cheng and Prusoff equation to correct for the concentration of the radioligand used in the assay. See reference 3 for detailed procedures.
The EC50 values were determined from the non-linear regression analysis of data collected from at least two independent experiments performed in duplicate. See reference 3 for detailed procedures.
[Total bound − Basal]/[Basal − Non-specific] × 100.
Concentration at 50% inhibition of muscle contraction at electrically stimulated isolated tissues (n = 4).
Inhibitory activity against the Substance P induced muscle contraction in the presence of 1 μM naloxone, Ke: concentration of antagonist needed to inhibit Substance P to half its activity (n = 4). See reference 3 for detailed procedures.
However, the opioid affinities and activities were rather improved in ligands with six amino-acid residues. Interestingly, compound 7 with Met in between the two message regions showed higher affinities for both DOR and MOR than those in 1 (IC50 = 2.9 and 23, respectively). The EC50 values of 7 in the GTPγS binding assays were 4.8 and 14 nM at DOR and MOR, respectively, consistent with its binding affinities. The delta opioid agonist activity in the MVD assay was at the similar level compared to 1 (26 nM), while its mu opioid agonist activity was improved (IC50 value in GPI assay was 45 nM). The binding affinities of 7 at the human and rNK1 receptors were nearly equivalent to those of 1. 8 has Leu in its fifth position and showed superior affinities in the radioligand binding assays and functional activities in the GTPγS binding assays at both DOR and MOR. 8 also showed two-fold higher IC50 value in the MVD assay, but the IC50 value in the GPI assay for mu opioid agonist activity was comparable to that of 1. The binding affinities and substance P antagonist activity in the GPI assay were improved from those of 1. While ligand 9 possessing Pro6 showed lower binding affinities and functional activities for delta opioid agonist (IC50 = 5.7 nM, EC50 = 76 nM, IC50 = 85 nM for radioligand binding assay, GTPγS binding assay and MVD assay, respectively). The IC50 value of 9 at hNK1 was nearly equivalent to that of 1, but its Ki value for rNK1 was 5-fold decreased from 1.
It should be noted that these three contracted chimeric peptides 7-9 displayed delta selectivity (2.6 to 55-fold) in their opioid agonist activities, but compound 10 with Gly6 showed 16 fold selectivity for the mu receptor opioid. This mu-selectivity of 10 was maintained in the GTPγS binding assays, but the isolated tissue-based assays displayed negligible selectivity. The higher Emax value at DOR (87%) than at MOR (36%) could have some effect on this shift in the delta/mu opioid selectivity. 10 showed 14-fold higher Ki value at hNK1 receptor than that of 1, but its Ki value at the rNK1 was decreased. The IC50 value in the GPI assay against substance P stimulation was 59 nM, which is within three-fold difference from the IC50 values in MVD and GPI assays for opioid agonist activities. These results in the isolated tissue-based assays imply that ligand 10 could work as a well-balanced and potent delta and mu opioid agonist as well as a substance P antagonist in vivo. This potent and balanced trio of activities motivated us to perform in vivo animal studies using compound 10, to confirm its in vivo analgesic efficacy.
Compound 10 was evaluated for in vivo antinociceptive activity in non-injured rats following spinal administration. Rats treated with 10 (10μg/5μL) withdrew the hind paw from a radiant heat source (Hargreaves) at significantly longer latencies than those treated with vehicle 15 min after compound injection (p<0.05) (Fig. 2). Vehicle was 10% dimethyl sulfoxide (DMSO) in 90% distilled water. Next, 10 was evaluated for antiallodynic activity (von Frey) after spinal administration in spinal nerve ligated (SNL) rats. Following intrathecal injection, 10 induced significant attenuation of SNL induced tactile allodynia at 3, 30, and 30μg/5μL when compared to vehicle (p < 0.05). The duration of action of compound 10 was dose-dependent. However, no significant reversal of SNL induced tactile or thermal hypersensitivity was observed following oral administration of 10 compared to vehicle, indicating its limited oral bioavailability (data not shown). These in vivo experiments were performed as previously reported.2a
Figure 2.
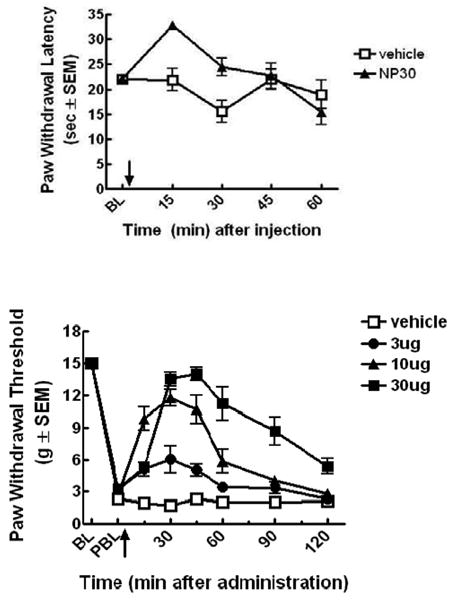
Antinociceptive (top) and antiallodynic (bottom) effect of bifunctional peptide derivative 10 after intrathecal injection. Arrow indicated the time of injection. PBL means post-spinal nerve ligated baseline.
In this report, we performed optimization for truncation of our lead peptide-derived ligand 1 in Met5-Pro6-Leu7, which is a address sequence for both opioid agonist and NK1 antagonist pharmacophores. Among the synthesized derivatives, 10, contracted to six amino acid residues, showed mu-selective opioid and effective NK1 antagonist affinities, together with balanced and potent opioid agonist as well as substance P antagonist activities in isolated tissue-based assays. Moreover, 10 showed significant antinociceptive and antiallodynic activities in vivo. Since the truncated peptide is a good template to design novel peptide-mimetic small molecules, these results indicate that compound 10 could be considered as an important research tool to develop a novel analgesic drugs, possibly with oral bioavailability.
Acknowledgments
The work was supported by grants from the USDHS, National Institute on Drug Abuse, DA-13449 and DA-06284. We thank Dr. Yeon Sun Lee and Dr. Eva Varga for scientific discussion, and Ms. Magdalena Kaczmarska for culturing cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Mantyh PW, Allen CJ, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE. Proc Natl Acad Sci U S A. 1995;92:2622. doi: 10.1073/pnas.92.7.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kalso E. Eur J Pain. 2005;9:131. doi: 10.1016/j.ejpain.2004.05.007. [DOI] [PubMed] [Google Scholar]; (c) King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Neurosignals. 2005;14:194. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 2.(a) Largent-Milnes TM, Yamamoto T, Nair P, Moulton JW, Hruby VJ, Lai J, Porreca F, Vanderah TW. Br J Pharmacol. 2010;161(5):986. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hruby VJ. J Org Chem. 2009;74:9245. doi: 10.1021/jo901767e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Yamamoto T, Nair P, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. J Med Chem. 2007;50:2779. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto T, Nair P, Vagner J, Davis P, Ma SW, Navratilova E, Moye M, Tumati S, Vanderah TW, Lai J, Porreca F, Yamamura HI, Hruby VJ. J Med Chem. 2008;51:1369. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, Navratilova E, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. J Med Chem. 2008;51:6334. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, Davis P, Ma SW, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Med Chem. 2009;52:5164. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nair P, Yamamoto T, Kulkarni V, Moye S, Navratilova E, Davis P, Largent T, Ma SW, Yamamura HI, Vanderah T, Lai J, Porreca F, Hruby VJ. Adv Exp Med Biol. 2009;611:537. doi: 10.1007/978-0-387-73657-0_235. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yamamoto T, Nair P, Ma SW, Davis P, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. Bioorg Med Chem. 2009;17(20):7337. doi: 10.1016/j.bmc.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Yamamoto T, Nair P, Jacobsen NE, Vagner J, Kulkarni V, Davis P, Ma SW, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Med Chem. 2009;52(16):5164. doi: 10.1021/jm900473p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yamamoto T, Nair P, Jacobsen NE, Kulkarni V, Davis P, Ma SW, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Med Chem. 2010;53(15):5491. doi: 10.1021/jm100157m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yamamoto T, Nair P, Largent-Milnes TM, Jacobsen NE, Davis P, Ma SW, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. J Med Chem. 2011;54(7):2029. doi: 10.1021/jm101023r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horan PJ, Mattia A, Bilsky EJ, Weber S, Davis TP, Yamamura HI, Malatynska E, Appleyard SM, Slaninova J, Misicka A, Lipowski AW, Hruby VJ, Porreca F. J Pharmacol Exp Ther. 1993;265:1446. [PubMed] [Google Scholar]
- 5.(a) Cascieri MA, Macleod AM, Underwood D, Shiao LL, Ber E, Sadowski S, Yu H, Merchant KJ, Swain CJ, Strader CD, Fong TM. J Biol Chem. 1994;269:6587. [PubMed] [Google Scholar]; (b) MacLeod AM, Merchant KJ, Cascieri MA, Sadowski S, Ber E, Swain CJ, Baker R. J Med Chem. 1993;36:2044. doi: 10.1021/jm00066a015. [DOI] [PubMed] [Google Scholar]; (c) Millet R, Goossens L, Goossens JF, Chavatte P, Bertrand-Caumont K, Houssin R, Henichart JP. J Pept Sci. 2001;7:323. doi: 10.1002/psc.326. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hruby VJ, Nair P, Yamamoto T. U.S. Patent 8026218. 2007; (b) Ballet S, Feytens D, Buysse K, Chung NN, Lemieux C, Tumati S, Keresztes A, Van Duppen J, Lai J, Varga E, Porreca F, Schiller PW, Vanden Broeck J, Tourwé D. J Med Chem. 2011;54(7):467. doi: 10.1021/jm1016285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipkowski AW, Misicka A, Davis P, Stropova D, Janders J, Lachwa M, Porreca F, Yamamura HI, Hruby VJ. Bioorg Med Chem Lett. 1999;9:2763. doi: 10.1016/s0960-894x(99)00464-3. [DOI] [PubMed] [Google Scholar]