

Abstract
Quantifying the concentration and purity of a target protein is essential for high-throughput protein expression test and rapid screening of highly soluble proteins. However, conventional methods such as PAGE and dot blot assay generally involve multiple time-consuming tasks requiring hours or do not allow instant quantification. Here, we demonstrate a new method based on the Photoactive yellow protein turn Off/On Label (POOL) system that can instantly quantify the concentration and purity of a target protein. The main idea of POOL is to use Photoactive Yellow Protein (PYP), or its miniaturized version, as a fusion partner of the target protein. The characteristic blue light absorption and the consequent yellow color of PYP is absent when initially expressed without its chromophore, but can be turned on by binding its chromophore, p-coumaric acid. The appearance of yellow color upon adding a precursor of chromophore to the co-expressed PYP can be used to check the expression amount of the target protein via visual inspection within a few seconds as well as to quantify its concentration and purity with the aid of a spectrometer within a few minutes. The concentrations measured by the POOL method, which usually takes a few minutes, show excellent agreement with those by the BCA Kit, which usually takes ∼1 h. We demonstrate the applicability of POOL in E. coli, insect, and mammalian cells, and for high-throughput protein expression screening.
Keywords: protein expression test, high-throughput protein expression screening, protein purification, color tag, protein quantification, protein purity estimation
Introduction
Biophysical and biochemical investigations of protein structure and function often rely on the availability of a large amount of a protein sample. To meet this demand, proteins are commonly overexpressed by recombinant technology and purified using affinity chromatography techniques.1–11 During such processes, considerable time and effort is spent to estimate the quantity and purity of the expressed target protein.12 A standard procedure for protein purification involves running PAGE multiple times to measure the expression rate, the concentration of the target protein, and the purities of all the fractions eluted from chromatography to identify the fractions containing the target protein.13 PAGE requires a variety of equipment and involves many time-consuming tasks.14 Herein, we report a complementary method that facilitates expression test and quantification of concentration and purity without running several PAGEs.
The main idea of the new method is to use Photoactive Yellow Protein (PYP), or its truncated version, as a fusion partner with an affinity tag [Fig. 1(A)]. Although making the gene construct of a target protein, one can use PYP with an affinity tag as a tagging partner. PYP is a small (∼14 kDa) and highly soluble protein manifesting in Halorhodospira halophila and carries out the first step of phototaxis signal transduction via changing its structure upon absorbing blue light.15–30 Because PYP appears in only one species, there is no need to make PYP-deficient cells, and thus PYP is appropriate for global tagging. PYP has p-coumaric acid as its chromophore. In the absence of chromophore, the apo PYP has no absorption in the visible region, but the holo PYP bound with the chromophore strongly absorbs in the visible region, exhibiting intense yellow color. This property makes PYP a useful Off/On system31–33 that we named PYP turn Off/On Label (POOL). During the protein expression, chromophore-deficient apo-PYP is co-expressed with the target protein. The appearance of yellow color after adding a precursor of chromophore to the co-expressed PYP can be used to check the expression of the target protein via visual inspection within a few seconds. In addition, the extinction coefficient of PYP is high and well established (45,500 M−1 cm−1 at 446 nm for the wild type and 53,800 M−1 cm−1 at 460 nm for the E46Q mutant),34 and thus it is possible to detect the exact amount of target protein by measuring the UV–VIS spectrum. Moreover, POOL is capable of estimating the purity of the target protein based on the ratio of absorbance at 280 nm and 460 nm (see below).
Figure 1.
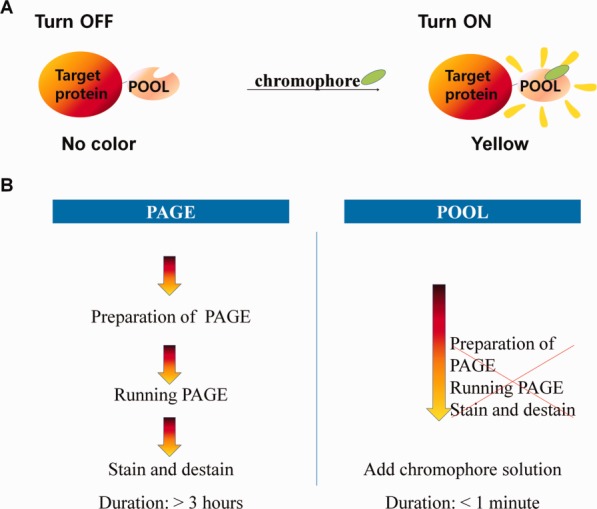
Schematic representation of the POOL system and comparison with the conventional PAGE method. (A) POOL without chromophore is co-expressed with target protein. Upon addition of a precursor of chromophore, POOL exhibits yellow color, thereby signaling the expression of the target protein. (B) POOL reduces time-consuming steps that take over several hours when using PAGE.
Results
Instant colorimetric inspection of protein expression level and amount of target protein during purification
The usefulness of POOL was tested for a number of model target proteins. E46Q mutant PYP was used instead of the wild type because the higher extinction coefficient of E46Q35 provides slightly higher sensitivity than the wild type. The first model protein was Small Ubiquitin like Modifier protein (SUMO) that has 98 amino acids (10,780 Da).36,37 A precursor of chromophore, anhydride p-coumaric acid,31 was added to the SUMO-POOL-expressed cell lysate to check the appearance of yellow color. As shown in the Figure 2(A), prior to the addition of the precursor of chromophore, the cell lysate (1 mL of cultured medium) had no visible color. Upon the addition of the precursor of chromophore, the sample immediately exhibited a yellowish color. One can even approximate the amount of the target protein by comparing the color with those of standard solutions containing PYP at various known concentrations. We could clearly see that the concentration of the target protein is between 10 and 60 μM as shown in Figure 2(C). To enhance the reliability of the practice, as a control, we added the precursor of chromophore to the E. coli lysate in which only the enterokinase38,39 was expressed. The fraction exhibited no visible color, even after the addition of the precursor of chromophore.
Figure 2.
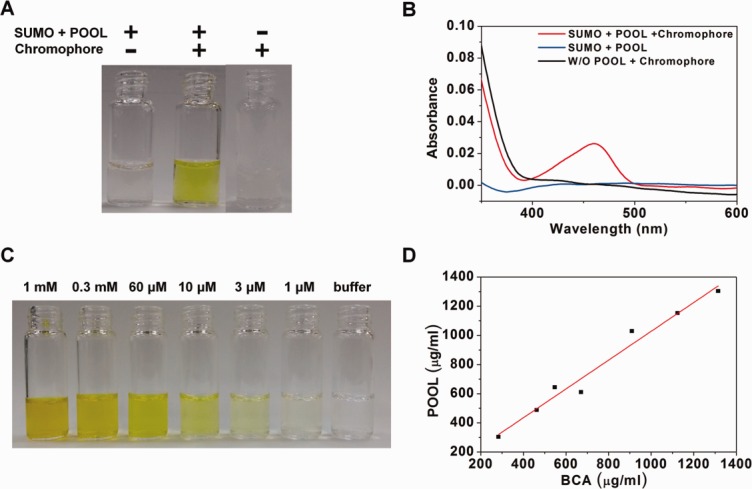
Quantification of the target protein with/without the POOL system by observing the color change and UV–Visible spectroscopy. This figure shows a comparison of the color difference and UV–Visible spectra before and after adding a precursor of chromophore. (A) The color change with/without POOL. The first corresponds to SUMO–POOL fusion protein before adding a precursor of chromophore. The second corresponds to SUMO–POOL fusion protein after adding a precursor of chromophore. The third corresponds to a target protein not fused with POOL after the addition of a precursor of chromophore. (B) UV–Visible spectra corresponding to (A). The blue, red and black curves correspond to the first, second and third cases of (A), respectively. (C) Standard reference solutions used for estimating the concentration. (D) Comparison of the protein concentration measured by the BCA method and POOL method. The slope is 0.99 with a mean error of ∼4%.
This advantage of POOL is even more pronounced in the protein purification steps. Each fraction from various protein separation procedures, such as affinity chromatography or ion-exchange chromatography, can be directly examined by POOL. Figure 3 shows several eluted fractions after the ion exchange chromatography and a comparison between the results of PAGE and POOL. The presence of strong yellow color in the eluted fractions indicates the presence of the target protein (SUMO in this case), which is double confirmed by PAGE gels and UV–Visible spectra. The fraction without yellow color does not have the target protein and the fraction with yellow color has high concentration of the target protein. The UV–Visible spectra show consistent results; the fraction without yellow color has no peak near 460 nm and the fraction with yellow color has a dominant peak near 460 nm. Therefore one can select the fractions containing the target protein on the basis of the presence of yellow color, and thus can save time because the collection of the fractions can be immediately stopped as soon as the fraction lacking yellow color starts to be eluted.
Figure 3.
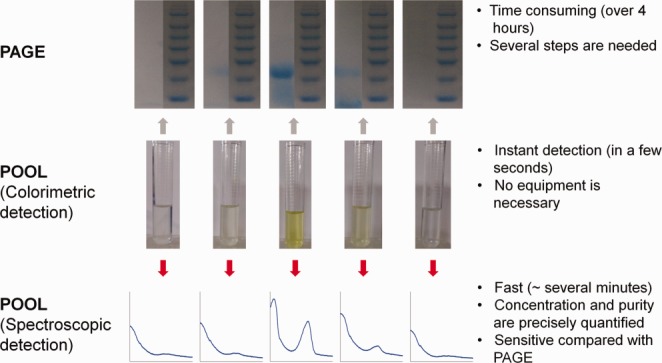
Eluted fractions (middle) of SUMO–POOL fusion protein via ion-exchange chromatography, their corresponding PAGE gels (upper) and UV–Visible spectra (bottom), and the merits and disadvantages of PAGE and POOL.
Rapid determination of the target protein concentration with a UV–Visible spectrometer
More accurate and sensitive quantification of the target protein concentration can be achieved by measuring the UV–Visible spectrum instead of the visual colorimetric detection. After the addition of the precursor of chromophore, the lysate containing a POOL fusion protein showed a distinguishable peak near the 460 nm region of the UV–Visible spectrum. The concentration can be easily calculated by using the following simple relation based on Bear-Lambert law,
| (1) |
where A460 and B460 are the absorbances at 460 nm after and before chromophore addition and the path length of the cell is assumed to be 1 cm. B460 is taken into account because E. coli extract solution sometimes have absorption at 460 nm. In the SUMO-POOL case, the absorbances at 460 nm of the solution diluted by a factor of 60 were 0 and 0.022, respectively, before and after chromophore addition. Thus, according to Eq. (1), the concentration is 0.0245 mM, which is consistent with the result obtained from the colorimetric method. To provide quantitative evidence for the accuracy of the POOL-assisted quantification, we measured the protein concentration by using a commercially available tool, the BCA kit (Pierce, Thermo Scientific, Rockford, IL) and compared the results with those of POOL. For this, we purified SUMO-PYP without chromophore and prepared solutions of various concentrations. Their concentrations were measured by both the BCA Kit and the POOL method. As can be seen in Figure 2(D), the resulting concentrations show satisfactory correlation with the slope of 0.99. We note that the time consumed for the measurements were ∼1 h and a few minutes for the BCA Kit and POOL, respectively. The concentration determined by POOL shows ∼4% deviation compared with that by the BCA Kit.
Rapid determination of the target protein purity during protein purification
After purification by chromatography, the purity, as well as the concentration of the target protein, can be easily estimated by using a UV–Visible spectrometer or a microplate absorbance reader using the following equation,
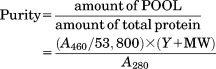 |
(2) |
A460 is the absorbance at 460 nm, A280 is the absorbance at 280 nm, Y is the molecular weight of PYP or it mutant, and MW is the molecular weight of the target protein. Here, the rather crude approximation that A280 roughly corresponds to the concentration (mg/mL) of all proteins in the solution is used.40 If one selects several fractions from chromatography where the yellow color begins and ends and measures the absorbance at 280 and 460 nm to identify their corresponding purities, it becomes very easy to determine the fractions containing the target protein with enough concentration and purity. Thus, running PAGE for every single eluted fraction to identify the fractions containing the target protein becomes unnecessary. If a microplate absorbance reader capable of simultaneously measuring the UV–Visible spectra of several wells is used, the entire process would take only a few minutes.
Application of POOL for high-throughput protein expression screening
We applied POOL to high-throughput expression screening to obtain optimal conditions for protein expression for human calmodulin, mercury binding peptide (MerP),41 and horse myoglobin (Mb). We used a microplate absorbance reader and measured the absorbance at 460 nm to determine the degree of target protein expression and calculated the concentration (Fig. 4); the testing yielded the results consistent with those obtained by the colorimetric method. Figure 4 shows that the addition of a chromophore precursor induces an immediate color change for the target protein. Therefore, it is possible to obtain an approximate measure of quantity of the expressed protein through the instant colorimetric method and thus to determine the optimal expression condition for the target protein. Under our testing conditions, we can see that the human calmodulin expression reaches its maximum at 18°C with the IPTG concentration of 0.2 mM using pQE80L vector. Furthermore, mercury binding peptides are well expressed under all conditions and the horse myoglobin shows a similar trend to that of human calmodulin. These tests prove that the simplistic nature of the POOL method allows convenient high-throughput protein expression screening to obtain optimal conditions for soluble protein expression.
Figure 4.
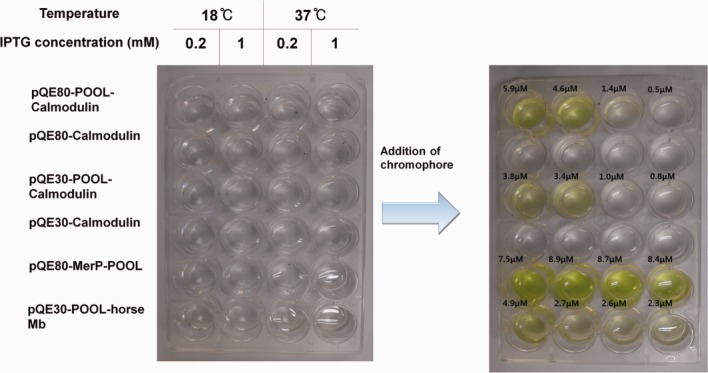
Optimization of the expression condition by high-throughput protein expression screening aided by POOL for human calmodulin. To determine the best expression condition of human calmodulin and to study the efficiency of POOL, the E. coli lysate with the constructs with and without POOL were tested. The color change is not observed in the wells containing cell lysate without POOL. The optimal expression condition can be identified simply by the addition of a precursor of chromophore. The measured concentration is shown for each well.
Application of POOL to eukaryote cells
POOL is also applicable to target proteins expressed in eukaryote cells. We checked the expression of the membrane protein CD40 in the mammalian 293 cell line and the truncated TLR3 (toll-like receptor 3 without transmembrane domain) in the insect Sf9 cell line42,43 (Supporting Information Fig. S4). In both cases, we could easily perform an expression test simply by adding the chromophore precursor.
Making mini-PYP for POOL
Although the full-length PYP is highly soluble and does not interfere with the expression of other proteins, a tag of a smaller size is preferred because it is generally expected to interfere less with the expression of the target protein.44 Through recombinant-biotechnology approaches, we have identified the most basic segment of PYP capable of binding to deprotonated p-coumaric acid and thus being used as a POOL tag. We tested various N-terminal and C-terminal deletion mutants of PYP fused with SUMO (Fig. 5). First, we produced eight types of N-terminal deletion mutants [deletion of residues 1–5 (N5), 1–10 (N10), 1–18 (N18), 1–27 (N27), 1–31 (N31), 1–40 (N40), 1–42 (N42), and 1–46 (N46)] and three types of C-terminal deletion mutants [deletion of residues 73–125 (C53), 107–125 (C18), and 117–125 (C8)]. Of all the deletion mutants, three mutants, N15, N10 and N18, show consistent color change, whereas the rest initially show yellow color but gradually lose their color within several minutes. Actually, all the tested mutants can be used to check expressions as the color persisted for at least more than half a minute. We also designed truncated PYP by removing both the N-terminal and C-terminal residues (deletion of residues 1–27 and 107–125 (N27C18)).44 This mini-PYP that has the 28–106 residues of the original PYP can be also used as a POOL tag. However, to obtain an accurate value of concentration and purity, at least the N18 mutant should be used.
Figure 5.

Results of calorimetric screening of PYP deletion mutants. The color change of 12 deletion mutants upon adding a precursor of chromophore is shown. Persistent color change was observed for N5, N10, and N18, but the rest of the N-terminal deletion mutants and all C-terminal deletion mutants showed weak color change that gradually faded. The N27C18 deletion mutant can be still used to check expression of the target protein as it shows color change for a few minutes.
Discussion
The genetically engineered POOL enables us to instantly quantify the concentration and purity of a target protein, thereby aiding the protein expression and purification. This confirms that, with POOL, one can check the amount of protein expression by simply observing the color change, and thus significantly reduces the time required for checking the amount of protein expression. For example, on average the usage of PAGE for checking protein expression requires several hours, whereas the POOL approach requires only a few seconds. UV–Visible absorbance measurement allows determination of the concentration and purity of the target protein within a few minutes. The concentrations measured by the POOL method, which usually takes a few minutes, show an excellent correlation with those by the BCA Kit, which usually takes ∼1 h [Fig. 2(D)]. POOL also allows instant approximation of the amount of protein in the fractions after chromatography by simply comparing colors and easy selection of the fractions containing the highly pure target protein. POOL can be used in eukaryotic cells as well as E. coli. For example, even if the membrane protein CD40 has a low expression rate, we could detect color change in the cell lysate upon addition of the chromophore precursor.
The instant turn/on property of POOL makes it an excellent method for high-throughput expression screening. Conventional methods for high-throughput sample detection45–49 and quantification such as affinity columns, PAGEs, and immune-detection of dot blots via usage of antibodies50 demand significant time and effort or lack accuracy. Compared to those methods, POOL affords easy fast and accurate high-throughput screening. Using POOL together with a microplate absorbance reader can also yield accurate measurements since a spectrometer possesses high sensitivity. The high sensitivity of POOL eliminates the need for the step of concentration via capture, such as affinity column chromatography. POOL may be comparable to the high-throughput protein expression screening51,52 using GFP as a fluorescence tag. In addition, the turn-on property and the smaller size (see below) of POOL may provide additional advantages.
When compared with the other protein tag techniques, POOL has the following advantages. First, POOL is very soluble, has a small size of about half the size of GFP (26.9 kDa), a well-known tag,53–55 and does not disturb the expression and solubility of a target protein. Furthermore, even smaller versions of PYP can be employed by utilizing the minimal segment of PYP. For example, POOL sometimes appears to assist the expression rate and solubility (Supporting Information Fig. S2) of a target protein. This characteristic of POOL may be associated with high expression rate and solubility of recombinant PYP in the E. coli cells. For example, a recombinant PYP construct can produce about 50 mg of soluble protein per liter of E. coli culture.19,56 Second, if we have to tag other proteins that have inherent visible color, such as GFP, LOV2, BLUF, and cytochrome c, it is difficult to distinguish whether the color is due to contamination, or due to other proteins or pigments expressed in the cell, or by the target protein itself. However, using the turn/on property of POOL upon addition of the chromophore, one can precisely distinguish whether the color change is due to the fused tag protein or some other factors. Nevertheless, POOL has the following limitations. First, if the function of the target protein is greatly interfered by external amino acids, POOL, which acts as external amino acids, can cause problems. This problem is also commonly observed in other protein tags. Second, as in the case of other protein tags, we have to use a protease to cleave the target protein from the POOL product. However, this could be overcome by using an autoprocessing enzyme tag.57 Using the POOL approach requires construction of the initial target-POOL fusion protein and cleavage step afterwards. However in the case where a fusion tag such as MBP,58–61 and TRX62,63 has to be used for the purification purpose, incorporating POOL does not impose any extra effort. In addition, the vectors designed for incorporating any new target protein gene with POOL are already constructed and available (see Materials and Methods). In conclusion we expect that POOL can be a useful method for many applications such as protein expression test, purity check, purification and high throughput protein expression screening.
Materials and Methods
Cloning for application of POOL
The gene of a target protein was cloned to a recombinant vector along with the PYP gene in either the N-terminal or C-terminal side. The gene cloning was performed in a manner shown in Supporting Information Figure S1. We used the pQE or pET vector systems, but any vector can be used as the base. The PYP gene was inserted into the base vector without the stop codon. In addition to this, a restriction enzyme site, which is used for subcloning both the protease recognition site and the target protein simultaneously, is inserted into either the left or right side of the PYP gene depending on the relative position of the target protein gene with respect to the PYP gene. Any typical protease recognition site can be introduced. We used a protease having a tag (in our case, the his tag) identical to the tag of PYP so that both the protease and the cut PYP can be removed by a single affinity chromatography. We tested the usefulness of POOL by inserting calmodulin, SUMO, horse myoglobin, and mercury binding peptide (MerP) into the pQE80 and pQE30 vectors. All the cloning procedures were conducted using the EZ-cloning kit (enzynomics™) according to the EZ-cloning kit manual. We have made two vectors optimized for cloning (pQE80L + His tag + POOL + protease recognition site + cloning site and pET15b + cloning site + protease recognition site + POOL + His tag) (Supporting Information Fig. S1), which can be provided upon request. The details of the cloning experiments performed in this study are described in Supporting Information.
High-throughput protein expression screening
High-throughput protein expression test was conducted by growing small scale cell cultures on two 24-well plates. The expression condition of a target protein was tested by using various combinations of IPTG concentration (1 and 0.2 mM), temperature (37 and 18°C) and vector (pQE80 and pQE30). After sonication of cultured cells, the cell lysate was centrifuged (HANIL Science Industrial Co. Ltd, Combi-514R) and the supernatant was transferred to a new 24-well plate. The absorbances at 280 and 460 nm of the supernatant were measured using a microplate absorbance reader (Bio-RAD Laboratories, INC. xMark™). Then the precursor of chromophore was added to the supernatant for inspecting the color change. The concentration of the expressed target protein at each well can be easily estimated by comparing its yellow color with those of the standard reference solutions of known concentrations. For more quantitative measurement of concentration and purity, the absorbance at 460 nm has to be measured again using the microplate absorbance reader.
Mini-photoactive yellow protein expression and quantification
We checked the solubility and the expression of the SUMO-mini-POOL. All the constructs were transformed into the E. coli BL21 (DE3) cell line and transferred to a 24-well plate. The subsequent procedures are identical to those described for the preceding section (high throughput protein expression screening).
Acknowledgments
The authors thank Jie-Oh Lee, Geun-Young Cho and Hee-Sung Park for their helpful discussions.
Glossary
- GFP
green fluorescence protein
- Mb
myoglobin
- MerP
mercury binding peptide
- PAGE
Poly Acrylamide Gel Electrophoresis
- POOL
PYP turn Off/On Label
- PYP
Photoactive Yellow Protein
- SUMO
Small Ubiquitin like Modifier protein
- TLR3
toll-like receptor 3
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 3.Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 4.Tykvart J, Sacha P, Barinka C, Knedlik T, Starkova J, Lubkowski J, Konvalinka J. Efficient and versatile one-step affinity purification of in vivo biotinylated proteins: expression, characterization and structure analysis of recombinant human glutamate carboxypeptidase II. Protein Expr Purif. 2012;82:106–115. doi: 10.1016/j.pep.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shooltz DD, Alberts GL, Triezenberg SJ. One-step affinity purification of recombinant TATA binding proteins utilizing a modular protein interaction partner. Protein Expr Purif. 2008;59:297–301. doi: 10.1016/j.pep.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Cabanne C, Pezzini J, Joucla G, Hocquellet A, Barbot C, Garbay B, Santarelli X. Efficient purification of recombinant proteins fused to maltose-binding protein by mixed-mode chromatography. J Chromatogr A. 2009;1216:4451–4456. doi: 10.1016/j.chroma.2009.03.048. [DOI] [PubMed] [Google Scholar]
- 7.Lee KM, Ma KW, Shaw PC, Wong KB. A high-yield one-step purification method using copper-chelating chromatography for recombinant proteins fused with maltose-binding protein. Anal Biochem. 2006;358:152–154. doi: 10.1016/j.ab.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Keefe AD, Wilson DS, Seelig B, Szostak JW. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr Purif. 2001;23:440–446. doi: 10.1006/prep.2001.1515. [DOI] [PubMed] [Google Scholar]
- 9.Crowe J, Masone BS, Ribbe J. One-step purification of recombinant proteins with the 6xHis tag and Ni-NTA resin. Methods Mol Biol. 1996;58:491–510. doi: 10.1385/0-89603-402-X:491. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt TG, Skerra A. One-step affinity purification of bacterially produced proteins by means of the “Strep tag” and immobilized recombinant core streptavidin. J Chromatogr A. 1994;676:337–345. doi: 10.1016/0021-9673(94)80434-6. [DOI] [PubMed] [Google Scholar]
- 11.Mueller U, Bussow K, Diehl A, Bartl FJ, Niesen FH, Nyarsik L, Heinemann U. Rapid purification and crystal structure analysis of a small protein carrying two terminal affinity tags. J Struct Funct Genomics. 2003;4:217–225. doi: 10.1023/b:jsfg.0000016119.50040.a3. [DOI] [PubMed] [Google Scholar]
- 12.Graslund S, Nordlund P, Weigelt J, Hallberg BM, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, dhe-Paganon S, Park HW, Savchenko A, Yee A, Edwards A, Vincentelli R, Cambillau C, Kim R, Kim SH, Rao Z, Shi Y, Terwilliger TC, Kim CY, Hung LW, Waldo GS, Peleg Y, Albeck S, Unger T, Dym O, Prilusky J, Sussman JL, Stevens RC, Lesley SA, Wilson IA, Joachimiak A, Collart F, Dementieva I, Donnelly MI, Eschenfeldt WH, Kim Y, Stols L, Wu R, Zhou M, Burley SK, Emtage JS, Sauder JM, Thompson D, Bain K, Luz J, Gheyi T, Zhang F, Atwell S, Almo SC, Bonanno JB, Fiser A, Swaminathan S, Studier FW, Chance MR, Sali A, Acton TB, Xiao R, Zhao L, Ma LC, Hunt JF, Tong L, Cunningham K, Inouye M, Anderson S, Janjua H, Shastry R, Ho CK, Wang D, Wang H, Jiang M, Montelione GT, Stuart DI, Owens RJ, Daenke S, Schutz A, Heinemann U, Yokoyama S, Bussow K, Gunsalus KC. Protein production and purification. Nat Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith BJ. Quantification of proteins on polyacrylamide gels (nonradioactive) Methods Mol Biol. 1994;32:107–111. doi: 10.1385/0-89603-268-X:107. [DOI] [PubMed] [Google Scholar]
- 14.Chrambach A, Rodbard D. Polyacrylamide gel electrophoresis. Science. 1971;172:440–451. doi: 10.1126/science.172.3982.440. [DOI] [PubMed] [Google Scholar]
- 15.Kim TW, Lee JH, Choi J, Kim KH, van Wilderen LJ, Guerin L, Kim Y, Jung YO, Yang C, Kim J, Wulff M, van Thor JJ, Ihee H. Protein structural dynamics of photoactive yellow protein in solution revealed by pump-probe X-ray solution scattering. J Am Chem Soc. 2012;134:3145–3153. doi: 10.1021/ja210435n. [DOI] [PubMed] [Google Scholar]
- 16.Kumauchi M, Hara MT, Stalcup P, Xie A, Hoff WD. Identification of six new photoactive yellow proteins—diversity and structure-function relationships in a bacterial blue light photoreceptor. Photochem Photobiol. 2008;84:956–969. doi: 10.1111/j.1751-1097.2008.00335.x. [DOI] [PubMed] [Google Scholar]
- 17.McRee DE, Meyer TE, Cusanovich MA, Parge HE, Getzoff ED. Crystallographic characterization of a photoactive yellow protein with photochemistry similar to sensory rhodopsin. J Biol Chem. 1986;261:13850–13851. [PubMed] [Google Scholar]
- 18.Yeremenko S, van Stokkum IH, Moffat K, Hellingwerf KJ. Influence of the crystalline state on photoinduced dynamics of photoactive yellow protein studied by ultraviolet-visible transient absorption spectroscopy. Biophys J. 2006;90:4224–4235. doi: 10.1529/biophysj.105.074765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ihee H, Rajagopal S, Srajer V, Pahl R, Anderson S, Schmidt M, Schotte F, Anfinrud PA, Wulff M, Moffat K. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc Natl Acad Sci USA. 2005;102:7145–7150. doi: 10.1073/pnas.0409035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubinstenn G, Vuister GW, Mulder FA, Dux PE, Boelens R, Hellingwerf KJ, Kaptein R. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat Struct Biol. 1998;5:568–570. doi: 10.1038/823. [DOI] [PubMed] [Google Scholar]
- 21.Sprenger WW, Hoff WD, Armitage JP, Hellingwerf KJ. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J Bacteriol. 1993;175:3096–3104. doi: 10.1128/jb.175.10.3096-3104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heyne K, Mohammed OF, Usman A, Dreyer J, Nibbering ET, Cusanovich MA. Structural evolution of the chromophore in the primary stages of trans/cis isomerization in photoactive yellow protein. J Am Chem Soc. 2005;127:18100–18106. doi: 10.1021/ja051210k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borucki B, Kyndt JA, Joshi CP, Otto H, Meyer TE, Cusanovich MA, Heyn MP. Effect of salt and pH on the activation of photoactive yellow protein and gateway mutants Y98Q and Y98F. Biochemistry. 2005;44:13650–13663. doi: 10.1021/bi050991z. [DOI] [PubMed] [Google Scholar]
- 24.Joshi CP, Borucki B, Otto H, Meyer TE, Cusanovich MA, Heyn MP. Photoreversal kinetics of the I-1 and I-2 intermediates in the photocycle of photoactive yellow protein by double flash experiments with variable time delay. Biochemistry. 2005;44:656–665. doi: 10.1021/bi0481141. [DOI] [PubMed] [Google Scholar]
- 25.Meyer TE, Tollin G, Causgrove TP, Cheng P, Blankenship RE. Picosecond decay kinetics and quantum yield of fluorescence of the photoactive yellow protein from the halophilic purple phototrophic bacterium, Ectothiorhodospira halophila. Biophys J. 1991;59:988–991. doi: 10.1016/S0006-3495(91)82313-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan HY, Morgan SA, Brechun KE, Chen YY, Jaikaran AS, Woolley GA. Improving a designed photocontrolled DNA-binding protein. Biochemistry. 2011;50:1226–1237. doi: 10.1021/bi101432p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan SA, Woolley GA. A photoswitchable DNA-binding protein based on a truncated GCN4-photoactive yellow protein chimera. Photochem Photobiol Sci. 2010;9:1320–1326. doi: 10.1039/c0pp00214c. [DOI] [PubMed] [Google Scholar]
- 28.Morgan SA, Al-Abdul-Wahid S, Woolley GA. Structure-based design of a photocontrolled DNA binding protein. J Mol Biol. 2010;399:94–112. doi: 10.1016/j.jmb.2010.03.053. [DOI] [PubMed] [Google Scholar]
- 29.Jung YO, Lee JH, Kim J, Schmidt M, Moffat K, Srajer V, Ihee H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat Chem. 2013;5:212–220. doi: 10.1038/nchem.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran PL, Lovett JE, Carl PJ, Cammarata M, Lee JH, Jung YO, Ihee H, Timmel CR, van Thor JJ. The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J Am Chem Soc. 2011;133:9395–9404. doi: 10.1021/ja200617t. [DOI] [PubMed] [Google Scholar]
- 31.Imamoto Y, Ito T, Kataoka M, Tokunaga F. Reconstitution photoactive yellow protein from apoprotein and p-coumaric acid derivatives. FEBS Lett. 1995;374:157–160. doi: 10.1016/0014-5793(95)01096-w. [DOI] [PubMed] [Google Scholar]
- 32.Hori Y, Ueno H, Mizukami S, Kikuchi K. Photoactive yellow protein-based protein labeling system with turn-on fluorescence intensity. J Am Chem Soc. 2009;131:16610–16611. doi: 10.1021/ja904800k. [DOI] [PubMed] [Google Scholar]
- 33.Hori Y, Nakaki K, Sato M, Mizukami S, Kikuchi K. Development of protein-labeling probes with a redesigned fluorogenic switch based on intramolecular association for no-wash live-cell imaging. Angew Chem Int Ed Engl. 2012;51:5611–5614. doi: 10.1002/anie.201200867. [DOI] [PubMed] [Google Scholar]
- 34.Losi A, Gensch T, van der Horst MA, Hellingwerf KJ, Braslavsky SE. Hydrogen-bond network probed by time-resolved optoacoustic spectroscopy: photoactive yellow protein and the effect of E46Q and E46A mutations. Phys Chem Chem Phys. 2005;7:2229–2236. doi: 10.1039/b419079c. [DOI] [PubMed] [Google Scholar]
- 35.Imamoto Y, Koshimizu H, Mihara K, Hisatomi O, Mizukami T, Tsujimoto K, Kataoka M, Tokunaga F. Roles of amino acid residues near the chromophore of photoactive yellow protein. Biochemistry. 2001;40:4679–4685. doi: 10.1021/bi002291u. [DOI] [PubMed] [Google Scholar]
- 36.Li SJ, Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol. 2000;20:2367–2377. doi: 10.1128/mcb.20.7.2367-2377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gareau JR, Reverter D, Lima CD. Determinants of small ubiquitin-like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO-RanGAP1 binding activities of nucleoporin RanBP2. J Biol Chem. 2012;287:4740–4751. doi: 10.1074/jbc.M111.321141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simeonov P, Zahn M, Strater N, Zuchner T. Crystal structure of a supercharged variant of the human enteropeptidase light chain. Proteins. 2012;80:1907–1910. doi: 10.1002/prot.24084. [DOI] [PubMed] [Google Scholar]
- 39.Lu D, Futterer K, Korolev S, Zheng X, Tan K, Waksman G, Sadler JE. Crystal structure of enteropeptidase light chain complexed with an analog of the trypsinogen activation peptide. J Mol Biol. 1999;292:361–373. doi: 10.1006/jmbi.1999.3089. [DOI] [PubMed] [Google Scholar]
- 40.Stoscheck CM. Quantitation of protein. Methods Enzymol. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 41.Serre L, Rossy E, Pebay-Peyroula E, Cohen-Addad C, Coves J. Crystal structure of the oxidized form of the periplasmic mercury-binding protein MerP from Ralstonia metallidurans CH34. J Mol Biol. 2004;339:161–171. doi: 10.1016/j.jmb.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 42.Choe J, Kelker MS, Wilson IA. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science. 2005;309:581–585. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- 43.Bell JK, Botos I, Hall PR, Askins J, Shiloach J, Segal DM, Davies DR. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc Natl Acad Sci USA. 2005;102:10976–10980. doi: 10.1073/pnas.0505077102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, Hammarstrom M, Johansson I, Kotenyova T, Nilsson M, Nordlund P, Weigelt J. The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif. 2008;58:210–221. doi: 10.1016/j.pep.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen H, Martinez B, Oganesyan N, Kim R. An automated small-scale protein expression and purification screening provides beneficial information for protein production. J Struct Funct Genomics. 2004;5:23–27. doi: 10.1023/B:JSFG.0000029195.73810.86. [DOI] [PubMed] [Google Scholar]
- 46.Vincentelli R, Cimino A, Geerlof A, Kubo A, Satou Y, Cambillau C. High-throughput protein expression screening and purification in Escherichia coli. Methods. 2011;55:65–72. doi: 10.1016/j.ymeth.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Fan J, Heng J, Dai S, Shaw N, Zhou B, Huang B, He Z, Wang Y, Jiang T, Li X, Liu Z, Wang X, Zhang XC. An efficient strategy for high throughput screening of recombinant integral membrane protein expression and stability. Protein Expr Purif. 2011;78:6–13. doi: 10.1016/j.pep.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 48.Bordes F, Fudalej F, Dossat V, Nicaud JM, Marty A. A new recombinant protein expression system for high-throughput screening in the yeast Yarrowia lipolytica. J Microbiol Methods. 2007;70:493–502. doi: 10.1016/j.mimet.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Bursey EH, Kim CY, Yu M, Terwilliger TC, Hung LW. An automated high-throughput screening method for the identification of high-yield, soluble protein variants using cell-free expression and systematic truncation. J Struct Funct Genomics. 2006;7:139–147. doi: 10.1007/s10969-007-9017-4. [DOI] [PubMed] [Google Scholar]
- 50.Storey CC, Mearns G, Richmond SJ. Immune dot blot technique for diagnosing infection with Chlamydia trachomatis. Genitourin Med. 1987;63:375–379. doi: 10.1136/sti.63.6.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coleman MA, Lao VH, Segelke BW, Beernink PT. High-throughput, fluorescence-based screening for soluble protein expression. J Proteome Res. 2004;3:1024–1032. doi: 10.1021/pr049912g. [DOI] [PubMed] [Google Scholar]
- 52.Braud S, Moutiez M, Belin P, Abello N, Drevet P, Zinn-Justin S, Courcon M, Masson C, Dassa J, Charbonnier JB, Boulain JC, Menez A, Genet R, Gondry M. Dual expression system suitable for high-throughput fluorescence-based screening and production of soluble proteins. J Proteome Res. 2005;4:2137–2147. doi: 10.1021/pr050230i. [DOI] [PubMed] [Google Scholar]
- 53.Niswender KD, Blackman SM, Rohde L, Magnuson MA, Piston DW. Quantitative imaging of green fluorescent protein in cultured cells: comparison of microscopic techniques, use in fusion proteins and detection limits. J Microsc. 1995;180:109–116. doi: 10.1111/j.1365-2818.1995.tb03665.x. [DOI] [PubMed] [Google Scholar]
- 54.Misteli T, Spector DL. Applications of the green fluorescent protein in cell biology and biotechnology. Nat Biotechnol. 1997;15:961–964. doi: 10.1038/nbt1097-961. [DOI] [PubMed] [Google Scholar]
- 55.Gerdes HH, Kaether C. Green fluorescent protein: applications in cell biology. FEBS Lett. 1996;389:44–47. doi: 10.1016/0014-5793(96)00586-8. [DOI] [PubMed] [Google Scholar]
- 56.Mihara K, Hisatomi O, Imamoto Y, Kataoka M, Tokunaga F. Functional expression and site-directed mutagenesis of photoactive yellow protein. J Biochem. 1997;121:876–880. doi: 10.1093/oxfordjournals.jbchem.a021668. [DOI] [PubMed] [Google Scholar]
- 57.Shen A, Lupardus PJ, Morell M, Ponder EL, Sadaghiani AM, Garcia KC, Bogyo M. Simplified, enhanced protein purification using an inducible, autoprocessing enzyme tag. PLoS One. 2009;4:e8119. doi: 10.1371/journal.pone.0008119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pryor KD, Leiting B. High-level expression of soluble protein in Escherichia coli using a His(6)-tag and maltose-binding-protein double-affinity fusion system. Protein Expr Purif. 1997;10:309–319. doi: 10.1006/prep.1997.0759. [DOI] [PubMed] [Google Scholar]
- 59.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bornhorst JA, Falke JJ. Purification of proteins using polyhistidine affinity tags. App Chimeric Genes Hybrid Pro A. 2000;326:245–254. doi: 10.1016/s0076-6879(00)26058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carson M, Johnson DH, McDonald H, Brouillette C, DeLucas LJ. His-tag impact on structure. Acta Cryst. 2007;D63:295–301. doi: 10.1107/S0907444906052024. [DOI] [PubMed] [Google Scholar]
- 62.McCoy J, Lavallie E. Expression and purification of thioredoxin fusion proteins. Curr Protoc Mol Biol Chapter. 2001;16:Unit16 18. doi: 10.1002/0471142727.mb1608s28. [DOI] [PubMed] [Google Scholar]
- 63.Lavallie ER, Diblasio EA, Kovacic S, Grant KL, Schendel PF, Mccoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the Escherichia-coli cytoplasm. Bio-Technology. 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.