

Abstract
The retinal pigment epithelium (RPE) is a highly specialized CNS tissue that plays crucial roles in retinal homeostasis. Age-related morphological changes in the RPE have been associated with retinal degenerative disorders; our understanding of the underlying molecular mechanisms, however, remains incomplete. Here we report on a key role of Klotho (Kl), an aging-suppressor gene, in retinal health and RPE physiology. Kl−/− mice show RPE and photoreceptor degeneration, reduced pigment synthesis in the RPE, and impaired phagocytosis of the outer segment of the photoreceptors. Klotho protein (KL) is expressed in primary cultured human RPE, and regulates pigment synthesis by increasing the expression of MITF (microphthalmia transcription factor) and TYR (tyrosinase), two pivotal genes in melanogenesis. Importantly, KL increases phagocytosis in cultured RPE by inducing gene expression of MERTK/AXL/TYRO3. These effects of KL are mediated through cAMP-PKA-dependent phosphorylation of transcription factor CREB. In cultured human RPE, KL increases the l-3,4-dihydroxyphenylalanine synthesis and inhibits vascular endothelial growth factor (VEGF) secretion from basal membrane by inhibiting IGF-1 signaling and VEGF receptor 2 phosphorylation. KL also regulates the expression of stress-related genes in RPE, lowers the production of reactive oxygen species, and thereby, protects RPE from oxidative stress. Together, our results demonstrate a critical function for KL in mouse retinal health in vivo, and a protective role toward human RPE cells in vitro. We conclude that KL is an important regulator of RPE homeostasis, and propose that an age-dependent decline of KL expression may contribute to RPE degeneration and retinal pathology.
Introduction
The Klotho (Kl) gene was first described by Kuro-o et al. (1997), as a putative anti-aging gene, since Kl-null mice displayed phenotypes resembling human premature aging syndromes. Recent reports have suggested that Klotho protein (KL) activates the Ca2+ channel, transient receptor potential cation channel, superfamily V member 5 (TRPV5; Huang, 2012), influences intracellular signaling pathways including insulin-like growth factor-1 (IGF-1; Kurosu et al., 2005; Wolf et al., 2008), p53/p21 (de Oliveira, 2006), cAMP (Yang et al., 2003; Rakugi et al., 2007; Wang et al., 2012b), PKC (Imai et al., 2004), Wnt (Liu et al., 2007), and Na+/K+ ATPase; and regulates oxidative stress responses (Yamamoto et al., 2005; Sopjani et al., 2011). Circulating KL levels decline with aging, and thereby could increase the risk factor for age-related and chronic diseases (Yamazaki et al., 2010; Semba et al., 2011).
A recent study showed that Klotho enhances oligodendrocyte maturation and myelination of the optic nerve (Chen et al., 2013). Despite these recent advances, the effects of KL on retinal health and RPE physiology have not yet been examined.
The retinal pigment epithelium (RPE) is a monolayer of pigmented and polarized CNS tissue with crucial roles in retinal homeostasis, including the formation of the blood/retina barrier by tight junctions, transportation of nutrients such as glucose or vitamin A from blood to the photoreceptors, conveyance of water from subretinal space to the blood, establishment of immune privilege of the eye, and a constant ion composition in the subretinal space. The RPE participates in light absorption and isomerization of the retinol in the visual cycle, secretion of growth factors, and phagocytosis of the outer segments of the photoreceptors. The RPE is remarkable in its ability to transport fluid and secrete a variety of growth factors, neurotrophic factors, proinflammatory cytokines, and chemokines in a polarized manner. The apical site of RPE is in close proximity to the outer segments of photoreceptors that are phagocytosed, and the basolateral membrane is opposed to Bruch's membrane (BM) and the choroid blood supply (Holtkamp et al., 1998; Strauss, 2005; Maminishkis et al., 2006).
Numerous studies have reported on age-related morphological changes in RPE (Boulton and Dayhaw-Barker, 2001; Bonilha, 2008; Kozlowski, 2012) such as loss of melanin granules (Bonilha, 2008), microvilli atrophy and disorganization of the basal infoldings (Boulton and Dayhaw-Barker, 2001), mitochondrial DNA damage (Lin et al., 2011), elevated amyloid β production (Wang et al., 2012a), enhanced tissue factor expression (Cho et al., 2011), increased activity of acidic β-galactosidase suggestive of lysosomal dysfunction (Matsunaga et al., 1999; Kurz et al., 2000), and altered expression of several RPE structural proteins (Shelton et al., 1999; Gu et al., 2012), among others. However, to date the mechanisms causing the age-related changes in RPE morphology and function remain poorly understood.
Here we investigated the role played by KL in RPE physiology, focusing on signaling pathways that are impaired during aging and age-related retinal degeneration. We show that KL plays a protective role in RPE cells in vitro and in vivo, and, thus, a decline in KL expression may contribute to age-related retinal pathology.
Materials and Methods
Histological analysis of Kl wild-type and mutant retina
To compare the RPE from wild-type control (Kl+/+) and mutant Kl−/− mice, eyeballs from four 2-month-old Kl+/+ C57BL mice (n = 8) and age-matched Kl−/− mice (n = 8) on a C57BL/6 background, of either sex were obtained as a gift from Dr. Makoto Kuro-o (University of Texas Southwestern Medical Center, Dallas, TX). The eyeballs from one Kl+/+ and Kl−/− pair were fixed in 4% formalin buffered in 0.2 m phosphate buffer for 16 h at 4°C, cryosectioned at 20 μm, and used for histochemistry. The remaining three pairs of eyes were dehydrated in an ascending series of ethanol infusions, infiltrated with catalyzed JV-4 methacrylate resin, and embedded. Sections (1.5 μm thick) were cut with dry glass knives, dried down onto subbed slides, stained with 1% toluidine blue in acetate buffer (every seventh section was retained for mounting), and coverslipped. Tissue sections were viewed using a 100× (oil-immersion) objective. For electron microscopic (EM) imaging, tissues were double fixed in PBS-buffered glutaraldehyde (2.5%) and osmium tetroxide (0.5%), dehydrated, and embedded into Spurr's epoxy resin. Ultrathin sections (90 nm) were cut and double stained with uranyl acetate and lead citrate, and were analyzed with a JEOL JEM 1010 transmission electron microscope.
RPE cultures
Human primary cultures of RPE cells at passage 2 were purchased from ScienCell or established in our laboratory from the eyes of organ donors following a previously described protocol (Maminishkis et al., 2006). RPE were cultured in EpiCM epithelial cell medium (ScienCell), as described previously (Kokkinaki et al., 2011).
RPE functional assays
For the phagocytosis assays, the RPE cells were grown to confluency for 20 h on poly-d-lysine (2 μg/cm2, BD Biosciences) and laminin-coated (4 μg/ml, Sigma-Aldrich) 96-well plates (50,000 cells/well) in EpiCM medium without serum. Cultures were then incubated with recombinant Klotho protein (10 ng/ml; R&D Systems) for 30 min or 16 h. Phagocytosis was assayed by adding 200 μg of Alexa Fluor 488-conjugated zymosan (Invitrogen) for 1 h, and was quantified by fluorometry using the Vybrant Phagocytosis Assay (Invitrogen). RPE cell viability was measured with the PrestoBlue Cell Viability Reagent (Invitrogen) and quantified by fluorometry.
To measure KL-dependent changes in vascular endothelial growth factor (VEGF) secretion, the RPE cells were polarized for 4 weeks in six-well Transwell dishes (Corning) before treatment with recombinant KL (10 ng/ml) for 72 h, and the medium from both the upper and lower reservoirs was collected every 24 h and quantified by ELISA (ATCS).
To induce acute oxidative stress in the RPE, the cells were treated with 500 μm tertiary-butyl hydroperoxide (tBH; Sigma-Aldrich) for 2 h. RPE cells were grown on six-well plates (200,000 cells/well) and cultured for 20 h in EpiCM media without serum, followed by 16 h of incubation with KL (100 pm) in the same media. Two hours before the end of incubation with KL, 500 μm tBH was added to induce oxidative stress for 2 h, and then RNA was isolated for gene expression analysis by quantitative real-time PCR (qRT-PCR). RNA was isolated from each well and was analyzed separately by qRT-PCR. Each reaction was performed in triplicate (total of nine samples from each condition). Relative expression values for each gene were calculated using the ΔΔCt method after normalization to GAPDH.
To increase the intracellular levels of cAMP, the RPE were treated for 1 h with 100 μm isoproterenol, an α1- and β2-adrenoreceptor agonist and adenylylcyclase activator (Sigma-Aldrich) or with 1 mm 3-isobutyl-1-methylxanthine (IBMX, Sigma-Aldrich), a competitive nonselective phosphodiesterase inhibitor. Isoproterenol or IBMX was then removed, and the cells were cultured in EpiCM without serum for 16 h. To induce oxidative stress after isoproterenol or IBMX treatment, 500 μm tBH was added 2 h before the end of the 16 h period, followed by RNA isolation and qRT-PCR. Working concentrations for isoproterenol (100 μm) and IBMX (1 mm) were chosen based on established protocols (Montague and Cook, 1971; Zhang and Insel, 2001).
KL gene knockdown
siRNA specific for human KL transcript was purchased from Sigma-Aldrich (catalog #EHU131091). For each experiment, three independent transfections with 2 × 105 RPE cells and 200 pmol siRNA were performed using the high-efficiency I-013 protocol for the Amaxa Nucleofector II apparatus and the Basic Nucleofector Kit for Primary Mammalian Epithelial Cells Solution Mix (catalog #VPI-1005) from Lonza. To assess siRNA knockdown of the target protein (KL), cells were plated in 24-well plates (1 well per transfection) with 0.6 ml of EpiCM media per well and harvested for total RNA isolation at 24–48 h. To assess RPE phagocytotic activity after siRNA transfection, we used Lipofectamine RNAiMax Reagent (Invitrogen) as an alternative transfection method, as the process of nucleofection on its own already resulted in an increase in basal phagocytosis. The same number of cells and concentration of siRNA were used and the RPE cells were harvested at 24 h for analysis of gene expression or subjected to the phagocytosis assay at 48–72 h. A scrambled siRNA (Invitrogen) was used as a negative control in all assays involving gene expression knock-down experiments.
Quantitative real-time PCR
Total RNA was extracted with the RNeasy kit (Qiagen), treated with RNase-free DNase I (Qiagen), and reverse transcribed with oligo-dT using the SuperScript III cDNA synthesis kit (Invitrogen). Quantitative PCR was performed with the QuantiTect SYBR Green PCR Kit (Qiagen). Specific primers for each gene were designed with the PrimerQuest software (Integrated DNA Technologies), and the cDNA sequences of each gene (GenBank) were used to produce 100–250 bp PCR amplicons that span one or more exon/intron boundaries. For gene expression analysis of mouse genes, RNA was isolated from the retina of Kl−/−, Kl+/+, old and young mice of either sex.
Antibodies
Primary antibodies.
The primary antibodies used were as follows: rat anti-Klotho from TransGenic; mouse anti-MITF (microphthalmia transcription factor) from Millipore; mouse anti-ACTB, rabbit anti-α adducin, and rabbit anti-phospho (Ser481) adducin from Abcam; rabbit anti-TYR (tyrosinase) from Sigma-Aldrich; and rabbit anti-CREB, rabbit anti-phospho (Ser133) CREB, rabbit anti-VEGF receptor 2 (VEGFR2), rabbit anti-phospho (Tyr996) VEGFR2, rabbit anti-IGF-1 receptor (IGF-1R) β, mouse anti-phospho-tyrosine, rabbit anti-SHP-2 (Src homology region 2-containing protein tyrosine phosphatase-2), and rabbit anti-phospho (Tyr542) SHP-2, from Cell Signaling Technology.
Secondary antibodies.
The secondary antibodies used were as follows: donkey anti-rat TRITC-linked from Invitrogen; and goat anti-rabbit HRP-linked and goat anti-mouse HRP-linked from Cell Signaling Technology.
Immunostaining
RPE cells grown on plastic four-well chamber slides (Thermo Fisher Scientific) were stained using established protocols for the primary and secondary antibodies. Stained cells were mounted with anti-fading medium (Invitrogen), and images were captured by confocal microscopy (FV1000 Confocal Microscope, Olympus).
Immunoprecipitation
RPE cells were treated with 75 ng/ml IGF-1 and with increasing doses of KL (0, 100, 200, and 400 pm) for 15 min in serum-free EpiCM, and were lysed immediately for immunoprecipitation with IGF-1R antibody. RPE cells were homogenized in ice-cold immunoprecipitation buffer (1% NP-40 buffer, 25 mm Tris-HCl pH 7.4, 10 mm sodium orthovanadate, 10 mm sodium pyrophosphate, 100 mm sodium fluoride, 10 mm EDTA, and 10 mm EGTA) supplemented with Protease and Phosphatase Inhibitor Cocktail Tablets (Roche Applied Science), 1× Protease Inhibitor Cocktail Set I (EMD Millipore), and 1 mm phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich), and centrifuged for 10 min at 12,000 rpm. Homogenates were precleared with protein-A agarose beads following the manufacturer's instructions (Cell Signaling Technology). The protein concentration was measured by the Bradford assay (Bio-Rad) and was adjusted at 1 mg/ml. Homogenates containing 300 μg of total protein were incubated with antibody anti-IGF-1R (Cell Signaling Technology) diluted 1:100 for 16 h at 4°C, and then protein-A agarose beads were added for 2 h. The immunoprecipitates were rinsed in lysis buffer three times and then subjected to immunoblot analysis with anti-phospho-tyrosine (Cell Signaling Technology) or anti-IGF-1R antibodies. Total precipitated IGF-1R was also detected for normalization.
Immunoblot analysis
Protein samples were extracted in radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 0.5% sodium deoxycholate, and 1% SDS in 1× PBS), containing freshly added Protease and Phosphatase Inhibitor Cocktail Tablets (Roche Applied Science), 1× Protease Inhibitor Cocktail Set I (EMD Millipore), 1 mm sodium vanadate, 50 mm sodium fluoride, and 1 mm PMSF (Sigma-Aldrich). Protein concentrations were measured by Bradford assay (Bio-Rad). Protein samples were analyzed using the NuPAGE electrophoresis and XCell Western blot system (Invitrogen). Primary and secondary antibodies were used based on the manufacturer's instructions. Immunoreactive protein bands were visualized by the SuperSignal West Dura Chemiluminescent Substrate (Pierce) followed by x-ray film imaging.
For detection of phospho-VEGFR2, RPE cells were treated with 200 pm KL for 15 min in serum-free EpiCM. VEGFA was added in the last 2 min of the incubation with KL at a concentration of 100 ng/ml, followed by cell lysis and Western blot analysis with an antibody to phospho-(Tyr996) of VEGFR2. Equal amounts (35 μg) of total protein were loaded from each sample. Total VEGFR2 was also detected for normalization.
For detection of phospho-SHP-2, RPE cells were treated with increasing doses of KL (0, 100, 200, and 400 pm) for 48 h in serum-free EpiCM and lysed for Western blot analysis with an antibody to phospho-SHP-2. Total SHP-2 protein levels were detected for normalization.
Measurement of cAMP
RPE monolayers were cultured in 96-well plates in EpiCM without serum for 20 h and incubated with 10 ng/ml Klotho protein for various time intervals, as described in the Results. The levels of intracellular cAMP were measured using the cAMP-Glo Assay (Promega) following the manufacturer's protocol.
Measurement of l-dihydroxyphenylalanine by mass spectrometry
RPE monolayers were grown to confluency in 24-well plates. The RPE cells were starved in serum-free EpiCM for 20 h, before treatment with 2 nm KL protein (R&D Systems) for 72 h. After the treatment with KL, the cells were scraped, collected in 150 μl of water, and sonicated for 10 s for cell lysis. A sample of 5 μl was mixed with RIPA buffer before sonication and used for quantification of total protein concentration with Bradford assay. An internal standard [d3-l-3,4-dihydroxyphenylalanine (l-DOPA), Sigma-Aldrich] was added at a concentration of 250 nm and incubated for 10 min on ice. Samples were vortexed and spun at 13,000 rpm for 20 min at 4°C. The supernatant was dried down completely and reconstituted in 100 μl of water, which was used for the TQ-S mass spectrometry analysis. The endogenous l-DOPA concentration was determined by normalization to the internal standard and the protein concentrations.
Measurement of reactive oxygen species
The OxiSelect Intracellular ROS (reactive oxygen species) Assay Kit (Cell Biolabs) was used. RPE monolayers were cultured in 96-well plates with serum-free EpiCM for 20 h and then loaded with 1 mm cell-permeable fluorogenic probe 2′-7′-dichlorodihydrofluorescein diacetate for 1 h, followed by treatment with 2 nm KL for 24 h. During the last 2 h of KL treatment, 500 μm tBH (Sigma-Aldrich) was added to induce oxidative stress. The fluorescence intensity of each sample, proportional to the ROS levels, was measured against the fluorescence of the provided standard, using a Tecan Ultra 384 plate reader, following the manufacturer's protocol.
Results
Klotho knock-out mice exhibit an RPE degenerative phenotype
Melanin granules play an important role in light absorption and protection of the retina against light-induced toxicity and oxidative stress (Cai et al., 2000; Seagle et al., 2005a,b; Wang et al., 2006). Studies have shown that aging reduces the melanin content, and this decrease correlates with light-induced cell apoptosis (Schmidt and Peisch, 1986; Sarna et al., 2003). However, the mechanism by which aging induces loss of melanin in RPE cells is not known. Analysis of Kl−/− eyes by light microscopy revealed a decrease in pigmentation in the RPE cells of Kl−/− mice compared with Kl+/+ mice (Fig. 1A,B). Kl−/− RPE cells contained significantly fewer melanin granules (28.25 ± 1.45) than Kl+/+ RPE cells (36.0 ± 2.2; p < 0.04; Table 1). These morphological changes suggest that Kl plays a regulatory role in melanin synthesis in RPE cells in vivo. In addition, the layers of the choroid of Kl−/− mice appeared to be deformed by large blood vessels, as shown in Figure 1B.
Figure 1.
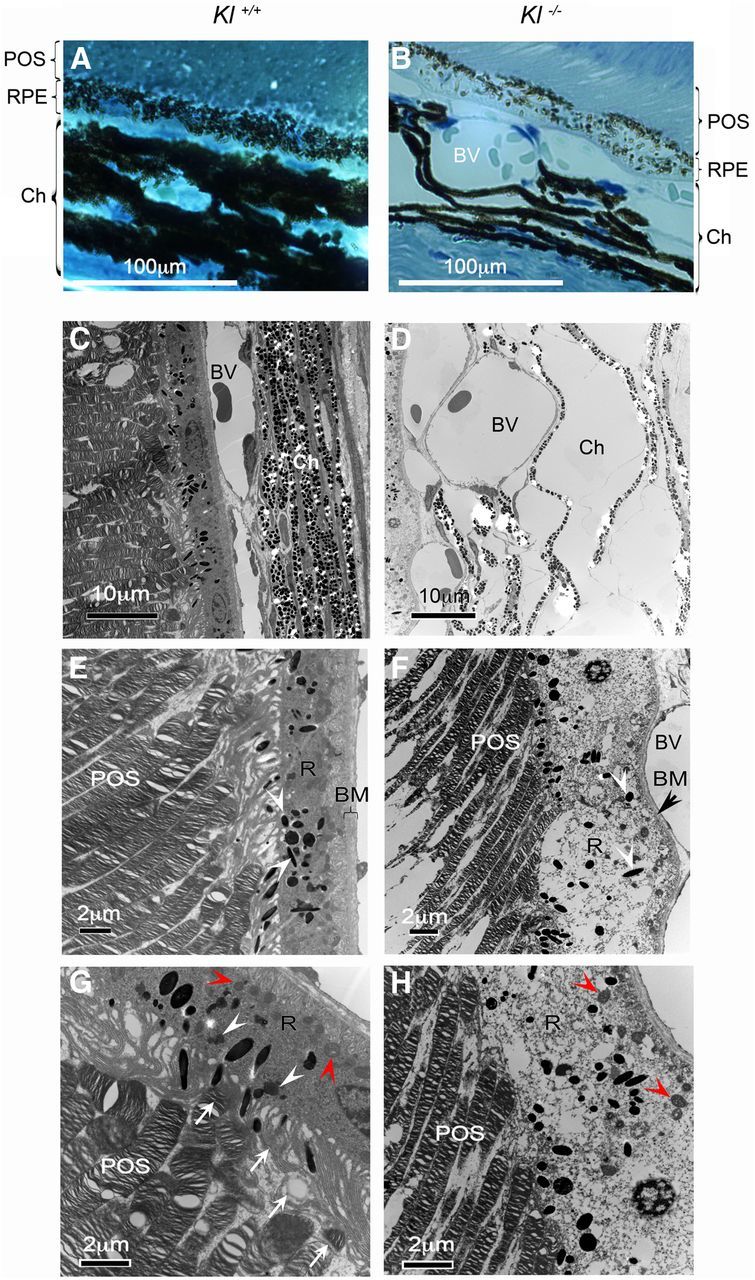
Klotho knock-out mice exhibit degenerative phenotypes in the retina. A, B, Light microscopy images of the retina of adult Kl+/+ and Kl−/− mice. The number of pigmentation granules in RPE cells is reduced by 30%. Eyes from four 6-week-old Kl−/− (B) and four age-matched Kl+/+ (A) mice were dissected for histological analysis. The number of melanin granules was counted in 10 RPE cells from each mouse; the statistical analysis of the results is presented in Table 1. The choroid (Ch) of the Kl−/− mice showed deformed layers separated by dilated blood vessels (BV; B). POS, Photoreceptor outer segments. C–H, Electron microscopy images of the retinal region of adult Kl+/+ and Kl−/− mice: choroid, RPE, and outer segments of the photoreceptors of wild-type mice (C); choroid and RPE of Kl−/− mice (D; the choroidal region appears grossly deformed with tissue layers that have been separated by severely dilated BVs; RPE (R) of Kl+/+ mice, showing normal cytoplasmic content, normal mitochondria, and Bruch's membrane (E); the RPE (R) of Kl−/− mice are degenerated with light cytoplasmic content, damaged mitochondria (H, red arrowheads), and melanosomes with disorganized distribution (F, white arrowheads). BM is thinner, deformed by dilated blood vessels, showing significant degeneration; F, black arrowhead). The POSs are thinner, showing signs of degeneration (F, H) compared with the Kl+/+ photoreceptors (E, G); phagocytosis of the POSs is observed in the RPE cells of Kl+/+ mice (G); arrows indicate presumptive phagocytotic vesicles at different stages of the phagocytosis process. White arrowheads show the mature lysosomes. Red arrowheads show normal mitochondria. H, No signs of phagocytosis of the outer segments of photoreceptors were observed in the RPE cells of Kl−/− mice. Red arrowheads show damaged mitochondria.
Table 1.
Comparison of numbers of melanin granules of RPE between Kl+/+ and Kl−/− mice
| Mouse 1 (10 cells) |
Mouse 2 (10 cells) |
Mouse 3 (10 cells) |
Mouse 4 (10 cells) |
Average ± SE |
Melanin granules |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kl+/+ | Kl−/− | Kl+/+ | Kl−/− | Kl+/+ | Kl−/− | Kl+/+ | Kl−/− | Kl+/+ | Kl−/− | Kl+/+ | Kl−/− | |
| Melanin granules per cell | 39.2 | 24.2 | 32.8 | 30.0 | 30.6 | 27.0 | 41.4 | 31.8 | 36.0 ± 2.2* | 28.25 ± 1.45 | 100% | 70% |
*p < 0.04 by t test.
Analysis of the retina and its supporting tissues at the ultrastructural level revealed several striking abnormalities in Kl−/− mice (Fig. 1C–H). At 6 weeks of age, the choroid in Kl+/+ mice appeared normal, with densely packed layers (Fig. 1C). In the Kl−/− mice, the choroid layers were deformed by severely dilated blood vessels (Fig. 1D). Moreover, in the Kl−/− mice the BM appeared thinner and deformed by the dilated choroidal vessels (Fig. 1F). The Kl−/− RPE appeared degenerated with a light cytoplasmic content (Fig. 1F) and disorganized melanosome distribution (Fig. 1F, white arrowheads) compared with Kl+/+ RPE (Fig. 1E, arrowheads). Higher magnification of the Kl−/− RPE also revealed damaged mitochondria (Fig. 1H, red arrowheads) compared with control RPE (Fig. 1G, red arrowheads). The outer segments of photoreceptor cells were thinner, showing signs of degeneration in the Kl−/− mice (Fig. 1F,H) compared with the outer segments of photoreceptors in Kl+/+ mice (Fig. 1E,G). The phagocytosis of the outer segment of photoreceptors by RPE appeared to be absent in the Kl−/− mice compared with Kl+/+ mice (Fig. 1H,G). Arrows in Figure 1G show various stages of phagocytosis; white arrowheads show mature lysosomal vesicles in the Kl+/+ RPE compared with the absence of these structures in the Kl−/− RPE (Fig. 1H). Collectively, these observations suggest that KL plays a key role in several important functions of the RPE and retina in vivo, including melanogenesis, phagocytosis of photoreceptor outer segments, and integrity of BM.
KL regulates melanogenesis and pigment synthesis in RPE
MITF is a master regulatory gene for survival of melanocytes (Tassabehji et al., 1994; Goding, 2000) and a key transcription factor that regulates the expression of TYR, the rate-limiting enzyme responsible for melanin biosynthesis in the RPE (Schraermeyer et al., 2006; Wang and Hebert, 2006). To examine the effect of KL on melanogenesis, we cultured primary human RPE cells. As shown in Figure 2A, cultured human RPE cells were immunoreactive for anti-Klotho, and labeling was primarily localized to the cell membrane, demonstrating KL expression in human RPE. To directly test a regulatory role of KL on human RPE pigmentation, RPE cells were cultured for 16 h in the presence or absence of a physiological concentration of recombinant KL protein (100 pm), and gene expression levels of MITF and TYR were measured by qRT-PCR. Bath-applied KL significantly increased MITF and TYR gene expression (Fig. 2B; p = 0.0003 and 0.0065, respectively). Conversely, transfection of human RPE cells with KL siRNA for 48 h significantly reduced the expression of MITF and TYR (p = 0.0034 and 0.0009, respectively), demonstrating that in RPE cells KL regulates genes involved in melanin biosynthesis (Fig. 2C). KL siRNA reduced KL mRNA expression to 10% of controls (Fig. 2C), while the cell viability remained at 80–90%. Western blot analysis of KL-treated RPE cells revealed a concentration-dependent increase in MITF (with 400 pm KL) and TYR (with 200 pm KL) protein levels at 24 and 48 h, respectively (Fig. 2D). MITF is strongly expressed in RPE, and that might explain the need of a higher KL concentration to increase the basal level of MITF in vitro. The delay in TYR increase might be explained by the fact that MITF is required before induction of TYR expression. Since both MITF and TYR genes are regulated through their cAMP response elements (Bertolotto et al., 1998; Widlund and Fisher, 2003; Wan et al., 2011) and MITF also regulates TYR gene expression (Reinisalo et al., 2012), we analyzed the ability of KL to regulate cAMP levels in RPE. The treatment of human RPE with bath-applied KL for various time intervals showed that cAMP rapidly increased and peaked after 10 min. This was followed by a decrease to basal cAMP levels after 20 min (Fig. 2E). We propose that the transient activation of cAMP induced by KL is sufficient to induce an increase in downstream gene expression after 16 h. To further test this hypothesis, we incubated human RPE cells for 1 h with 1 mm IBMX, a phosphodiesterase inhibitor, as an independent means to elevate intracellular cAMP levels. Similar to KL, IBMX increased MITF and TYR gene expression after 16 h (Fig. 2F), suggesting that KL induced MITF and TYR gene expression by increasing intracellular cAMP levels. Furthermore, treating RPE cells with isoproterenol, an α1- and β2-adrenoreceptor agonist known to stimulate RPE cAMP levels (Pierce et al., 2002), also increased MITF and TYR gene expression after 16 h (Fig. 2G), suggesting that a Gs-coupled GPCR may be activated by KL to elevate intracellular cAMP.
Figure 2.
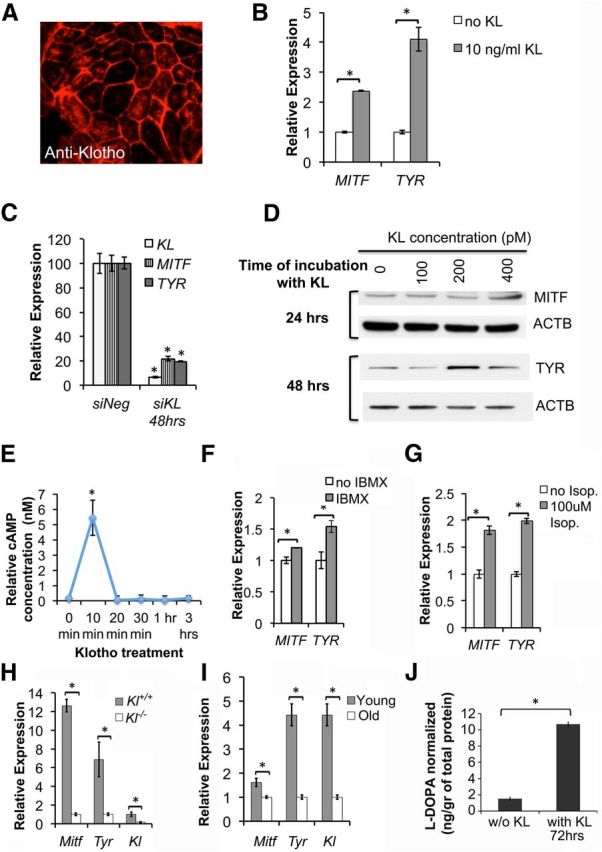
KL regulates melanogenesis and pigment synthesis in RPE cells. A, KL protein is expressed in primary human RPE, as shown by anti-Klotho immunolabeling (red). B, MITF and TYR gene expression in human RPE cultures is significantly increased following treatment with KL 10 ng/ml (100 pm) for 16 h. Relative gene expression was assessed by qRT-PCR analysis of RNA samples isolated from untreated RPE and RPE treated with KL. p = 0.0003 (MITF) and p = 0.0065 (TYR). C, The expression levels of MITF and TYR mRNAs were significantly reduced in human RPE transfected with specific siRNA for KL (siKL), compared with RPE transfected with scrambled siRNA (siNeg), as shown by qRT-PCR. p = 0.0027 (KL), p = 0.0034 (MITF), and 9 × 10−4 (TYR). Results in B and C represent the average of three independent experiments, and the qRT-PCR was performed in triplicate. RNA samples were normalized to human GAPDH, and the relative expression levels for each gene were calculated with the ΔΔCt method. D, Treatment of human RPE with KL protein increased the protein levels of MITF and TYR in a dose- and time-dependent manner, as shown by Western blot analysis with MITF and TYR antibodies. β-actin (ACTB) was used as a normalization control. E, Treatment of human RPE for 10 min with KL protein (100 pm) leads to a fivefold increase in cAMP levels. The cells were grown in 96-well plates (50,000 cells/well), and 6 wells were measured at each time point. The relative concentration of cAMP was calculated for each sample using a standard curve. The experiment was repeated twice, and the average concentrations for each time point are presented in the graph. The asterisk shows the increase in cAMP levels at 10 min of KL treatment, which is statistically significant as determined by t test (p = 0.0023). F, IBMX, an inhibitor of phosphodiesterases, induces MITF and TYR gene expression similarly to KL, by maintaining elevated levels of cellular cAMP. p = 0.0021 (MITF) and p = 0.0049 (TYR). G, Isoproterenol, an α1- and β2-adrenoreceptor agonist that raises intracellular cAMP levels, can also induce MITF and TYR gene expression similarly to KL. Human RPE cells were treated with 100 μm isoproterenol for 1 h, then isoproterenol was removed and RNA was isolated 16 h later for qRT-PCR. p = 0.0108 (MITF) and p = 0.0069 (TYR). H, A significant decline in the expression of Mitf and Tyr genes was observed in the retina of Kl−/− mice compared with the Kl+/+ mice. RNA samples isolated from the retinas of three Kl+/+ and three Kl−/− mice were used separately for the qRT-PCR analysis. Each sample was analyzed in triplicate, normalized to mouse β-actin (Actb), and the relative expression levels for each gene were calculated with the ΔΔCt method. The average relative expression ± SD for each sample is shown in the graph. p = 0.001 (Mitf), p = 0.030 (Tyr), and p = 0.001 (Kl). I, Age-dependent decline in the expression of Mitf, Tyr, and Kl genes is observed, as demonstrated by qRT-PCR on retinal samples of six young (3 months old) and six old (13 months old) mice. Total RNA was isolated from six C57BL/6 mice of each age group, and the qRT-PCR for each sample was performed in triplicate, as described in H. p = 0.025 (Mitf), p = 0.008 (Tyr), and p = 0.008 (Kl). Asterisks in B, C, F, G, H, and I indicate samples with significant differences in gene expression compared with the control, as determined by the t test (p < 0.05). J, l-DOPA synthesis in human RPE cells treated with 2 nm KL protein for 72 h showed significant increase (p = 0.01) as determined by mass spectrometry analysis, compared with l-DOPA synthesis in RPE cells not treated with KL in the same culture conditions. The mass spectrometry data were normalized based on an internal standard (d3-l-DOPA) and the protein concentration. The results of three independent experiments are presented. The asterisk indicates significant increase in the presence of KL, determined by t test.
To confirm the regulatory effect of Kl gene on Mitf and Tyr gene expression in vivo, the retinae of 6-week-old Kl−/− and Kl+/+ mice of either sex were isolated and analyzed by qRT-PCR. Our data showed a significant decrease in the level of Mitf (p = 0.001) and Tyr (p = 0.030) gene expression in the retina of the Kl−/− mice compared with wild-type controls (Fig. 2H).
It has been reported that circulating levels of KL are decreased during aging (Yamazaki et al., 2010). To test whether Kl expression declines with aging in the wild-type retina, we analyzed the Kl gene expression levels in the retina of six young (3 months old) and six old (13 months old) C57BL/6 wild-type mice of either sex. Figure 2I shows that retinal Kl gene expression decreases with aging (p = 0.008). In addition, to verify whether the decline in Kl gene expression translates into a decrease in Mitf and Tyr, and therefore to a decrease in melanin synthesis during aging, we compared the gene expression levels of Mitf and Tyr in the retinae of young and old mice. Our data confirmed a significant decrease in the Mitf (p = 0.025) and Tyr (p = 0.008) gene expression levels in the retinae of aged mice compared with those of young mice (Fig. 2I).
l-DOPA serves as a precursor to both melanins and catecholamines, acting along separate pathways (Slominski et al., 2004). It has been reported that both l-tyrosine and l-DOPA stimulate, induce, or regulate various elements of the melanogenic pathways (Slominski et al., 1988, 1989a,b; Slominski and Costantino, 1991a,b; Halaban et al., 2001, 2002). Since our data showed that KL regulates MITF and TYR expression and therefore affects pigmentation, we further investigated the role of KL on l-DOPA secretion in human RPE. The incubation of human RPE cells with 2 nm KL for 72 h followed by mass spectrometry showed a significant increase in the endogenous l-DOPA levels, as shown in Figure 2J.
Together, our studies with cultured RPE cells support the hypothesis that KL plays an important role in melanin synthesis and the maintenance of pigmentation in human and mouse RPE cells, and that a decrease in KL during aging might lead to impaired RPE function.
KL regulates phagocytosis in human and mouse RPE by upregulating MERTK (MER receptor tyrosine kinase) gene expression
Phagocytosis of the outer segment of photoreceptors is a crucial function of the RPE, and an aging-induced decline in phagocytosis is thought to contribute to age-related macular degeneration (AMD; Sun et al., 2007). To assess whether KL has a regulatory effect on human and mouse RPE phagocytosis, we performed an EM analysis of the RPE and the outer segment of photoreceptors of the Kl−/− and Kl+/+ retinae. The arrows in Figure 1G show various stages of the photoreceptor phagocytosis, and the white arrowheads point to mature lysosomes in the RPE of Kl+/+ mice, whereas these stages are absent in the Kl−/− RPE (Fig. 1H). To investigate the mechanism by which KL may regulate phagocytosis, we treated primary human RPE cells with recombinant KL for either a 30 min or for a 16 h interval and proceeded with the phagocytosis assay. The incubation of RPE cells with KL for 30 min decreased phagocytosis (p = 7.8 × 10−5), whereas long-term incubation (16 h) significantly increased (p = 1.3 × 10−6) phagocytosis in RPE cells (Fig. 3A). Conversely, transfection of RPE with KL siRNA significantly (p = 2.3 × 10−4) decreased phagocytosis at 72 h, further confirming a regulatory role of KL on phagocytosis (Fig. 3C). To verify the involvement of cAMP and GPCR signaling in the regulation of phagocytosis by KL, we treated the RPE cells with 1 mm IBMX or 100 μm isoproterenol and observed after both treatments the same pattern of an increase in phagocytosis efficiency similar to that observed with bath-applied KL treatment (Fig. 3A,B). However, it should be pointed out that since long-term incubation with isoproterenol or IBMX is toxic to the cells, we incubated the cells for only 1 h with isoproterenol or IBMX and measured phagocytosis 30 min and 16 h after removal of the drug. This might explain the lower increase in the phagocytosis rate with isoproterenol compared with KL (Fig. 3B). It has been reported that an increase in intracellular cAMP acutely reduces phagocytotic activity by β-adrenergic stimulation and by stimulation of adenosine A2 receptors (Hall et al., 1993; Gregory et al., 1994; Beitz et al., 1998). To further delineate the mechanism by which KL may regulate phagocytosis in RPE cells, we verified the expression of genes that are implicated in phagocytosis. MERTK is a member of the MER/AXL/TYRO3 receptor kinase family; mutations in the MERTK gene have been associated with disruption of the RPE phagocytosis pathway and retinitis pigmentosa (RP; D'Cruz et al., 2000; Gal et al., 2000). Expression of MER/AXL/TYRO3 was upregulated in RPE treated for 16 h with KL (Fig. 3D–E). The MER/AXL/TYRO3 promoter region contains a cAMP binding site (Korshunov, 2012). To further investigate the cAMP- and GPCR-dependent regulation of the phagocytosis by KL, we assayed the expression of MERTK, AXL, and TYRO3 genes 16 h after the removal of IBMX or isoproterenol. Similar to KL, IBMX and isoproterenol increased the gene expression levels of MER/AXL/TYRO3 (Fig. 3D,E). The lack of AXL induction by both IBMX and isoproterenol, as opposed to KL, might be explained by the fact that KL was present for 16 h in the RPE culture as opposed to the 1 h treatment with isoproterenol or IBMX.
Figure 3.

KL regulates phagocytosis in human and mouse RPE cells by upregulating MERTK gene expression. A, B, Treatment of RPE cells with 10 ng/ml (100 pm) recombinant KL protein results in a short-term (30 min) decrease and a long-term (16 h) increase in phagocytosis efficiency. Treatment with 1 mm IBMX (A) or with 100 μm isoproterenol (B) for 1 h has a similar effect on the phagocytosis efficiency 30 min and 16 h later. Results are the average ± SD of three independent experiments. RPE in six wells of a 96-well culture dish were used in each group. Asterisks indicate statistically significant differences in phagocytosis efficiency compared with the untreated control, as determined by the t test (p < 0.05). p values in A: 7.8 × 10−5 (KL 30 min); 1.3 × 10−6 (KL 16 h); 4.0 × 10−5 (IBMX 30 min); 2.1 × 10−5 (IBMX 16 h). p values in B: 2.0 × 10−4 (KL 30 min); 8.0 × 10−4 (KL 16 h); 4.6 × 10−5 [isoproterenol (Isop.) 30 min]; 2.7 × 10−4 (Isop. 16 h). C, RNAi knockdown of KL gene in RPE cells significantly reduces phagocytosis efficiency 72 h post-transfection. *p = 2.3 × 10−4 compared with scrambled siRNA (siNeg) control. Results represent two independent experiments, with 6 wells of a 96-well plate used in each group. D, E, Expression of genes important in phagocytosis in RPE cells (i.e., AXL, MERTK, TYRO3) is significantly increased by a 16 h treatment of RPE with KL protein, explaining the long-term effect of KL on phagocytosis. Treatment with 1 mm IBMX (D) or with 100 μm isoproterenol (E) for 1 h could also induce the expression of MERTK and TYRO3, but not of AXL, 16 h later. Asterisks indicate statistically significant differences between the untreated RPE cells and the RPE cells treated with IBMX, isoproterenol, or KL in the gene expression of the indicated genes, as determined by the t test (p < 0.05). Specifically, only KL treatment was able to increase the expression of AXL (D, E) compared with untreated RPE cells (p = 0.02) and RPE cells treated with IBMX (p = 0.05, D) or isoproterenol (p = 0.03, E). MERTK expression increased with KL treatment (p = 0.001, D, E), as well as with IBMX (p = 0.006, D) and isoproterenol (p = 0.026, E) treatments. IBMX treatment resulted in ∼50% higher levels of MERTK compared with the KL treatment (p = 0.01, D). Similarly, TYRO3 gene expression was significantly increased by KL (p = 0.002, D, E), IBMX (p = 9 × 10−4, D), and by isoproterenol (p = 0.009, E). KL treatment resulted in ∼30% higher TYRO3 levels compared with the TYRO3 increase caused by the IBMX treatment (p = 0.01, D). F, KL protein induces CREB phosphorylation in a concentration-dependent manner. RPE cells were treated with increasing doses of KL (0–400 pm) for 10 min and then lysed for Western blot analysis. A PKA-dependent increase in phosphorylation of CREB at Ser133 was revealed at 400 pm KL with a phospho-specific antibody. Levels of total CREB protein were not altered. G, KL protein induces PKA-dependent phosphorylation of adducin (at Ser481). RPE cells were treated with 100 pm KL and then lysed in RIPA buffer at the indicated time points for Western blot analysis. Phospho-adducin levels were detected by a specific antibody. Total adducin protein levels are also shown for normalization. The increase in phosphorylation of adducin caused by KL in 5–10 min may explain the inhibitory effect on RPE phagocytosis observed with short-term (30 min) treatment of RPE with 100 pm KL in Figure 3, A and B. H, I. Gene expression of phagocytosis factors (Axl, Mertk, Tyro3) is significantly reduced in the Kl−/− mouse retinas (H) compared with the Kl+/+ mouse retinas (H), and in old mouse retinae (13 months, I), compared with young mouse retinae (3 months, I). p values in H: 0.035 (Axl), 0.034 (Mertk) and 0.004 (Tyro3). p values in I: 0.004 (Axl), 0.015 (Mertk), and 0.002 (Tyro3). For the qRT-PCR analysis in H, RNA samples were isolated from the retinae of three Kl+/+ and three Kl−/− mice, and were analyzed separately. For the qRT-PCR in I, the RNA samples were isolated from the retinae of six young (3 months old) and six old (13 months old) C57BL/6 mice, and also were analyzed separately. The RNA samples in D and E were normalized to human GAPDH; in H and I, the mouse β-actin (Actb) was used for normalization, and the relative expression levels for each gene were calculated with the ΔΔCt method. The qRT-PCR analyses (D, E, H, and I) were performed in triplicate, and the average ± SD is shown for each sample.
To examine whether the regulatory effect of KL on phagocytosis is mediated through activation of cAMP-PKA-induced phosphorylation of CREB and downstream transcription regulation of MER/AXL/TYRO3, we analyzed CREB phosphorylation by Western blotting. As shown in Figure 3F, a 10 min treatment with KL increases CREB phosphorylation in cultured human RPE cells in a concentration-dependent manner. Together, these results suggest that the KL-induced increase of RPE phagocytosis is due to cAMP upregulation and cAMP-PKA-induced CREB phosphorylation that leads to upregulation of MER/AXL/TYRO3 expression. To understand the short-term inhibitory effect of KL incubation on RPE phagocytosis, in addition to the previously reported β-adrenergic stimulation and activation of A2 receptors (Hall et al., 1993; Gregory et al., 1994; Beitz et al., 1998), we hypothesized that deactivation of cytoskeleton proteins such as adducin by PKA phosphorylation might inhibit phagocytosis by preventing the spectrin–actin complex formation that could affect the membrane structures and therefore phagocytosis in RPE. Adducin is a membrane-skeletal protein that promotes association of spectrin with actin and caps the fast growing end of actin filaments (Matsuoka et al., 1996). Phosphorylation of α-adducin at Ser-408, -436, and -481 by PKA reduces the affinity of adducin for spectrin–F-actin complexes as well as the activity of adducin in promoting the binding of spectrin to F-actin (Gardner and Bennett, 1986; Matsuoka et al., 1996). We analyzed the phosphorylation of adducin in the RPE treated with KL for different time intervals. Interestingly, KL treatment of RPE induced adducin phosphorylation after 5 and 10 min of incubation, followed by a decrease in adducin phosphorylation after 15 min to 1 h incubation reaching the basal phosphorylation levels (Fig. 3G). This short-term phosphorylation is sufficient to inhibit adducin binding to spectrin and F-actin, and may explain the short-term inhibition of phagocytosis after 30 min of incubation with KL that was observed in our data (Fig. 3A,B).
To confirm the regulatory effect of KL on phagocytosis in vivo, we compared the gene expression levels of the Mer/Axl/Tyro3 in the retina of the Kl−/− and Kl+/+ mice. Our data showed a significant decrease in the levels of Mer/Axl/Tyro3 gene expression in the retinae of Kl−/− mice compared with those of Kl+/+ mice (Fig. 3H). Furthermore, to correlate the age-related decline in the KL protein in the retina and the MER/AXL/TYRO3 gene expression levels, we performed qRT-PCR in the retinae of old and young mice. Our data in Figure 3I show a significant decline in the gene expression levels of Mer/Axl/Tyro3 in the retinae of old mice compared with young mice.
It has been reported that Ca2+ signaling plays an important role in RPE function and in the regulation of phagocytosis (Rosenthal and Strauss, 2002; Nunes and Demaurex, 2010). The regulation of TRPV5 and Ca2+ homeostasis has been attributed to secreted α-KL protein (Imura et al., 2007; Cha et al., 2008; Lu et al., 2008). Further, it has been reported that TRPV5 is expressed in human RPE and can regulate Ca2+ entry (Kennedy et al., 2010). Therefore, we asked whether KL could activate TRPV5 channels in human RPE. Ca2+ imaging showed that KL treatment (24 h) did not increase [Ca2+] in human RPE (data not shown). This suggests that, unlike epithelial cells in the kidney, the TRPV5 channel in RPE cells is not regulated by KL protein.
KL inhibits VEGF secretion in human RPE
Choroidal neovascularization (CNV) is a hallmark of wet AMD and induces a progressive vision loss in patients (Nowak, 2006). VEGF, IGF-1, and IGF-1R have been implicated in CNV. It has been shown that IGF-1 signaling and VEGFR2 phosphorylation can induce VEGF secretion in RPE (Slomiany and Rosenzweig, 2004; Klettner et al., 2013), and that inhibitors of IGF-1R reduce VEGF secretion (Slomiany and Rosenzweig, 2004; Economou et al., 2008). Studies have demonstrated the inhibition of IGF-1 signaling with KL in rat hepatocellular carcinoma (hepatoma) cells (Kurosu et al., 2005) and human breast cancer cells (Wolf et al., 2008). In addition, a recent study showed that KL protein is associated with VEGFR-2/transient receptor potential canonical-1 (TRPC-1) channel in causing cointernalization and the regulation of TRPC-1-mediated Ca2+ entry to maintain endothelial integrity (Kusaba et al., 2010).
To examine the ability of KL to inhibit VEGF secretion in human RPE, we cultured human RPE in transwell plates for 4 weeks to obtain polarized RPE monolayer. Treatment of polarized RPE with 10 ng/ml KL for 48 h produced a significant decrease in VEGF secretion from both apical (p = 0.026) and basolateral membranes (p = 0.011) with a 50% decrease in secretion from the basolateral membrane (Fig. 4A). To delineate the mechanism by which KL regulates VEGF secretion, we have analyzed the effect of KL on tyrosine phosphorylation of IGF-1R by performing immunoprecipitation of RPE extracts after a 15 min treatment with different concentrations of KL and 75 ng/ml IGF-1, using an antibody to IGF-1R. Detection of phosphorylated IGF1-R was performed by Western blot analysis with an anti-phospho-tyrosine antibody. Our data in Figure 4B reveal an inhibition of tyrosine phosphorylation of IGF-1R by KL at 200 pm (20 ng/ml) and 400 pm (40 ng/ml), indicating that KL can inhibit VEGF secretion by inhibiting IGF-1 signaling in human RPE. The inhibition of IGF-1R signaling might also contribute to the cAMP activation in RPE shown above (Fig. 2E), since it has been shown that IGF-1 acutely reduces cAMP levels in astrocytes (Chesik et al., 2008). To investigate the effect of KL on VEGFR2 phosphorylation, we incubated human RPE with and without 200 pm (20 ng/ml) KL for 15 min and activated the phosphorylation of VEGFR2 by adding 100 ng/ml human recombinant VEGF for 2 min, followed by Western blot analysis using the phospho-tyrosine (Tyr996) VEGFR2 antibody. Our data in Fig. 4C show inhibition of VEGFR2 phosphorylation by KL confirming the inhibitory effect of KL on VEGFR2 phosphorylation and therefore VEGF secretion (Klettner et al., 2013).
Figure 4.

KL inhibits VEGF secretion in human RPE. A, Treatment of RPE with 10 ng/ml KL protein results in lower levels of VEGF secreted both from the apical and the basal sides, with 50% lower secretion from basal membrane. Three wells of a 24-well plate were used for each sample. Three independent experiments (total of nine wells per sample) were performed, and the average VEGF concentrations are presented in the graph. Asterisks indicate statistically significant differences in VEGF concentration between control and KL-treated RPE cells, as determined by the t test (p < 0.05). KL significantly reduced VEGF secretion both from the apical (p = 0.026) and from the basal (p = 0.011) sides. B, KL inhibits IGF-1 signaling in human RPE cells. The immunoprecipitated phospho-IGF-1R (shown by the arrow) was detected by Western blot analysis with the phospho-tyrosine antibody. Total precipitated IGF-1R was also detected for normalization. KL was able to inhibit the phosphorylation of IGF-1R at concentrations of 200 and 400 pm. C, KL inhibits VEGFR2 phosphorylation in human RPE. The phosphorylated VEGFR2 could not be detected when VEGFA was added in the presence of KL, compared with RPE cells treated with VEGFA alone, or untreated RPE cells, indicating an inhibitory effect of KL on VEGFR2 phosphorylation. Total VEGFR2 is shown for normalization. The relative densities of the bands were determined using ImageJ software. D, KL induces phosphorylation of SHP-2 in human RPE cells. KL was able to induce SHP-2 phosphorylation at 200 pm. Total SHP-2 protein levels are shown for normalization. IP, Immunoprecipitation; WB, Western blot.
Since our data demonstrated that KL regulates AXL gene expression (Fig. 3D,E), we were interested in examining whether KL regulates AXL-dependent pathways that contribute to VEGFR2 regulation. It has been shown that AXL tyrosine kinase receptor can inhibit VEGFR2 through the activation of SHP-2 in endothelial cells (Gallicchio et al., 2005). To further investigate the mechanisms by which KL regulates VEGFR2 phosphorylation, we analyzed the phosphorylation of SHP-2 at Tyr542 in human RPE cells in the presence and absence of KL. SHP-2 phosphorylation is thought to relieve basal inhibition and stimulate SHP-2 tyrosine phosphatase activity in living cells (Lu et al., 2001). Figure 4D shows SHP-2 phosphorylation by 200 pm KL after 48 h of treatment of RPE, indicating that the regulatory effect of KL on VEGF signaling also involves the AXL-dependent pathway. It should be noted that we have also assayed the phosphorylation of SHP-2 after 24 h of KL treatment and did not detect an increase in phosphorylation, possibly because a 24 h interval is required for AXL overexpression in the presence of KL (Fig. 3D,E) before the phosphorylation rate of SHP-2 is increased. Together, our data show that KL inhibits VEGF secretion in human RPE by reducing IGF-1-mediated phosphorylation of IGF-1R and by inhibiting VEGR2 phosphorylation. KL can also activate AXL gene expression that, in turn, can activate SHP-2 phosphorylation, which is responsible for AXL-mediated VEGFR2 inhibition.
Our in vitro studies help to explain the in vivo phenotypes observed in the choroid of the Kl−/− mice, where choroid tissue layers were separated by severely dilated blood vessels. Since KL exerts an inhibitory effect on VEGF signaling, we hypothesize that the absence of KL could cause excessive VEGF secretion from the basal membrane of RPE, increasing blood vessel formation and leading to the deformation of choroid layers, as observed in Figure 1D.
KL regulates the expression of drusen protein coding and AMD-associated genes, and decreases ROS production
RPE cells are constantly subjected to oxidative stress and high levels of peroxidized lipid membranes due to their high metabolic activity (Cai et al., 2000). Extended exposure to oxidative stress can disrupt RPE tight junctions inducing the breakage of the blood barrier and producing abnormal membrane bleb structures (Negi and Marmor, 1984; Feeney-Burns et al., 1987). Furthermore, impairment of RPE function in dry AMD can induce the formation of abnormal extracellular deposits called drusen that accumulate between the RPE and BM (Abdelsalam et al., 1999). KL has been shown to regulate oxidative stress by increasing the superoxide dismutase 2 (SOD2) gene expression levels (Yamamoto et al., 2005). To investigate the effect of KL on regulating the response to oxidative stress in RPE cells, we have established an in vitro acute stress condition by exposing RPE to 500 μm oxidative stress-inducing agent tBH for 2 h. We have cultured human RPE cells under normal and stress conditions in the presence and absence of KL, and have analyzed the expression of the stress-related genes by qRT-PCR. Human RPE cells grown under stress condition significantly increased the expression of the drusen protein-coding genes CRYAA and CRYGS. In addition, VEGFR2 and VEGFA, two genes expressed by RPE cells that have been associated with AMD, are increased by oxidative stress. Interestingly, incubating the RPE with KL for 16 h before and during the treatment with tBH significantly reduced the expression levels of the stress-related genes to control levels (Fig. 5A). Our data strongly suggest that KL plays a protective role against oxidative stress by regulating the expression of drusen-associated genes and genes associated with AMD. To examine the mechanism by which KL protects RPE against oxidative stress, we investigated the ability of KL in activating the expression of the mitochondrial manganese SOD2, which reduces oxidative stress and is the target gene upregulated by the forkhead family of transcription factors, which in turn are negatively regulated by IGF-1/IGF-1R signaling (Guo et al., 1999; Nakae et al., 1999; Rena et al., 1999). Our data showed that the incubation of RPE cells with KL for 16 h did not increase the SOD2 gene expression under normal culture conditions. However, KL was able to restore the SOD2 gene expression to normal levels under oxidative stress conditions, where SOD2 levels were significantly downregulated (p = 0.0016; Fig. 5B). On the contrary, KL siRNA significantly reduced (p = 0.036) the SOD2 gene expression in RPE cells (Fig. 5C), further supporting the role of KL in maintaining normal levels of SOD2.
Figure 5.

KL protects RPE cells from oxidative stress. A, Oxidative stress was induced in RPE cells in the absence and presence of KL. Three wells were used for each of the four conditions (No stress, no KL; No Stress, +KL; Stress, no KL; and Stress, +KL). RNA was isolated from each well and was analyzed separately by qRT-PCR. Each reaction was performed in triplicate (total of nine samples from each condition). Relative expression values for each gene were calculated using the ΔΔCt method after normalization to GAPDH. The average of the three experiments is presented in the graph. KL reduced the expression of all four stress-related genes that were tested under the stress condition, supporting its protective role against oxidative stress in RPE cells. Asterisks indicate samples with statistically significant differences in gene expression based on the t test (p < 0.05). Specifically, stress in the absence of KL significantly increased the expression of CRYAA (p = 0.005), CRYGS (p = 0.031), VEGFR2 (p = 0.035), and VEGFA (p = 0.006), whereas KL under stress decreased the expression of CRYAA (p = 0.005), CRYGS (p = 0.001), and VEGFR2 (p = 0.013) genes to the normal (No Stress, no KL) levels or lower. VEGFA gene expression was also decreased under stress by KL, but did not reach normal levels (p = 0.019). B, KL increases SOD2 gene expression under the stress condition. RPE cells were cultured and treated as described in A, and SOD2 gene expression was assayed. KL did not affect SOD2 mRNA levels when added in normal culture condition, but, when combined with stress, where SOD2 mRNA was decreased significantly, KL was able to restore the SOD2 expression to normal levels. *p = 0.0016. C, KL RNAi knockdown in RPE cells results in a 60% decrease of SOD2 gene expression levels, as shown by qRT-PCR, further supporting a regulatory role of KL on SOD2. The graph represents the average of three experiments, and the asterisks indicate statistically significant differences in gene expression (p = 0.011 for KL, p =0.036 for SOD2). D, KL significantly decreased the production of ROS under oxidative stress in primary cultures RPE1 and RPE2. The experiments were performed in triplicate, in 96-well plates. The statistical significance of the decrease in ROS levels between KL treated and untreated cells was verified by t test, resulting in p values < 0.05 for each time point. 5 min: p = 0.005 (RPE1) and p = 0.001 (RPE2); 15 min: p = 0.005 (RPE1) and p = 0.001 (RPE2); 30 min: p = 0.011 (RPE1) and p = 0.004 (RPE2); 1 h: p = 0.020 (RPE1) and p = 0.0005 (RPE2). E, F, The cAMP agonist isoproterenol (E) and the cAMP and cGMP phosphodiesterase nonspecific inhibitor IBMX (F) can partially protect RPE cells from oxidative stress by reducing the mRNA levels of two of four stress-related genes (CRYAA and VEGFR2 reduced by isoproterenol; CRYAA and CRYGS reduced by IBMX) back to normal levels under the stress condition. Statistically significant differences in gene expression (p < 0.05) for each gene between the control [No isoproterenol (Isop.), No Stress, E; and No IBMX, No Stress, F) and the experimental conditions are indicated by the asterisks. Specifically, stress in the absence of isoproterenol (E) significantly increased the expression of CRYAA (p = 0.013), CRYGS (p = 0.031), VEGFR2 (p = 0.003), and VEGFA (p = 0.017), whereas the presence of isoproterenol under the stress condition reduced CRYAA and VEGFR2 to normal levels, but not CRYGS (p = 0.015) and VEGFA (p = 0.003). Furthermore, stress in the absence of IBMX (F) significantly increased the expression of CRYAA (p = 0.026), CRYGS (p = 0.01), and VEGFR2 (p = 0.01), whereas the presence of IBMX under the stress condition reduced CRYAA and CRYGS to normal levels or lower, but increased VEGFR2 (p = 0.0074) and VEGFA (p = 0.002).
To confirm that KL can protect RPE against oxidative stress, we measured the levels of ROS under oxidative stress conditions in the presence and absence of KL. As shown by our data in Figure 5D, the human RPE cells produced increased levels of ROS under oxidative stress conditions in the absence of KL, whereas the ROS production was decreased to the normal (i.e., no stress) levels in the presence of KL (Fig. 5D). These observations further support the protective role of KL against oxidative stress in RPE cells.
Finally, to test a possible role for cAMP signaling in mediating the KL effect on the stress response genes, we treated RPE cells with 100 μm isoproterenol or 1 mm IBMX for 1 h, removed the isoproterenol or IBMX, treated the cells with tBH 16 h after removal of the drugs, and compared the expression of stress response genes under stress and normal conditions, with or without treatment with isoproterenol or IBMX. For CRYAA (Cvekl et al., 1995) and VEGFR2 genes, isoproterenol treatment alone could decrease the gene expression to normal levels under stress (Fig. 5E), which is similar to the effect of KL treatment (Fig. 5A). For CRYGS and VEGFA, although isoproterenol reduced their gene expression under stress, it could not reduce it back to the normal levels (Fig. 5E). CRYAA and CRYGS gene expression was reduced to normal levels by IBMX under the stress condition (Fig. 5F), similar to the effect of KL on these genes (Fig. 5A). However, the effect of IBMX on the expression of VEGFR2 and VEGFA genes was different than that of KL. IBMX increased VEGFA gene expression under oxidative stress (Fig. 5F). In addition, IBMX increased VEGFR2 expression levels both in normal and stress conditions (by 16-fold and 70-fold, respectively), suggesting that the expression of these genes is regulated by additional signals in addition to cAMP. Because IBMX is a nonspecific inhibitor of both cAMP and cGMP phosphodiesterases, it is expected to have a broader effect on gene expression than that of isoproterenol (Fig. 5E). Therefore, the precise signaling pathways by which KL regulates the drusen-coding genes and AMD-related genes need to be further investigated.
Discussion
Our studies demonstrate that the anti-aging protein KL plays a critical role in RPE physiology and retinal health. In the wild-type mouse retina, we observed an age-dependent decline in Kl expression. Germline ablation of Kl leads to numerous structural changes in the adult retina in vivo, and most prominently affects Bruch's membrane, the RPE, photoreceptor outer segments, and retinal supporting tissue, the choroid. Functional studies with primary cultured human RPE cells identified signaling pathways regulated by KL, which have been implicated in melanogenesis, phagocytosis, oxidative stress responses, and vascularization. Specifically, we find that KL regulates the expression of genes important for melanin synthesis, upregulates MERTK expression and promotes phagocytosis, inhibits VEGF secretion, increases the expression of gene products that protect from oxidative stress, and reduces ROS production under oxidative stress. Based on these findings, we propose that KL has a protective function in the retina and is an important regulator of RPE physiology. Therefore, it is tempting to speculate that the downregulation of KL during aging may impair RPE function and contribute to pathologic changes observed in age-related retinal degeneration, such as AMD.
RPE pigmentation is regulated by KL
Studies in humans, examining eyes over several decades of life, revealed an age-dependent ∼25% decline in the number of melanin granules in the RPE and the choroid (Feeney-Burns et al., 1984; Weiter et al., 1986; Sarna et al., 2003; Bonilha, 2008). The loss of Kl expression has been associated with premature tissue aging and reduced lifespan (Kuro-o et al., 1997). Interestingly, we found that Kl−/− mice exhibit a decline in the number of pigment granules in RPE cells. To investigate the mechanism by which KL might regulate melanogenesis, we used primary human RPE cells and analyzed the effect of KL on gene and protein expression of MITF and TYR, two pivotal genes in melanogenesis. We found that KL can regulate MITF and therefore TYR through a cAMP-dependent signaling pathway. Consistent with our findings, two previous studies with endothelial cells showed that KL elevates intracellular cAMP (Yang et al., 2003; Rakugi et al., 2007). In the aging mouse retina, Kl gene expression is significantly reduced and correlates with a decline in Mitf and Tyr gene expression in old (13 months of age) compared with young (3 months of age) mice, providing a possible explanation for the age-dependent decline in RPE pigmentation. The more pronounced decline in Mitf and Tyr gene expression in the retina of Kl−/− compared with Kl+/+ mice is consistent with the idea that the loss of Kl leads to decreased RPE pigmentation.
Moreover, two independent studies reported a decline in circulating soluble α-Klotho levels in aging humans (Yamazaki et al., 2010; Semba et al., 2011), further supporting our hypothesis that an age-related decrease in KL synthesis and availability could lead to a decline in RPE pigmentation.
Whether KL gene expression and protein levels decline during aging in human RPE cells and compromise pigmentation in vivo remains to be investigated.
Kl deficiency induces retinal degeneration
At 6 weeks of age, we observed severe morphological abnormalities in the choroid and Bruch's membrane. In addition, we observed signs of RPE and photoreceptor degeneration, including mitochondrial damage and cellular atrophy. Since the RPE is an integral part of the retinal blood barrier, damage of Bruch's membrane and the RPE may lead to neovascularization. Moreover, RPE degradation can translate into photoreceptor degeneration and the gradual loss of vision as observed in age-related retinal degeneration (Matsunaga et al., 1999; Kozlowski, 2012).
A recent study reported on the upregulation of α-Klotho in degenerating photoreceptors of rd1, rd2, P23H, and S334ter animal models of RP (Farinelli et al., 2013). The same study found that exogenous application of α-Klotho to explant cultures of wild-type and rd1 retinae did not affect the number of apoptotic cells but caused disorganization at the edge of retinal explants compared with untreated specimens. In the present study, we provide independent lines of evidence that Klotho plays a protective function in mouse retinae in vivo and toward cultured human RPE cells in vitro. We used a Kl−/− mouse model and found that lack of Kl expression induces RPE, photoreceptor, and Bruch's membrane degeneration. We also used primary cultures of human RPE cells and observed a protective role for KL in regulating oxidative stress genes and ROS production. Farinelli et al. (2013) used whole retinal explants of rd1 mice and found no evidence for a protective role of bath-applied KL toward photoreceptors. Important differences between the two studies are the use of different animal models to study retinal pathology (Kl−/− vs rd1) and the concentration of bath-applied KL (Farinelli et al., 2013; 100 pm vs 1–2.5 nm). Also, KL may only protect against oxidative damage but not retinal damage caused by the rd1 mutation. Additional studies are needed to determine whether the increased expression of KL in rd1 animals observed by Farinelli et al. (2013) directly contributes to photoreceptor cell apoptosis or is part of an adaptive response to counteract retinal cell death.
Regulation of RPE phagocytosis by KL
Lifelong phagocytosis of the outer segment of photoreceptors by the postmitotic RPE eliminates the photoreceptor cell wastes and retains the recyclable cellular material. This RPE function is crucial for maintenance of photoreceptors, continuously generating new outer segments from their base (Kevany and Palczewski, 2010). Lipofuscin is a complex aggregate of material that occurs in a variety of metabolically active postmitotic cells such as RPE (Yin, 1996; Boulton et al., 2004) and mainly derives from the ingestion of photoreceptor outer segments. Pronounced accumulation of lipofuscin during aging induces phototoxicity, which is attributed to the decline in RPE function (Sundelin et al., 1998).
Our analysis of Kl−/− RPE by EM, revealed an apparent absence of phagocytosis of the outer segments of the photoreceptors, whereas various stages of phagocytosis were observed in the retina of the Kl+/+ mice.
Consistent with our ultrastructural findings, in vitro studies with RPE demonstrate that KL induces phagocytosis in RPE by regulating the expression of MERTK. Using KL siRNA to knock down KL gene expression, we further confirmed a regulatory role for KL in RPE phagocytosis, demonstrating that KL downregulation inhibits phagocytosis in RPE cells. In vivo, MERTK expression is significantly reduced in the retina of Kl−/− compared with Kl+/+ mice. A direct correlation between the decrease of KL protein in human eyes during aging and lipofuscin formation should be a focus of future investigations.
VEGF secretion in RPE is inhibited by KL
RPE cells secrete several growth factors that support the survival of photoreceptors, and ensure a structural basis for the transport and supply of nutrients (Strauss, 2005). VEGF is secreted at low concentration by the RPE cells in the healthy eye to ensure an intact endothelium of the choriocapillaries and to prevent endothelial cell apoptosis (Burns and Hartz, 1992; Strauss, 2005), whereas in AMD, VEGF is secreted at a higher rate, resulting in CNV (Kliffen et al., 1997). The ultrastructural analysis of the Kl−/− retinal region revealed severely dilated blood vessels in the choroid separating and deforming the choroid tissue layers, therefore suggesting an important role for KL in regulating VEGF secretion. Our in vitro data demonstrated that KL regulates VEGF secretion in RPE cells by the downregulation of IGF-1 signaling through the inhibition of IGF-R tyrosine phosphorylation and by the inhibition of VEGFR2 phosphorylation. Our results also showed that KL induces the phosphorylation of SHP-2, which in turn can inhibit VEGFR2 activation (Gallicchio et al., 2005). These observations suggest possible therapeutic applications for KL protein for the treatment of CNV observed in wet AMD.
The expression of drusen protein-coding genes, AMD-associated genes, and ROS production are modulated with KL
KL has been reported to have antioxidant activity in human endothelial cells (Carracedo et al., 2012) and rat aorta smooth muscle cells (Wang et al., 2012b). Here we experimentally established a stress condition to induce the expression of drusen and stress-related genes. We found that KL has the ability to modulate the gene expression of drusen protein-coding genes and genes associated with AMD. Interestingly, KL appears to exert a protective effect against oxidative stress by regulating the expression of these genes back to control levels. We provide evidence that KL can regulate oxidative stress responses by inhibiting IGF-1 signaling and increasing the SOD2 gene expression under stress in human RPE. In addition to regulating the SOD2 expression, KL can regulate ROS production under oxidative stress by reducing ROS to normal levels. Our findings could have important implications for AMD pathology and may be used for the development of new therapeutic strategies.
A model for KL function in RPE and retinal health
Consistent with previous studies on KL and premature aging (Kuro-o et al., 1997; Kuro-o, 2009), we observed an accelerated degenerative phenotype in the retina of Kl−/− mice. Our studies with primary human RPE cultures showed that KL increased cAMP levels via a Gs-coupled GPCR and induced CREB phosphorylation, resulting in the upregulation of pivotal genes for pigmentation and phagocytosis. KL inhibited VEGF secretion by downregulating IGF-1R and VEGFR2 phosphorylation in RPE. KL also regulated stress response genes and reactive oxygen species production in human RPE in vitro. Together, our in vivo and in vitro studies suggest an important role for KL in RPE homeostasis and protection against oxidative stress. Additional studies are needed to fully delineate the regulatory role of Klotho on signaling pathways important for RPE function and retinal health in vivo.
Footnotes
We thank Dr. Makoto Kuro-o (University of Texas Southwestern, TX) for generously providing the Kl−/− mouse eyes and a pair of Kl+/− mice; Dr. Roman Giger (University of Michigan Medical School) for critical review of the manuscript; and Drs. Haohua Qian and Yichao Li at NIH/NEI for technical support. The Georgetown-Lombardi Comprehensive Cancer Center Shared Resource facilities were used for qRT-PCR and confocal microscopy.
The authors declare no competing financial interests.
References
- Abdelsalam A, Del Priore L, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999;44:1–29. doi: 10.1016/S0039-6257(99)00072-7. [DOI] [PubMed] [Google Scholar]
- Beitz E, Volkel H, Guo Y, Schultz JE. Adenylyl cyclase type 7 is the predominant isoform in the bovine retinal pigment epithelium. Acta Anat (Basel) 1998;162:157–162. doi: 10.1159/000046481. [DOI] [PubMed] [Google Scholar]
- Bertolotto C, Abbe P, Hemesath TJ, Bille K, Fisher DE, Ortonne JP, Ballotti R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J Cell Biol. 1998;142:827–835. doi: 10.1083/jcb.142.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL. Age and disease-related structural changes in the retinal pigment epithelium. Clin Ophthalmol. 2008;2:413–424. doi: 10.2147/OPTH.S2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton M, Dayhaw-Barker P. The role of the retinal pigment epithelium: topographical variation and ageing changes. Eye (Lond) 2001;15:384–389. doi: 10.1038/eye.2001.141. [DOI] [PubMed] [Google Scholar]
- Boulton M, Rozanowska M, Rozanowski B, Wess T. The photoreactivity of ocular lipofuscin. Photochem Photobiol Sci. 2004;3:759–764. doi: 10.1039/b400108g. [DOI] [PubMed] [Google Scholar]
- Burns MS, Hartz MJ. The retinal pigment epithelium induces fenestration of endothelial cells in vivo. Curr Eye Res. 1992;11:863–873. doi: 10.3109/02713689209033484. [DOI] [PubMed] [Google Scholar]
- Cai J, Nelson KC, Wu M, Sternberg P, Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/S1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Carracedo J, Buendía P, Merino A, Madueño JA, Peralbo E, Ortiz A, Martín-Malo A, Aljama P, Rodríguez M, Ramírez R. Klotho modulates the stress response in human senescent endothelial cells. Mech Ageing Dev. 2012;133:647–654. doi: 10.1016/j.mad.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro-O M, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105:9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Sloane JA, Li H, Aytan N, Giannaris EL, Zeldich E, Hinman JD, Dedeoglu A, Rosene DL, Bansal R, Luebke JI, Kuro-o M, Abraham CR. The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci. 2013;33:1927–1939. doi: 10.1523/JNEUROSCI.2080-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesik D, Wilczak N, De Keyser J. IGF-1 regulates cAMP levels in astrocytes through a beta2-adrenergic receptor-dependant mechanism. Int J Med Sci. 2008;5:240–243. doi: 10.7150/ijms.5.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Cao X, Shen D, Tuo J, Parver LM, Rickles FR, Chan CC. Evidence for enhanced tissue factor expression in age-related macular degeneration. Lab Invest. 2011;91:519–526. doi: 10.1038/labinvest.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Kashanchi F, Sax CM, Brady JN, Piatigorsky J. Transcriptional regulation of the mouse alpha A-crystallin gene: activation dependent on a cyclic AMP-responsive element (DE1/CRE) and a Pax-6-binding site. Mol Cell Biol. 1995;15:653–660. doi: 10.1128/mcb.15.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- de Oliveira RM. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway. FEBS Lett. 2006;580:5753–5758. doi: 10.1016/j.febslet.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Economou MA, Wu J, Vasilcanu D, Rosengren L, All-Ericsson C, van der Ploeg I, Menu E, Girnita L, Axelson M, Larsson O, Seregard S, Kvanta A. Inhibition of VEGF secretion and experimental choroidal neovascularization by picropodophyllin (PPP), an inhibitor of the insulin-like growth factor-1 receptor. Invest Ophthalmol Vis Sci. 2008;49:2620–2626. doi: 10.1167/iovs.07-0742. [DOI] [PubMed] [Google Scholar]
- Farinelli P, Arango-Gonzalez B, Volkl J, Alesutan I, Lang F, Zrenner E, Paquet-Durand F, Ekstrom PA. Retinitis Pigmentosa: over-expression of anti-ageing protein Klotho in degenerating photoreceptors. J Neurochem. 2013 doi: 10.1111/jnc.12353. doi: 10.1111/jnc.12353. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Feeney-Burns L, Hilderbrand ES, Eldridge S. Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Invest Ophthalmol Vis Sci. 1984;25:195–200. [PubMed] [Google Scholar]
- Feeney-Burns L, Gao CL, Tidwell M. Lysosomal enzyme cytochemistry of human RPE, Bruch's membrane and drusen. Invest Ophthalmol Vis Sci. 1987;28:1138–1147. [PubMed] [Google Scholar]
- Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- Gallicchio M, Mitola S, Valdembri D, Fantozzi R, Varnum B, Avanzi GC, Bussolino F. Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood. 2005;105:1970–1976. doi: 10.1182/blood-2004-04-1469. [DOI] [PubMed] [Google Scholar]
- Gardner K, Bennett V. A new erythrocyte membrane-associated protein with calmodulin binding activity. Identification and purification. J Biol Chem. 1986;261:1339–1348. [PubMed] [Google Scholar]
- Goding CR. Mitf from neural crest to melanoma: signal transduction and transcription in the melanocyte lineage. Genes Dev. 2000;14:1712–1728. doi: 10.1101/gad.14.14.1712. [DOI] [PubMed] [Google Scholar]
- Gregory CY, Abrams TA, Hall MO. Stimulation of A2 adenosine receptors inhibits the ingestion of photoreceptor outer segments by retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1994;35:819–825. [PubMed] [Google Scholar]
- Gu X, Neric NJ, Crabb JS, Crabb JW, Bhattacharya SK, Rayborn ME, Hollyfield JG, Bonilha VL. Age-related changes in the retinal pigment epithelium (RPE) PLoS One. 2012;7:e38673. doi: 10.1371/journal.pone.0038673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–17192. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- Halaban R. Pigmentation in melanomas: changes manifesting underlying oncogenic and metabolic activities. Oncol Res. 2002;13:3–8. doi: 10.3727/096504002108747908. [DOI] [PubMed] [Google Scholar]
- Halaban R, Cheng E, Svedine S, Aron R, Hebert DN. Proper folding and endoplasmic reticulum to golgi transport of tyrosinase are induced by its substrates, DOPA and tyrosine. J Biol Chem. 2001;276:11933–11938. doi: 10.1074/jbc.M008703200. [DOI] [PubMed] [Google Scholar]
- Hall MO, Abrams TA, Mittag TW. The phagocytosis of rod outer segments is inhibited by drugs linked to cyclic adenosine monophosphate production. Invest Ophthalmol Vis Sci. 1993;34:2392–2401. [PubMed] [Google Scholar]
- Holtkamp GM, Van Rossem M, de Vos AF, Willekens B, Peek R, Kijlstra A. Polarized secretion of IL-6 and IL-8 by human retinal pigment epithelial cells. Clin Exp Immunol. 1998;112:34–43. doi: 10.1046/j.1365-2249.1998.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL. Regulation of ion channels by secreted Klotho. Adv Exp Med Biol. 2012;728:100–106. doi: 10.1007/978-1-4614-0887-1_7. [DOI] [PubMed] [Google Scholar]
- Imai M, Ishikawa K, Matsukawa N, Kida I, Ohta J, Ikushima M, Chihara Y, Rui X, Rakugi H, Ogihara T. Klotho protein activates the PKC pathway in the kidney and testis and suppresses 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression. Endocrine. 2004;25:229–234. doi: 10.1385/ENDO:25:3:229. [DOI] [PubMed] [Google Scholar]
- Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, Obuse C, Togashi K, Tominaga M, Kita N, Tomiyama K, Iijima J, Nabeshima Y, Fujioka M, Asato R, Tanaka S, Kojima K, Ito J, Nozaki K, Hashimoto N, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- Kennedy BG, Torabi AJ, Kurzawa R, Echtenkamp SF, Mangini NJ. Expression of transient receptor potential vanilloid channels TRPV5 and TRPV6 in retinal pigment epithelium. Mol Vis. 2010;16:665–675. [PMC free article] [PubMed] [Google Scholar]
- Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klettner A, Westhues D, Lassen J, Bartsch S, Roider J. Regulation of constitutive vascular endothelial growth factor secretion in retinal pigment epithelium/choroid organ cultures: p38, nuclear factor kappaB, and the vascular endothelial growth factor receptor-2/phosphatidylinositol 3 kinase pathway. Mol Vis. 2013;19:281–291. [PMC free article] [PubMed] [Google Scholar]
- Kliffen M, Sharma HS, Mooy CM, Kerkvliet S, de Jong PT. Increased expression of angiogenic growth factors in age-related maculopathy. Br J Ophthalmol. 1997;81:154–162. doi: 10.1136/bjo.81.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinaki M, Sahibzada N, Golestaneh N. Human iPS-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized VEGF secretion and gene expression pattern similar to native RPE. Stem Cells. 2011;29:825–835. doi: 10.1002/stem.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov VA. Axl-dependent signalling: a clinical update. Clin Sci (Lond) 2012;122:361–368. doi: 10.1042/CS20110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski MR. RPE cell senescence: a key contributor to age-related macular degeneration. Med Hypotheses. 2012;78:505–510. doi: 10.1016/j.mehy.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. Klotho and aging. Biochim Biophys Acta. 2009;1790:1049–1058. doi: 10.1016/j.bbagen.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- Kusaba T, Okigaki M, Matui A, Murakami M, Ishikawa K, Kimura T, Sonomura K, Adachi Y, Shibuya M, Shirayama T, Tanda S, Hatta T, Sasaki S, Mori Y, Matsubara H. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc Natl Acad Sci U S A. 2010;107:19308–19313. doi: 10.1073/pnas.1008544107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, Boulton ME, Godley BF. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:3521–3529. doi: 10.1167/iovs.10-6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Lu P, Boros S, Chang Q, Bindels RJ, Hoenderop JG. The beta-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol Dial Transplant. 2008;23:3397–3402. doi: 10.1093/ndt/gfn291. [DOI] [PubMed] [Google Scholar]
- Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell. 2001;8:759–769. doi: 10.1016/S1097-2765(01)00369-0. [DOI] [PubMed] [Google Scholar]
- Maminishkis A, Chen S, Jalickee S, Banzon T, Shi G, Wang FE, Ehalt T, Hammer JA, Miller SS. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Invest Ophthalmol Vis Sci. 2006;47:3612–3624. doi: 10.1167/iovs.05-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga H, Handa JT, Aotaki-Keen A, Sherwood SW, West MD, Hjelmeland LM. Beta-galactosidase histochemistry and telomere loss in senescent retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40:197–202. [PubMed] [Google Scholar]
- Matsuoka Y, Hughes CA, Bennett V. Adducin regulation. Definition of the calmodulin-binding domain and sites of phosphorylation by protein kinases A and C. J Biol Chem. 1996;271:25157–25166. doi: 10.1074/jbc.271.41.25157. [DOI] [PubMed] [Google Scholar]
- Montague W, Cook JR. The role of adenosine 3′:5′-cyclic monophosphate in the regulation of insulin release by isolated rat islets of Langerhans. Biochem J. 1971;122:115–120. doi: 10.1042/bj1220115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–15985. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- Negi A, Marmor MF. Experimental serous retinal detachment and focal pigment epithelial damage. Arch Ophthalmol. 1984;102:445–449. doi: 10.1001/archopht.1984.01040030359038. [DOI] [PubMed] [Google Scholar]
- Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006;58:353–363. [PubMed] [Google Scholar]
- Nunes P, Demaurex N. The role of calcium signaling in phagocytosis. J Leukoc Biol. 2010;88:57–68. doi: 10.1189/jlb.0110028. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Rakugi H, Matsukawa N, Ishikawa K, Yang J, Imai M, Ikushima M, Maekawa Y, Kida I, Miyazaki J, Ogihara T. Anti-oxidative effect of Klotho on endothelial cells through cAMP activation. Endocrine. 2007;31:82–87. doi: 10.1007/s12020-007-0016-9. [DOI] [PubMed] [Google Scholar]
- Reinisalo M, Putula J, Mannermaa E, Urtti A, Honkakoski P. Regulation of the human tyrosinase gene in retinal pigment epithelium cells: the significance of transcription factor orthodenticle homeobox 2 and its polymorphic binding site. Mol Vis. 2012;18:38–54. [PMC free article] [PubMed] [Google Scholar]
- Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- Rosenthal R, Strauss O. Ca2+-channels in the RPE. Adv Exp Med Biol. 2002;514:225–235. doi: 10.1007/978-1-4615-0121-3_13. [DOI] [PubMed] [Google Scholar]
- Sarna T, Burke JM, Korytowski W, Rózanowska M, Skumatz CM, Zareba A, Zareba M. Loss of melanin from human RPE with aging: possible role of melanin photooxidation. Exp Eye Res. 2003;76:89–98. doi: 10.1016/S0014-4835(02)00247-6. [DOI] [PubMed] [Google Scholar]
- Schmidt SY, Peisch RD. Melanin concentration in normal human retinal pigment epithelium. Regional variation and age-related reduction. Invest Ophthalmol Vis Sci. 1986;27:1063–1067. [PubMed] [Google Scholar]
- Schraermeyer U, Kopitz J, Peters S, Henke-Fahle S, Blitgen-Heinecke P, Kokkinou D, Schwarz T, Bartz-Schmidt KU. Tyrosinase biosynthesis in adult mammalian retinal pigment epithelial cells. Exp Eye Res. 2006;83:315–321. doi: 10.1016/j.exer.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Seagle BL, Rezai KA, Kobori Y, Gasyna EM, Rezaei KA, Norris JR., Jr Melanin photoprotection in the human retinal pigment epithelium and its correlation with light-induced cell apoptosis. Proc Natl Acad Sci U S A. 2005a;102:8978–8983. doi: 10.1073/pnas.0501971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagle BL, Rezai KA, Gasyna EM, Kobori Y, Rezaei KA, Norris JR., Jr Time-resolved detection of melanin free radicals quenching reactive oxygen species. J Am Chem Soc. 2005b;127:11220–11221. doi: 10.1021/ja052773z. [DOI] [PubMed] [Google Scholar]
- Semba RD, Cappola AR, Sun K, Bandinelli S, Dalal M, Crasto C, Guralnik JM, Ferrucci L. Plasma klotho and mortality risk in older community-dwelling adults. J Gerontol A Biol Sci Med Sci. 2011;66:794–800. doi: 10.1093/gerona/glr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–945. doi: 10.1016/S0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- Slomiany MG, Rosenzweig SA. IGF-1-induced VEGF and IGFBP-3 secretion correlates with increased HIF-1 alpha expression and activity in retinal pigment epithelial cell line D407. Invest Ophthalmol Vis Sci. 2004;45:2838–2847. doi: 10.1167/iovs.03-0565. [DOI] [PubMed] [Google Scholar]
- Slominski A, Costantino R. Molecular mechanism of tyrosinase regulation by L-dopa in hamster melanoma cells. Life Sci. 1991a;48:2075–2079. doi: 10.1016/0024-3205(91)90164-7. [DOI] [PubMed] [Google Scholar]
- Slominski A, Costantino R. L-tyrosine induces tyrosinase expression via a posttranscriptional mechanism. Experientia. 1991b;47:721–724. doi: 10.1007/BF01958826. [DOI] [PubMed] [Google Scholar]
- Slominski A, Moellmann G, Kuklinska E, Bomirski A, Pawelek J. Positive regulation of melanin pigmentation by two key substrates of the melanogenic pathway, L-tyrosine and L-dopa. J Cell Sci. 1988;89:287–296. doi: 10.1242/jcs.89.3.287. [DOI] [PubMed] [Google Scholar]
- Slominski A, Moellmann G, Kuklinska E. L-tyrosine, L-dopa, and tyrosinase as positive regulators of the subcellular apparatus of melanogenesis in Bomirski Ab amelanotic melanoma cells. Pigment Cell Res. 1989a;2:109–116. doi: 10.1111/j.1600-0749.1989.tb00170.x. [DOI] [PubMed] [Google Scholar]
- Slominski A, Jastreboff P, Pawelek J. L-tyrosine stimulates induction of tyrosinase activity by MSH and reduces cooperative interactions between MSH receptors in hamster melanoma cells. Biosci Rep. 1989b;9:579–586. doi: 10.1007/BF01119801. [DOI] [PubMed] [Google Scholar]
- Slominski A, Tobin DJ, Shibahara S, Wortsman J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev. 2004;84:1155–1228. doi: 10.1152/physrev.00044.2003. [DOI] [PubMed] [Google Scholar]
- Sopjani M, Alesutan I, Dërmaku-Sopjani M, Gu S, Zelenak C, Munoz C, Velic A, Föller M, Rosenblatt KP, Kuro-o M, Lang F. Regulation of the Na+/K+ ATPase by Klotho. FEBS Lett. 2011;585:1759–1764. doi: 10.1016/j.febslet.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Sun K, Cai H, Tezel TH, Paik D, Gaillard ER, Del Priore LV. Bruch's membrane aging decreases phagocytosis of outer segments by retinal pigment epithelium. Mol Vis. 2007;13:2310–2319. [PubMed] [Google Scholar]
- Sundelin S, Wihlmark U, Nilsson SE, Brunk UT. Lipofuscin accumulation in cultured retinal pigment epithelial cells reduces their phagocytic capacity. Curr Eye Res. 1998;17:851–857. doi: 10.1080/02713689808951268. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8:251–255. doi: 10.1038/ng1194-251. [DOI] [PubMed] [Google Scholar]
- Wan P, Hu Y, He L. Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors. Mol Cell Biochem. 2011;354:241–246. doi: 10.1007/s11010-011-0823-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Ohno-Matsui K, Morita I. Elevated amyloid beta production in senescent retinal pigment epithelium, a possible mechanism of subretinal deposition of amyloid beta in age-related macular degeneration. Biochem Biophys Res Commun. 2012a;423:73–78. doi: 10.1016/j.bbrc.2012.05.085. [DOI] [PubMed] [Google Scholar]
- Wang N, Hebert DN. Tyrosinase maturation through the mammalian secretory pathway: bringing color to life. Pigment Cell Res. 2006;19:3–18. doi: 10.1111/j.1600-0749.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kuro-o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell. 2012b;11:410–417. doi: 10.1111/j.1474-9726.2012.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Dillon J, Gaillard ER. Antioxidant properties of melanin in retinal pigment epithelial cells. Photochem Photobiol. 2006;82:474–479. doi: 10.1562/2005-10-21-RA-725. [DOI] [PubMed] [Google Scholar]
- Weiter JJ, Delori FC, Wing GL, Fitch KA. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest Ophthalmol Vis Sci. 1986;27:145–152. [PubMed] [Google Scholar]
- Widlund HR, Fisher DE. Microphthalamia-associated transcription factor: a critical regulator of pigment cell development and survival. Oncogene. 2003;22:3035–3041. doi: 10.1038/sj.onc.1206443. [DOI] [PubMed] [Google Scholar]
- Wolf I, Levanon-Cohen S, Bose S, Ligumsky H, Sredni B, Kanety H, Kuro-o M, Karlan B, Kaufman B, Koeffler HP, Rubinek T. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene. 2008;27:7094–7105. doi: 10.1038/onc.2008.292. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Miyoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP, Kuro-o M. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280:38029–38034. doi: 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, Hasegawa H, Yamashita T, Nakatani K, Saito Y, Okamoto N, Kurumatani N, Namba N, Kitaoka T, Ozono K, Sakai T, Hataya H, Ichikawa S, Imel EA, Econs MJ, et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem Biophys Res Commun. 2010;398:513–518. doi: 10.1016/j.bbrc.2010.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Matsukawa N, Rakugi H, Imai M, Kida I, Nagai M, Ohta J, Fukuo K, Nabeshima Y, Ogihara T. Upregulation of cAMP is a new functional signal pathway of Klotho in endothelial cells. Biochem Biophys Res Commun. 2003;301:424–429. doi: 10.1016/S0006-291X(02)03056-5. [DOI] [PubMed] [Google Scholar]
- Yin D. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic Biol Med. 1996;21:871–888. doi: 10.1016/0891-5849(96)00175-X. [DOI] [PubMed] [Google Scholar]
- Zhang L, Insel PA. Bcl-2 protects lymphoma cells from apoptosis but not growth arrest promoted by cAMP and dexamethasone. Am J Physiol Cell Physiol. 2001;281:C1642–C1647. doi: 10.1152/ajpcell.2001.281.5.C1642. [DOI] [PubMed] [Google Scholar]