

Abstract
The substitution of Ser187, a residue located far from the active site of human liver peroxisomal alanine:glyoxylate aminotransferase (AGT), by Phe gives rise to a variant associated with primary hyperoxaluria type I. Unexpectedly, previous studies revealed that the recombinant form of S187F exhibits a remarkable loss of catalytic activity, an increased pyridoxal 5′-phosphate (PLP) binding affinity and a different coenzyme binding mode compared with normal AGT. To shed light on the structural elements responsible for these defects, we solved the crystal structure of the variant to a resolution of 2.9 Å. Although the overall conformation of the variant is similar to that of normal AGT, we noticed: (i) a displacement of the PLP-binding Lys209 and Val185, located on the re and si side of PLP, respectively, and (ii) slight conformational changes of other active site residues, in particular Trp108, the base stacking residue with the pyridine cofactor moiety. This active site perturbation results in a mispositioning of the AGT-pyridoxamine 5′-phosphate (PMP) complex and of the external aldimine, as predicted by molecular modeling studies. Taken together, both predicted and observed movements caused by the S187F mutation are consistent with the following functional properties of the variant: (i) a 300- to 500-fold decrease in both the rate constant of L-alanine half-transamination and the kcat of the overall transamination, (ii) a different PMP binding mode and affinity, and (iii) a different microenvironment of the external aldimine. Proposals for the treatment of patients bearing S187F mutation are discussed on the basis of these results. Proteins 2013; 81:1457–1465. © 2013 Wiley Periodicals, Inc.
Keywords: alanine:glyoxylate aminotransferase, Primary Hyperoxaluria Type I, pyridoxal 5′-phosphate, crystal structure, pathogenic variant, molecular modeling
INTRODUCTION
Human alanine:glyoxylate aminotransferase (AGT) is a Fold-type I pyridoxal 5′-phosphate (PLP)-dependent enzyme, which catalyses the conversion of L-alanine and glyoxylate to pyruvate and glycine in liver peroxisomes.1 The protein is a 86 kDa-homodimer and each subunit comprises a 21-residues long N-terminal extension that wraps over the surface of the neighboring subunit and is important for the attainment of the correct conformation of the holo-protein,2 a 260-residues large domain containing most of the active site and of the dimerization interface, and a 110-residues C-terminal domain containing regions involved in the recognition and binding to the Pex5p peroxisomal receptor.3, 4 The AGT aminotransferase reaction follows a ping-pong bi–bi mechanism that relies on the formation of the external aldimine intermediate, in which the amino group of L-alanine replaces the ɛ-amino group of Lys209, the PLP-binding residue. Subsequent proton abstraction from Cα yields the quinonoid intermediate, and reprotonation at the C4′ of the coenzyme produces the ketimine intermediate. Hydrolysis of the ketimine intermediate yields pyruvate and the pyridoxamine-5′-phosphate (PMP) form of the enzyme. The second half-reaction in reverse direction with glyoxylate gives the enzyme in its initial PLP form and glycine.
Inherited mutations of the AGXT gene lead to the rare metabolic disorder Primary Hyperoxaluria Type I (PH1) (MIM #259900).5 In the absence of functional AGT, glyoxylate is converted to oxalate by cytosolic lactate dehydrogenase (LDH). Oxalate is an end-product of metabolism and is excreted in the urine. The increased production of oxalate in PH1 leads to the formation and deposition of calcium oxalate crystals in the urinary tract and kidneys causing urolithiasis and nephrocalcinosis. The subsequent renal failure results in the systemic deposition of calcium oxalate crystals.6, 7
More than 150 pathogenic mutations of the AGXT gene are associated with PH1 and the vast majority of them are missense point mutations that lead to the synthesis of an aberrant gene product.8 The molecular mechanisms by which missense pathogenic mutations cause AGT deficiency include the loss of catalytic activity, the reduced stability, the aberrant folding, the aggregation propensity, and the mislocalization of the protein.1,9–;22 Some mutations co-segregate and functionally interact with the minor allele of the AGXT gene encoding a non-pathogenic polymorphic form, which differs from that encoded by the more frequent major allele by a P11L and a I340M amino acid substitutions. Considering this heterogeneity of PH1 in terms of genotype and enzymatic phenotype, a crucial point for the understanding of the disease pathogenesis is the elucidation of the defect of each disease-causing variant at a molecular level. A fundamental step in this direction is the definition of the structural perturbations induced by the amino acid substitution on the AGT molecule and of the consequent alterations of the enzyme structural and/or functional properties.
The C→T mutation at position 682 in the major allele of the AGXT gene, leading to the S187F amino acid substitution, has been initially found in a PH1 patient showing less than 1% AGT catalytic activity with respect to normal subjects, as well as the nearly complete loss of AGT immunoreactive protein in liver biopsy. The genetic analysis revealed that the patient was heterozygous for the mutation, with the allele bearing the C682→T mutation producing an mRNA of normal size and abundance, while the other allele did not produce detectable amounts of mRNA.23
Ser187 is a large-domain residue conserved in the mammalian AGTs and belongs to a random-coil region that is not part of the active site.4 Cell-free expression studies have revealed that the S187F variant has reduced dimer stability and increased sensitivity to proteasomal degradation and that PLP is able to increase the stability of the protein.14, 15 We recently showed that the S187F variant in the recombinant purified form displays a reduced thermal stability in the apo-form with respect to normal AGT and that the presence of bound PLP is able to relieve the protein instability.18 Nevertheless, we found that the Ser-to-Phe substitution at position 187 also affects the AGT functional properties. In fact, the purified recombinant S187F variant has a reduced catalytic efficiency, an affinity for PLP increased of at least 27-fold, and a different coenzyme binding mode with respect to normal AGT.18 This implies that the variant might be characterized by structural changes remote from the mutation site with effects on the topography of the active site.
In order to have a deeper understanding of the structural and functional effects of the S187F mutation on AGT, we have solved the X-ray crystal structure of the S187F variant to a resolution of 2.9 Å. In addition, the putative binding mode of PMP and of the L-alanine external aldimine has been determined by molecular modeling studies. By comparing the properties of S187F with those of the normal protein, we showed that the Ser187-to-Phe substitution in human AGT leads to (i) active site modifications mainly consisting in the mispositioning of the PLP-binding lysine, (ii) an altered binding mode of pyridoxamine 5′-phosphate (PMP) and L-alanine external aldimine, and (iii) a reduced rate of the L-alanine half-transamination that could account for the reduced rate of the overall transamination of the variant.
MATERIALS AND METHODS
Protein expression and purification
The S187F variant in its His-tagged form was expressed in Escherichia coli and purified with the procedure already described.24 The apo form of the variant was prepared as previously described.24 The protein concentration in the AGT samples was determined by absorbance spectroscopy using an extinction coefficient of 9.54 × 104M−1cm-1at 280 nm.9
Crystallization and X-ray structure determination
S187F was concentrated to 5 mg/ml. Crystals were obtained by mixing 1 μl protein with 1 μl reservoir solution, comprising 0.1M HEPES (pH 7.5), 0.1M LiSO4, 23 % [w/w] PEG4000 and submitting to hanging drop vapor diffusion at 20 °C.
X-ray data were collected at X13 at the DORIS III synchrotron radiation storage ring, Hamburg, Germany. Data were processed with XDS25 and scaled with SCALA.26 Five percent of the reflections were randomly selected for cross validation. Initial analysis of the data suggested an orthorhombic space group, as all three unit cell angles were close to 90°. However, our attempts to solve the structure were unsuccessful. Consequently, we used Pointless27 to determine the correct space group (P21). The structure of the S187F variant was solved by molecular replacement with Phaser28 using normal AGT as starting model (PDB entry: 1H0C).4 REFMAC29 was used to refine the initial structural model, applying translation/libration/srew parameterization, and including restraints from the non-crystallographic symmetry between the AGT chains. Manual building and structure analysis were carried out in COOT.30 The structure quality was assessed with MolProbity.31 Programs of the CCP4 package32 were used for average B-factor calculation (Fig. S1), structure manipulation, analysis, and validation.
Kinetic studies
The half-transamination reaction of holoS187F (15 μM) with L-alanine (5–500 mM) was performed in 100 mM potassium phosphate buffer, pH 7.4, at 25°C in a total volume of 250 μl. The decrease in the 413 nm absorbance signal was measured as a function of time and fitted to the following equation:
| (1) |
where At is the absorbance at time t, ΔA1 is the amplitude of the fast phase, ΔA2 is the amplitude of the slow phase, kobs1 is the observed rate constant of the fast phase, kobs2 is the observed rate constant of the slow phase, and A∞ is the final absorbance.
The kmax and apparent Kmapp values for each phase of the L-alanine half-reaction were determined by plotting the observed rate constants versus L-alanine concentrations and fitting the data to Eq. (2):
| (2) |
The coupling of the L-alanine half-transamination to LDH was carried out by performing the reaction in the presence of rabbit muscle LDH (0.5 mg/ml) and NADH (200 μM).
The detection and quantification of the PLP and PMP content during the L-alanine half-transamination was carried out using a previously described HPLC method.9
The kinetic parameters of the S187F mutant for the overall transamination of the L-alanine/glyoxylate pair in the absence and presence of 5 mM pyruvate were determined by measuring the amount of glyoxylate consumed by HPLC after derivatization with dinitrophenylhydrazine as previously described.33 Substrate saturation curves were fitted using Eq. (3), and dead-end inhibition data were fitted using Eq. (4) for competitive inhibition:
| (3) |
| (4) |
where v is the initial velocity, V is the maximum velocity, A is substrate concentration, Ka is the Km for substrate A, and Kis is the slope constant.34
Spectroscopic measurements
Absorption measurements were made with a Jasco V-550 spectrophotometer with a 1-cm path length quartz cuvette at a protein concentration of 15 μM.35 Visible circular dichroism spectra were recorded on a Jasco J-710 spectropolarimeter, by using 1-cm path length quartz cuvettes at an enzyme concentration of 10 μM. Routinely, three spectra were recorded at a scan speed of 50 nm min−1 with a bandwidth of 2 nm and averaged automatically. Both the instruments were equipped with a thermostatically controlled compartment at 25°C.
Molecular modeling
The three-dimensional coordinates of the S187F mutant in the internal aldimine form were used as a starting point to generate the external aldimine and PMP bound forms of the enzyme, by means of energy minimization. These enzymatic forms of normal AGT and variant were generated using the BIOPOLYMER package from InsightII (V.2000, MSI, Los Angeles). PMP and external aldimine and were initially positioned into the active site of S187F, initially following the binding mode of PLP. Atomic potentials, partial and formal charges were defined using Cff91, a broadly used and already validated forcefield,36 whose analytical function and set of constants have been derived to best fit ab initio computed molecular orbital energy surfaces for a number of functional groups that are present in macromolecules (acids, alcohols, alkanes, alkenes, amides, amines, aromatics, esters, ethers, etc.). Validation of proper potentials, partial and formal charges was performed on PMP, internal and external aldimines, since after modifying the internal aldimine as previously described, the Cff91 potentials and charges were still unassigned. To this end, unrecognized atoms were manually identified and their potentials and charges fixed, after having manually verified: (i) type, based on Cff91 force field (this was done for polar hydrogen bonded to N of the pyridine ring, h*, and protonated nitrogen in 6-membered ring, nh+); (ii) hybridization (sp2 aromatic carbons); (iii) valences; (iv) formal charges; and (v) partial charges, re-computed with a semi-empirical method (PM3). Each complex was then subjected to further energy minimization. The minimization protocol adopted was based on a multi-step approach: first, all atoms except added hydrogens were fixed to allow hydrogens to adjust to the atomic environment. To this purpose, 1000 steepest descents steps were performed, a distance-dependent dielectric constant and a cut-off distance of 40 Å were used during each simulation, until the maximum energy derivative was less than 41.8 kJ·mol−1Å−1. Then main-chain atoms were fixed and side chains of every residue comprised in a sphere of 5 Å from the coenzyme were subjected to a gradually decreasing tethering force (from 4180 kJ·Å−2 to 418 kJ·Å−2) using conjugated gradients, until maximum derivative was less than 4.18 kJ·mol−1Å−1. Finally, a decreasing tethering force (until the system was totally relaxed) was applied on PMP and external aldimine, and every side-chain atom comprised in a sphere of 5 Å from PLP, using conjugated gradients until the maximum derivative was less than 0.0004 kJ·mol−1Å−1. Discover 2.9 and Analysis package of InsightII were used for minimization.
Data analysis
The kinetic experiments were performed at least in duplicate, and in each case, the SEM was less than 10%. All data analysis was performed by non-linear curve fitting using Origin® 7.03 (OriginLab), and the errors indicated result from fitting to the appropriate equations.
RESULTS AND DISCUSSION
The S187F mutation affects the enzyme active site topology
To understand the molecular background of PH1 caused by the single amino acid mutation S187F, we have determined the crystal structure of the S187F variant at 2.9 Å resolution (PDB entry 4I8A, Table 1) (Fig. 1., Fig. S2). The asymmetric unit contains two AGT dimers, of which the complete polypeptide chain is visible in the final electron density, except for the N-terminal residues 1–5 (molecules A and C) or 1–4 (molecules B and D), and the C-terminal residues 391–392 of molecules B, C and D. Interestingly, the complete C-terminus, known as a peroxisomal targeting signal type 1 (PTS1), is visible in chain A, possibly due to the proximity of the N-terminal domain of molecule C that stabilizes the C-terminal loop (Fig. S3).
Table I.
X-ray Structure Determination
| AGT(S187F) | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 78.9, 101.6,116.8 |
| α, β, γ (°) | 90.0, 90.64, 90.0 |
| Resolution (Å) | 20.09–2.90 (3.06–2.90) |
| Rmerge | 19.5 (47.6) |
| I/σI | 4.5 (1.9) |
| Completeness (%) | 99.2 (99.9) |
| Redundancy | 3.2 (3.2) |
| Wilson-plot B value | 57.9 |
| Measured reflections | 130,322 |
| Unique reflections | 40,642 |
| Unique reflections (Rfree) | 2041 |
| Refinement | |
| Resolution (Å) | 18.07–2.90 |
| Number of reflections | 38 503 |
| Molecules/ASU | |
| Protein | 4 |
| Glycerol | 13 |
| Rwork/Rfree | 25.3/27.8 |
| Number of TLS groups | 20 |
| Number of atoms | |
| Protein | 11,851 |
| Glycerol | 78 |
| B factors | |
| Protein | 30.9 |
| LYS-PLP | 30.4 |
| Glycerol | 44.1 |
| RMS deviations | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.466 |
| MolProbity validation | |
| Ramachandran favored (%) | 98.4 |
| Ramachandran outliers (%) | 0.0 |
| Rotamer outliers (%) | 2.77 |
Figure 1.
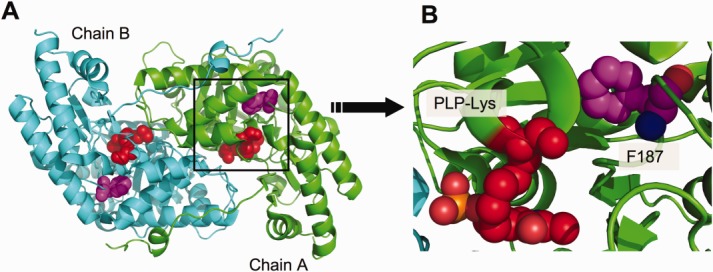
(a) Cartoon representation of the S187F variant dimer. The mutated amino acid (magenta) and the PLP-binding lysine (red) is represented by spheres. (b) Magnification of the area around the PLP-binding lysine. Because of the position of the phenylalanine mutation relative to the co-factor binding site, a conformational change is induced in the close proximity of the PLP-binding lysine. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The S187F structure superimposes well onto the normal AGT counterpart, with a root-mean-square deviation of 0.48 Å. Although the overall conformation of the two proteins is virtually identical, significant structural changes can be observed near the mutated amino acid (Fig. 2) as well as at the active site of the protein (Fig. 3). As a result of the mutation, the loop that contains Phe187 substantially changes its conformation, and moves Phe187 into the position of Leu188 of normal AGT. Because of this displacement, the Val185 residue of the mutant protein also changes position, and its side chain moves away from the PLP. The small hydrophobic pocket that is formed by Phe187 and Leu188 can accommodate Ala210, which eventually causes a shift of Ala210 and Lys209 towards Phe187 (Fig. 2). Although these conformational changes slightly alter the position of the PLP cofactor and of some active site residues (Trp108, Ser158, His83, and Asp183), they result in a significant displacement of Lys209 and the loop 183-188 (Fig. 2). In fact, the distance between the Cα of Lys209 and the C3 of the PLP methylpyridine ring decreases from 8.0 Å in the normal AGT enzyme to 6.5 Å in the mutant enzyme (Fig. 3), and the distance between the Cα of Val185 and the C3 of the PLP methylpyridine ring increases from 4.9 Å in the normal AGT enzyme to 6.7 Å in the mutant. Moreover, as calculated by Pocket Finder,37 in S187F the PLP cavity has a volume of 1073 Å3, while in normal AGT it has a volume of 949 Å3. These structural data allow us to explain how the mutation of a residue whose Cα is located at 10.5 Å from the C4′ of the coenzyme can affect the AGT functional properties so strongly. The shortened conformation of the lysine side chain observed could account for the reduced catalytic activity of the S187F variant, since Lys209 is a key residue taking part in the AGT catalytic mechanism, while the slightly altered position of several active site residues could account for the different PLP binding mode of S187F. Moreover, although it is difficult to understand the increased PLP binding affinity of the variant with respect to normal AGT,18 we speculate that it can be imputed to the increased volume of the PLP-binding pocket that could allow a better coenzyme accommodation.
Figure 2.
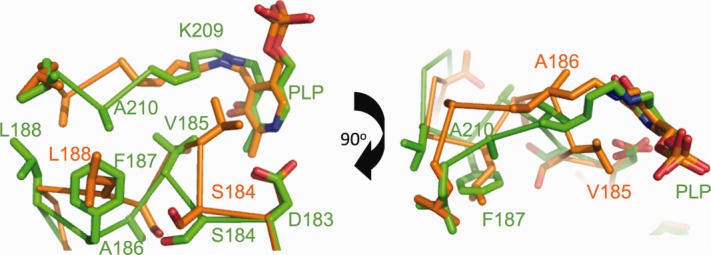
Superposition of the structure of the S187F variant (green) with that of normal AGT (orange) in the area of the cofactor binding site. Residues are labeled with the respective colors. The hydrophobic cleft that is formed by residues 185-188 triggers a conformational change in the loop where the PLP-binding lysine is located. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.
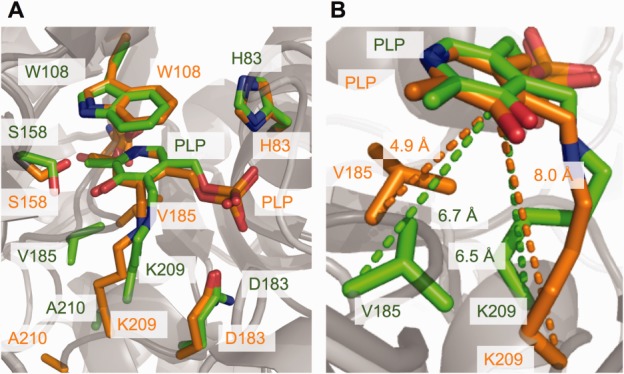
(a) Stick representation of the active site in the S187F variant (green) and normal AGT (orange). Because of the mutation, the main chain atoms of Lys209 in the mutant enzyme move closer to the PLP, which results in a shortened side chain conformation of Lys209. Other active site residues are only slightly affected by the mutation-induced structural changes. (b) Changes in the PLP-Lys209 and PLP-Val185 distances are represented by dashed lines in the respective colors. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The S187F mutation affects the coenzyme binding mode and affinity as well as the external aldimine microenvironment
We investigated if the active site conformational changes of the S187F variant revealed by its crystal structure could affect the binding of other reaction intermediates besides the PLP coenzyme.18 We found that even a prolonged incubation time of apoS187F with PMP (up to 350 μM) does not result in the appearance of the positive dichroic signal at 340 nm, typical of the AGT-PMP complex formation.9 However, a direct measurement of bound PMP after 1 h incubation of the apo form of the mutant (10 μM) with 350 μM PMP in 100 mM potassium phosphate buffer, pH 7.4 (final volume of 500 μl) followed by concentration to about 10 μl by a Microcon device and four washing with 500 μl of the above buffer revealed that the retentate contains about 8 μM PMP. Thus, since only 40% of the variant is in the PMP bound form, the KD(PMP) would be of ∼10 μM, at least 100-fold higher than that of normal AGT (data not shown).9 We have also monitored the spectral changes occurring in the S187F variant upon addition of D-alanine, an unproductive substrate analogue that mimics the formation of the external aldimine intermediate. The CD spectrum of S187F in the presence of a saturating concentration of D-alanine displays a positive band at 415 nm, 10-nm blue shifted with respect to that of normal AGT. Altogether, these data indicate that the variant is characterized by an altered PMP binding mode and affinity as well as by a different microenvironment of the external aldimine.
To have insights about the possible impact of the S187F mutation on the active site topography of these reaction intermediates, we compared docking models indicative of the putative binding mode of PMP and of the external aldimine with L-alanine [Fig. 4(a, b)] with those of normal AGT.9 The putative position of the manually docked molecules and their neighboring residues was relaxed by energy minimization means. Besides small changes related to the position of Tyr260, the most striking feature that distinguishes the S187F variant from normal AGT is the relative positioning of the coenzyme moiety and of Trp108. In fact, in S187F, both the AGT-PMP complex and the external aldimine are tilted by approximately 25° with respect to PLP towards Trp108 and Asp183 and the Trp108 side chain (on the re side of PLP) undergoes a remarkable conformational change, flipping by approximately 45° around the Cα–Cβ bond. The movement predicted is probably due to the need to accommodate the amino group of PMP or the substrate’s moiety and to avoid steric clashes with Lys209, and it is energetically impaired in normal AGT by the location of Val185 on the si side of the coenzyme.9 Although these represent simulated conformations, the predicted changes would result in loss of stabilization of PMP and the external aldimine on both re and si side of the pyridine ring. This is consistent with our experimental data and could account for the changes in both PMP binding mode and affinity and in the microenvironment of the external aldimine with D-alanine with respect to normal AGT.
Figure 4.
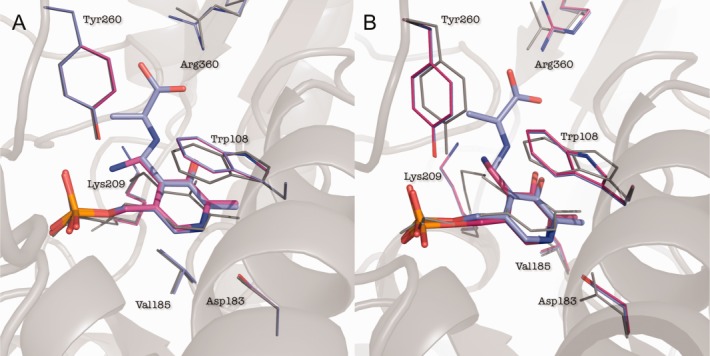
Modeling of the active site of the PMP- (pink) and L-alanine external aldimine- (cyan) bound forms of normal (a) and S187F (b) AGT. The internal aldimine (grey) is also shown for reference. Oxygen, nitrogen and phosphorous atoms are colored red, blue, and orange, respectively.
The S187F mutation decreases the catalytic efficiency of the L-alanine half-transamination
We found that the S187F variant in the absence of exogenous coenzyme has values of kcat for the alanine/glyoxylate pair, KmL-alanine and KmGlyoxylate, equal to 0.147 ± 0.009 s−1, 52 ± 2 mM and 0.038 ± 0.008 mM, respectively. In order to investigate if the decrease in the catalytic efficiency was due to an impact of the mutation on the first and/or the second half-transamination reaction, we compared the kinetics of the L-alanine half-transamination of the variant (15 μM) with that of normal AGT. Similar to what is already observed in normal AGT,9 we could not detect the external aldimine with L-alanine and the quinonoid intermediates of the S187F variant. However, while both the decrease of the 429 nm absorbance band and the concomitant PLP → PMP conversion are single exponential processes in normal AGT,9 they display a biphasic behavior in the variant (Fig. 5). Both the apparent first-order rate constants, kobs, of the fast and the slow phases show a hyperbolic dependence on L-alanine concentration, with kmax and Kmapp values of 0.15 ± 0.02 s−1 and 64 ± 15 mM and of 0.015 ± 0.002 s−1 and 132 ± 38 mM for the fast and the slow phase, respectively. Thus, the S187F mutation strongly affects the first half-reaction by causing a ∼550-fold decrease of its catalytic efficiency with respect to normal AGT.9 This is not unexpected, considering the mispositioning of Lys209, as revealed by the crystal structure of the S187F mutant, and the role of the PLP-binding lysine in aminotransferases on Cα proton abstraction and C4′ reprotonation.38 According to this view, the replacement of a lysine in position 209 by an arginine residue completely abolishes the AGT catalytic activity.39 Since Lys209 is supposed to act as an acid/base catalyst in both half-reactions of AGT, an effect of the S187F mutation on the ketoacid half-transamination would also be expected. Unfortunately, this reaction cannot be monitored, because the apoenzyme of the S187F variant is unstable and prone to aggregation at the high enzyme concentration required to obtain an apo-PMP complex (at least 100 μM).
Figure 5.
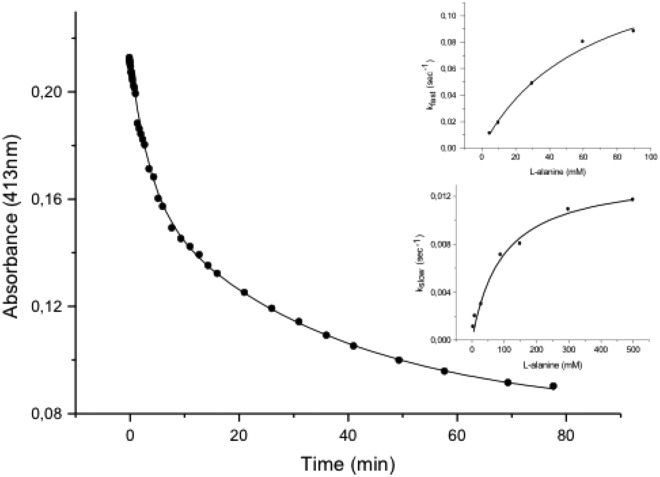
Time-dependent L-alanine half-transamination of the S187F variant. Plot of the 413 nm absorbance with time for the reaction of 15 μM S187F with 5 mM L-alanine. The line represents a two exponential fit. Insets: dependence of the rate constants (kfast and kslow) as a function of L-alanine concentration. The lines represent the data fitted to Eq. (2).
How can we explain the biphasic kinetics of the L-alanine half-transamination of the S187F mutant? The finding that the PLP binding kinetics is a monophasic process and that the kinetic constants of the L-alanine half-transamination do not change upon addition of D-alanine allow us to exclude the possibility that the S187F variant is found in solution as a mixture of a fast and a slow reacting form. Moreover, the fact that about 10 μM pyruvate is formed at the end of the fast phase the variant exhibits a Ki value for the competitive inhibition of pyruvate against glyoxylate identical to that of normal AGT9 (2.2 ± 1.3 mM) suggests that the slow phase of the half-transamination is not due to pyruvate inhibition. Nevertheless, we found that (i) the L-alanine half-transamination becomes a single exponential process (with a kobs value comparable to that of the fast phase) when it is coupled to the LDH/NADH reaction system, (ii) 100% of the PMP formed during the fast phase is bound at the AGT active site, and (iii) a 40% of bound PMP is present at the end of the slow phase. These results indicate that pyruvate is not released in solution in two kinetically distinguishable processes and that the slow phase is due to the accumulation of bound PMP that, in the presence of pyruvate, leads to an equilibrium between the forward and the reverse reaction. Consistent with this idea, a gradual recovery of the 413 nm absorbance is clearly detected at the end of the slow phase, indicative of the occurrence of the reversal transamination reaction.
CONCLUSIONS
As already reported for other diseases involving PLP enzymes,40, 41 PH1 is characterized by a remarkable heterogeneity in terms of enzymatic phenotype. Thus, the best treatment strategy would depend on the specific molecular mechanism involved in the AGT mutation considered. Until now, the effect of many PH1-causing missense mutations has been rationalized in terms of the position of the mutated residue in the crystal structure of normal AGT.4 However, this approach does not always guarantee the elucidation of the molecular defect of each variant, as we show here with the S187F variant. The fact that the AGT catalytic activity and expression level in the liver biopsy of a patient bearing the S187F mutation was very low,23 along with the fact that the Ser187 residue is not part of the active site, could be mistaken as indications that this mutant only has a structural defect. In which case, treatment with pharmacological chaperones could potentially stabilize the folded conformation of the variant and would represent the best therapeutic strategy for patients bearing the S187F mutation. However, we had previously found that replacement of Ser187 by Phe in the recombinant protein caused both structural (reduced thermostability of the apo form) and functional (reduced catalytic efficiency and altered PLP binding mode and affinity) defects.18 Additionally, in the present work, we present the crystal structure of the variant paired with bioinformatic studies and show that the mutation causes a remarkable perturbation of the active site resulting in a mispositioning of (i) the PLP-binding lysine, (ii) the AGT–PMP complex, and (iii) the external aldimine. These data are consistent with biochemical analyses indicating a reduced rate of L-alanine half-transamination, a different PMP binding mode and affinity as well as an altered microenvironment of the external aldimine with D-alanine in comparison with normal AGT. The fact that even the folded variant exhibits structural defects at the active site responsible for a low transaminase activity, leads us to conclude that a therapy with pharmacological chaperones would probably only have limited effects for patients bearing the S187F mutation. The therapeutic strategies most likely to be effective in this case would be based on gene therapy or enzyme administration.
Acknowledgments
K.F. is a Bolyai Research Fellow of the Hungarian Academy of Sciences. The authors declare no conflict of interest.
Glossary
- AGT
alanine:glyoxylate aminotransferase
- CD
circular dichroism
- KD(PLP)
equilibrium dissociation constant for PLP
- KD(PMP)
equilibrium dissociation constant for PMP
- PLP
pyridoxal 5′-phosphate
- PH1
Primary Hyperoxaluria Type I
- PMP
pyridoxamine 5′-phosphate.
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Danpure CJ. Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol. 2004;98:e39–44. doi: 10.1159/000080254. [DOI] [PubMed] [Google Scholar]
- 2.Montioli R, Fargue S, Lewin J, Zamparelli C, Danpure CJ, Borri Voltattorni C, Cellini B. The N-terminal extension is essential for the formation of the active dimeric structure of liver peroxisomal alanine:glyoxylate aminotransferase. Int J Biochem Cell Biol. 2012;44:536–546. doi: 10.1016/j.biocel.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Fodor K, Wolf J, Erdmann R, Schliebs W, Wilmanns M. Molecular requirements for peroxisomal targeting of alanine-glyoxylate aminotransferase as an essential determinant in primary hyperoxaluria type 1. PLoS Biol. 2012;10:e1001309. doi: 10.1371/journal.pbio.1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Roe SM, Hou Y, Bartlam M, Rao Z, Pearl LH, Danpure CJ. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J Mol Biol. 2003;331:643–652. doi: 10.1016/s0022-2836(03)00791-5. [DOI] [PubMed] [Google Scholar]
- 5.Danpure CJ, Jennings PR. Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–24. doi: 10.1016/0014-5793(86)80563-4. [DOI] [PubMed] [Google Scholar]
- 6.Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, Fargue S, Groothoff J, Harambat J, Hoppe B, Jamieson NV, Kemper MJ, Mandrile G, Marangella M, Picca S, Rumsby G, Salido E, Straub M, van Woerden CS. OxalEurope. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant. 2012;27:1729–1736. doi: 10.1093/ndt/gfs078. [DOI] [PubMed] [Google Scholar]
- 7.Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012;1822:1453–1464. doi: 10.1016/j.bbadis.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Williams EL, Acquaviva C, Amoroso A, Chevalier F, Coulter-Mackie M, Monico CG, Giachino D, Owen T, Robbiano A, Salido E, Waterham H, Rumsby G. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat. 2009;30:910–917. doi: 10.1002/humu.21021. [DOI] [PubMed] [Google Scholar]
- 9.Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri Voltattorni C. Human wild-type alanine:glyoxylate aminotransferase and its naturally occurring G82E variant: functional properties and physiological implications. Biochem J. 2007;408:39–50. doi: 10.1042/BJ20070637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cellini B, Lorenzetto A, Montioli R, Oppici E, Voltattorni CB. Human liver peroxisomal alanine:glyoxylate aminotransferase: Different stability under chemical stress of the major allele, the minor allele, and its pathogenic G170R variant. Biochimie. 2010;92:1801–1811. doi: 10.1016/j.biochi.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Cellini B, Montioli R, Paiardini A, Lorenzetto A, Maset F, Bellini T, Oppici E, Voltattorni CB. Molecular defects of the glycine 41 variants of alanine glyoxylate aminotransferase associated with primary hyperoxaluria type I. Proc Natl Acad Sci U S A. 2010;107:2896–2901. doi: 10.1073/pnas.0908565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cellini B, Montioli R, Paiardini A, Lorenzetto A, Voltattorni CB. Molecular insight into the synergism between the minor allele of human liver peroxisomal alanine:glyoxylate aminotransferase and the F152I mutation. J Biol Chem. 2009;284:8349–8358. doi: 10.1074/jbc.M808965200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cellini B, Oppici E, Paiardini A, Montioli R. Molecular insights into primary hyperoxaluria type 1 pathogenesis. Front Biosci. 2012;17:621–634. doi: 10.2741/3948. [DOI] [PubMed] [Google Scholar]
- 14.Coulter-Mackie MB, Lian Q. Consequences of missense mutations for dimerization and turnover of alanine:glyoxylate aminotransferase: study of a spectrum of mutations. Mol Genet Metab. 2006;89:349–359. doi: 10.1016/j.ymgme.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 15.Coulter-Mackie MB, Lian Q. Partial trypsin digestion as an indicator of mis-folding of mutant alanine:glyoxylate aminotransferase and chaperone effects of specific ligands. Study of a spectrum of missense mutants. Mol Genet Metab. 2008;94:368–374. doi: 10.1016/j.ymgme.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 16.Danpure CJ. Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways. Biochim Biophys Acta. 2006;1763:1776–1784. doi: 10.1016/j.bbamcr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 17.Hopper ED, Pittman AM, Fitzgerald MC, Tucker CL. In vivo and in vitro examination of stability of primary hyperoxaluria-associated human alanine:glyoxylate aminotransferase. J Biol Chem. 2008;283:30493–30502. doi: 10.1074/jbc.M803525200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oppici E, Montioli R, Lorenzetto A, Bianconi S, Borri Voltattorni C, Cellini B. Biochemical analyses are instrumental in identifying the impact of mutations on holo and/or apo-forms and on the region(s) of alanine:glyoxylate aminotransferase variants associated with primary hyperoxaluria type I. Mol Genet Metab. 2012;105:132–140. doi: 10.1016/j.ymgme.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pey AL, Salido E, Sanchez-Ruiz JM. Role of low native state kinetic stability and interaction of partially unfolded states with molecular chaperones in the mitochondrial protein mistargeting associated with primary hyperoxaluria. Amino Acids. 2011;41:1233–1245. doi: 10.1007/s00726-010-0801-2. [DOI] [PubMed] [Google Scholar]
- 20.Pittman AM, Lage MD, Poltoratsky V, Vrana JD, Paiardini A, Roncador A, Cellini B, Hughes RM, Tucker CL. Rapid Profiling of Disease Alleles Using a Tunable Reporter of Protein Misfolding. Genetics. 2012 doi: 10.1534/genetics.112.143750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santana A, Salido E, Torres A, Shapiro LJ. Primary hyperoxaluria type 1 in the Canary Islands: a conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc Natl Acad Sci U S A. 2003;100:7277–7282. doi: 10.1073/pnas.1131968100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cellini B, Montioli R, Voltattorni CB. Human liver peroxisomal alanine:glyoxylate aminotransferase: characterization of the two allelic forms and their pathogenic variants. Biochim Biophys Acta. 2011;1814:1577–1584. doi: 10.1016/j.bbapap.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Minatogawa Y, Tone S, Allsop J, Purdue PE, Takada Y, Danpur CJ, Kido R. A serine-to-phenylalanine substitution leads to loss of alanine:glyoxylate aminotransferase catalytic activity and immunoreactivity in a patient with primary hyperoxaluria type 1. Hum Mol Genet. 1992;1:643–644. doi: 10.1093/hmg/1.8.643. [DOI] [PubMed] [Google Scholar]
- 24.Cellini B, Montioli R, Bianconi S, Lopez-Alonso JP, Voltattorni CB. Construction, purification and characterization of untagged human liver alanine-glyoxylate aminotransferase expressed in Escherichia coli. Protein Pept Lett. 2008;15:153–159. doi: 10.2174/092986608783489580. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans PR. SCALA. Joint CCP4 and ESF-EAMBC Newletter on Protein Crystallography. 1997;22:2. [Google Scholar]
- 27.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. COOT: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Cellini B, Bertoldi M, Borri Voltattorni C. Treponema denticola cystalysin catalyzes beta-desulfination of L-cysteine sulfinic acid and beta-decarboxylation of L-aspartate and oxalacetate. FEBS Lett. 2003;554:306–310. doi: 10.1016/s0014-5793(03)01178-5. [DOI] [PubMed] [Google Scholar]
- 34.Daidone F, Montioli R, Paiardini A, Cellini B, Macchiarulo A, Giardina G, Bossa F, Borri Voltattorni C. Identification by virtual screening and in vitro testing of human DOPA decarboxylase inhibitors. PLoS One. 2012;7:e31610. doi: 10.1371/journal.pone.0031610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montioli R, Oppici E, Cellini B, Roncador A, Dindo M, Voltattorni CB. S250F variant associated with aromatic amino acid decarboxylase deficiency: molecular defects and intracellular rescue by pyridoxine. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt011. [DOI] [PubMed] [Google Scholar]
- 36.Maple JR, Dinur U, Hagler AT. Derivation of force fields for molecular mechanics and dynamics from ab initio energy surfaces. Proc Natl Acad Sci U S A. 1988;85:5350–5354. doi: 10.1073/pnas.85.15.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–363. doi: 10.1016/s1093-3263(98)00002-3. 389. [DOI] [PubMed] [Google Scholar]
- 38.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 39.Lumb MJ, Danpure CJ. Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. J Biol Chem. 2000;275:36415–36422. doi: 10.1074/jbc.M006693200. [DOI] [PubMed] [Google Scholar]
- 40.Zhu W, Lin A, Banerjee R. Kinetic properties of polymorphic variants and pathogenic mutants in human cystathionine gamma-lyase. Biochemistry. 2008;47:6226–6232. doi: 10.1021/bi800351a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cellini B, Montioli R, Oppici E, Voltattorni CB. Biochemical and computational approaches to improve the clinical treatment of dopa decarboxylase-related diseases: an overview. Open Biochem J. 2012;6:131–138. doi: 10.2174/1874091X01206010131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.