

Abstract
Deacetylases such as sirtuins (SIRTs) convert NAD to nicotinamide (NAM). Nicotinamide phosphoribosyl transferase (Nampt) is the rate-limiting enzyme in the NAD salvage pathway responsible for converting NAM to NAD to maintain cellular redox state. Activation of AMP-activated protein kinase (AMPK) increases SIRT activity by elevating NAD levels. As NAM directly inhibits SIRTs, increased Nampt activation or expression could be a metabolic stress response. Evidence suggests that AMPK regulates Nampt mRNA content, but whether repeated AMPK activation is necessary for increasing Nampt protein levels is unknown. To this end, we assessed whether exercise training- or 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR)-mediated increases in skeletal muscle Nampt abundance are AMPK dependent. One-legged knee-extensor exercise training in humans increased Nampt protein by 16% (P < 0.05) in the trained, but not the untrained leg. Moreover, increases in Nampt mRNA following acute exercise or AICAR treatment (P < 0.05 for both) were maintained in mouse skeletal muscle lacking a functional AMPK α2 subunit. Nampt protein was reduced in skeletal muscle of sedentary AMPK α2 kinase dead (KD), but 6.5 weeks of endurance exercise training increased skeletal muscle Nampt protein to a similar extent in both wild-type (WT) (24%) and AMPK α2 KD (18%) mice. In contrast, 4 weeks of daily AICAR treatment increased Nampt protein in skeletal muscle in WT mice (27%), but this effect did not occur in AMPK α2 KD mice. In conclusion, functional α2-containing AMPK heterotrimers are required for elevation of skeletal muscle Nampt protein, but not mRNA induction. These findings suggest AMPK plays a post-translational role in the regulation of skeletal muscle Nampt protein abundance, and further indicate that the regulation of cellular energy charge and nutrient sensing is mechanistically related.
Key points
NAD is a substrate for sirtuins (SIRTs), which regulate gene transcription in response to specific metabolic stresses.
Nicotinamide phosphoribosyl transferase (Nampt) is the rate-limiting enzyme in the NAD salvage pathway.
Using transgenic mouse models, we tested the hypothesis that skeletal muscle Nampt protein abundance would increase in response to metabolic stress in a manner dependent on the cellular nucleotide sensor, AMP-activated protein kinase (AMPK).
Exercise training, as well as repeated pharmacological activation of AMPK by 5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR), increased Nampt protein abundance. However, only the AICAR-mediated increase in Nampt protein abundance was dependent on AMPK.
Our results suggest that cellular energy charge and nutrient sensing by SIRTs may be mechanistically related, and that Nampt may play a key role for cellular adaptation to metabolic stress.
Introduction
Mitochondrial oxidative ATP synthesis is tightly coupled to the cycling of NAD between oxidised (NAD) and reduced (NADH) forms. The contribution of NAD to other cellular processes has long been assumed (Rechsteiner et al. 1976), and the discovery that NAD acts as a required substrate in signalling pathways critical in maintaining cellular metabolic homeostasis (Cantóet al. 2009) has heightened interest in NAD metabolism.
Sirtuins (SIRTs) were first recognised for their potential role in promoting longevity in response to caloric restriction by a mechanism that involves modulation of mitochondrial respiration capacity (Lin et al. 2000, 2002; Dali-Youcef et al. 2007). NAD acts as a substrate for SIRTs (designated in mammals as SIRT1–SIRT7), resulting in SIRT-dependent histone deacetylation and modulation of other proteins. During this reaction, NAD is converted to nicotinamide (NAM). Because NAM inhibits SIRT activity (Bitterman et al. 2002), NAM must be reconverted to NAD to maintain SIRT activity and mitochondrial metabolism. The rate-limiting enzyme in the NAD salvage pathway is nicotinamide phosphoribosyl transferase (Nampt; Revollo et al. 2004; Garten et al. 2009). Thus, Nampt may influence the cellular response to a variety of metabolic stresses such as caloric restriction or exercise via regulation of NAM biosynthesis.
SIRT1, the most intensively studied SIRT to date, deace-tylates non-histone proteins such as peroxisome prolife-rator-activated receptor γ-coactivator-1α (PGC-1α), a key element in the adaptive response to metabolic stress-induced mitochondrial biogenesis (Puigserver et al. 1998; Nemoto et al. 2005; Rodgers et al. 2005), as well as p53 (Luo et al. 2001), p300 (Bouras et al. 2005) and MyoD (Fulco et al. 2008). Although the role of SIRT1 in mediating exercise-induced increases in mitochondrial biogenesis has been challenged (Philp et al. 2011), SIRT1-dependent responses to exercise and fasting are compromised in AMP-activated protein kinase (AMPK)-deficient skeletal muscle (Cantóet al. 2010). AMPK is a heterotrimeric protein consisting of multiple isoforms of catalytic (α1, α2) and regulatory (β1, β2 and γ1, γ2, γ3) subunits, which mainly functions as a major sensor of cellular fuel status (Koh et al. 2008). In human and rodent skeletal muscle, AMPK trimers containing α2 catalytic subunits are dominant (Wojtaszewski et al. 2005; Treebak et al. 2009). Thus, a signalling network containing AMPK, Nampt and SIRT1 may interact at the level of PGC-1α to mediate transcriptional responses.
AMPK activation raises intracellular NAD concentrations and activates SIRT1 (Cantóet al. 2009), possibly via augmented Nampt activity or protein abundance. Skeletal muscle Nampt protein abundance is increased with endurance exercise training in humans (Costford et al. 2010), but whether these effects are specific to contracting muscle or secondary to improvements in the whole-body metabolic milieu concurrent with training is unclear. Interestingly, exercise- and fasting-induced increases in Nampt mRNA levels are blunted in skeletal muscle of AMPK γ3 knockout (KO) mice (Cantóet al. 2010). Moreover, Nampt expression is increased during glucose restriction in C2C12 mouse myoblasts and mouse skeletal muscle in an AMPK-dependent manner (Fulco et al. 2008; Wang et al. 2012). Collectively, these findings suggest that cellular fuel sensing and downstream alterations in metabolism may be mechanistically connected via AMPK and Nampt.
Here we assessed the effect of one-legged exercise training on skeletal muscle Nampt protein abundance in healthy volunteers. Because of the apparent functional connection between the cellular energy level and SIRT activity with AMPK and Nampt functioning as potentially critical intermediates, we hypothesised that increases in skeletal muscle Nampt protein are dependent on AMPK signalling. To address this, we studied several mouse models of reduced skeletal muscle AMPK activity to determine the effect of exercise and AMPK activators (5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide (AICAR) and metformin) on muscle Nampt protein abundance. Because AMPK is required for the ability of PGC-1α to function as a transcriptional co-activator (Jäger et al. 2007), we also tested the hypothesis that Nampt protein is regulated by PGC-1α in response to exercise training and repeated AMPK activation using PGC-1α-deficient mice.
Methods
Ethical approval
All animal experiments were approved by the Danish Animal Experimental Inspectorate, and complied with the European Convention for the protection of Vertebrate Animals used for Experiments and Other Scientific Purposes (Council of Europe 123, Strasbourg, France, 1985). Protocols for experiments conducted at Joslin Diabetes Center were in agreement with guidelines of the Institutional Animal Care and Use Committee of the Joslin Diabetes Center, and the National Institutes of Health. In addition, experiments conformed to the principles of UK regulations as previously described (Drummond, 2009). The number of animals used for each experiment is stated in each specific section. The human training experiment was approved by the local ethics committee and performed in agreement with the Declaration of Helsinki. All subjects provided informed consent before participating in the study.
Generation of cell lines
To assess the validity of Nampt antibodies used in this study, C2C12 mouse myoblasts were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen # 41965-062) containing 10% foetal bovine serum (FBS, Sigma # F7524) and 0.05 μg mL−1 penicillin streptomycin (P/S; Invitrogen # 15070–063) at 37°C, 5% CO2. For overexpression of FLAG-tagged Nampt, mouse Nampt was cloned into p3xflag-cmv-9-10_G903 vector (Sigma # 4401), and C2C12 myoblasts were transfected with 2 μg well−1 (9.6 cm2) using Lipofectamine 2000 (Invitrogen #11668-027) and OptiMem (Invitrogen # 51985-026) according to manufacturer's instructions. Cells were harvested the following day by washing once using ice-cold phosphate-buffered saline and adding lysis buffer (in mm: Hepes, 50, pH 7.4; 10% glycerol; 1% IGEPAL; NaCl, 150; NaF, 10; EDTA, 1; EGTA, 1; sodium pyrophosphate, 20; sodium orthovanadate, 2; protease inhibitors (SigmaFast, Sigma Aldrich) according to manufacturer's instructions). Protein concentration was determined via bicinchoninic acid assay (Thermo Scientific # 23223).
Stable Nampt knockdown C2C12 cells were generated using short hairpin (sh)RNA delivered by lentiviral infection. Human embryonic kidney 293FT cells (60% confluent, 9.6 cm2 plate) were co-transfected with pLKO (shRNA-containing plasmid), psPAX2 packaging plasmid and pMD2.G envelope plasmid (ratio: 2 μg pLKO; 1.5 μg psPAX2; 0.5 μg pMD2.G) using Superfect transfection reagent (Qiagen) and OptiMem. Two separate shRNA sequences (Nampt_Sh1, Nampt_Sh2; Open Biosystems # RMM3981–201818874, RMM3981–201824136) and a scrambled control (Sigma # SHC202) were used. The medium was replaced the following morning with DMEM containing 10% FBS and P/S. Forty-eight hours later, virus-containing media was collected and used to infect 40% confluent proliferating C2C12 myoblasts. Cells were changed to media containing 2.5 μg mL−1 puromycin (Sigma) 24 h after infection. Cells were maintained in selection media until immediately prior to an experiment.
Exercise training – humans
Skeletal muscle samples from eight young male subjects were obtained from a previous study (Frøsig et al. 2004). The training programme consisted of 15 sessions of one-legged knee extensor endurance training over the course of 3 weeks. Subjects performed four sessions in week 1, five sessions in week 2, and six sessions in week 3. The duration of training sessions started at 1 h per session and was gradually and consistently increased to 2 h per session for all subjects. Needle biopsies were obtained before training from the vastus lateralis muscle of the rested and exercised leg under local anaesthesia (2% lidocaine), and again 15 h after the final exercise bout.
Assessment of Nampt protein abundance in non-stimulated mouse skeletal muscle
To assess the importance of AMPK on Nampt protein abundance, we studied three different transgenic mouse strains (n= 5–11 per strain) and corresponding wild-type (WT) littermates (n= 6–9 per strain). Tibialis anterior muscles from skeletal muscle-specific LKB1 KO mice (LKB1 KO; the major activating kinase of AMPK), transgenic mice carrying a muscle-specific inactive AMPK α2 isoform (AMPK α2i) and from transgenic mice with elevated muscle AMPK activity due to a muscle-specific AMPK-activating R70Q γ1 mutation in the AMPK γ1 subunit (AMPK γ1 TG) were removed following cervical dislocation and immediately frozen in liquid nitrogen. These models have been described in detail previously (Koh et al. 2006; Barréet al. 2007; Fujii et al. 2008).
Acute exercise – mice
For familiarisation to treadmill running prior to experiments, mice were exercised for approximately 5 min day−1 for three consecutive days at speeds of approximately 5–10 m min−1. Male AMPK α2 KO mice (n= 32–40) and WT littermates (n= 32–40; Viollet et al. 2003) were run on a motorised treadmill for 90 min (10 min at 13 m min−1, 80 min at 17 m min−1), as previously described (Jørgensen et al. 2005). Mice were killed by cervical dislocation, and quadriceps muscles were removed from sedentary animals and immediately following, 1 h and 3 h following exercise cessation.
Exercise training – mice
Female mice overexpressing a kinase dead (KD) α2 AMPK subunit (Mu et al. 2001; n= 28) and WT littermates (n= 28) underwent 6.5 weeks of endurance exercise training or served as sedentary controls. Mice had free access to wheel cages for voluntary running 7 days week−1. Running distance was recorded using a cycling odometer (BC1009, Sigma, Germany). In addition, mice were exercised 1 h day−1 at 16 m min−1 on a motorised treadmill (Columbus Instruments) on weekdays. During the first week of the training period, treadmill exercise was performed for 10 min on day 1, 20 min on day 2, 30 min on day 3, 40 min on day 4 and 50 min on day 5. The morning following the final exercise bout, mice were anaesthetised by an intraperitoneal injection using Avertin (250 mg tribromoethanol kg−1 body weight). Quadriceps muscles were removed, frozen in liquid nitrogen and stored at −80°C until further analysis. Following a similar combined treadmill and wheel-cage training protocol, PGC-1α KO and WT mice (Lin et al. 2004) were exercised for 5 weeks. Quadriceps muscle samples from this experiment have previously been used for other analyses (Leick et al. 2008).
Acute AICAR treatment
Following a 6 h fast, 36 female C57BL/6J mice were injected subcutaneously with either saline or AICAR (500 mg kg−1 body weight) to determine the time course of AICAR-mediated Nampt induction. Mice were killed by cervical dislocation 2, 4 and 8 h after injection, and quadriceps muscles were removed, frozen in liquid nitrogen and analysed for Nampt mRNA expression. Following this experiment, female AMPK α2 KD and control animals received a single intraperitoneal injection of 500 mg kg−1 body weight AICAR or 0.9% NaCl solution. Eight hours following the injection, mice were killed by cervical dislocation, and quadriceps muscles were removed, frozen in liquid nitrogen and stored at −80°C.
Repeated AICAR treatment
Two genetic mouse models were used to study the effect of repeated AICAR administration on skeletal muscle Nampt expression. Male AMPK α2 KD (n= 15) and control mice (n= 16) received daily subcutaneous injections of 500 mg kg−1 body weight AICAR or 0.9% NaCl solution for 4 weeks. Mice were anaesthetised by an intraperitoneal injection using Avertin (250 mg kg−1 body weight) 24 h after the last injection, and quadriceps muscles were removed, frozen in liquid nitrogen and stored at −80°C. Samples were also obtained from a previously published study of PGC-1α KO and WT mice that were treated under the same conditions and as previously described (Leick et al. 2010).
Metformin treatment
AMPK α2 KD (n= 24) and control mice (n= 22) were treated with an oral dosage of 150 mg kg−1 metformin twice per day (i.e. a total dose of 300 mg kg−1 per day) or saline for 2 weeks. Samples were obtained from a previously published study (Kristensen et al. 2013). Metformin or saline solutions were administered via oral gavage. The final dose of metformin or saline was administered on the afternoon preceding the experimental day. Mice were anaesthetised by an intraperitoneal injection of pentobarbital (100 mg kg−1 body weight). Gastrocnemius muscles were removed, separated into white and red portions, frozen in liquid nitrogen, and stored at −80°C.
Western blot analysis
Muscle samples were processed in ice-cold lysis buffer (in mm: Hepes, 50, pH 7.4; 10% glycerol; 1% IGEPAL; NaCl, 150; NaF, 10; EDTA, 1; EGTA, 1; sodium pyrophosphate, 20; sodium orthovanadate, 2; protease inhibitors (SigmaFast, Sigma Aldrich) according to manufacturer's instructions), resolved using SDS–PAGE, and transferred as previously described (Frøsig et al. 2004). Aliquots were loaded in a balanced manner, with samples from all experimental conditions present on all gels. Following transfer, mouse samples were subjected to immunoblot analysis to detect Nampt protein (Bethyl, A300–372A). Exercise training-induced adaptation in skeletal muscle was confirmed by immunoblot analysis for hexokinase II protein (Cell Signalling, 2687). Human samples were subjected to immunoblot analysis to detect Nampt protein (Bethyl, A300–779A). Samples from C2C12 cells overexpressing a Nampt-FLAG were subjected to immunoblot analysis using an anti-FLAG antibody (Sigma, 7425). Western blots were visualised using a BioRad ChemiDoc chemiluminescence system, and densitometry analyses were performed using ImageLab software version 3.0 (Bio-Rad, Hercules, CA, USA).
Quantitative polymerase chain reaction (qPCR)
Total RNA from 20–30 mg of mouse muscle or C2C12 samples were extracted using Trizol (Qiagen). RNA (1 μg) was reverse-transcribed with a high-capacity complementary DNA (cDNA) reverse transcription kit (Applied Biosystems). Realtime PCR was performed, starting with 12.5 ng of cDNA and both sense and antisense oligonucleotides (300 nm each) in a final volume of 10 μl with the SYBR Green PCR Master Mix (Applied Biosystems). Fluorescence was monitored and analysed in a CFX96 Realtime system (BioRad). The obtained cycle threshold (Ct) values reflecting the initial content of the specific transcript in the samples were converted to an arbitrary amount by using standard curves obtained from a serial dilution of a pooled sample made from all samples. Gene expression was normalised by housekeeping gene (TBP or glyceraldehyde 3-phosphate dehydrogenase (GAPDH)) expression performed in parallel or by unit of input cDNA (Qubit ssDNA assay kit, Invitrogen). Amplification of specific transcripts was confirmed by analysing melting curve profiles at the end of each PCR experiment. Primer sequences for Nampt were: FP: 5′-CTCTTCGCAAGAGACTGCTGG-3′; RP: 5′-GAGCAATTCCCGCCACAGTATC-3′. TBP primers used were: FP: ACCCTTCACCAATGACTCCTATG; RP: TGACTGCAGCAAATCGCTTGG. A standard commercial GAPDH assay (Applied Biosystems) was used.
Statistics
Data are reported as means ± SEM. Samples from the human training study (Fig. 2) were analysed using a 2 × 2 repeated-measures ANOVA. Differences in Nampt protein abundance between mouse models of altered AMPK activity (Fig. 3) were analysed via unpaired, two-tailed t tests. The effect of acute exercise on Nampt mRNA (Fig. 4) was analysed using a 2 × 4 ANOVA (genotype by time point). For exercise training and AICAR studies in mice (Figs 5, 6B and C and 7), data were analysed using a 2 × 2 ANOVA (genotype by time point). The effect of acute AICAR on Nampt mRNA (Fig. 6A) was analysed via 2 × 3 (treatment by time point) ANOVA. For metformin studies (Fig. 8), data were analysed using a 2 × 2 × 2 ANOVA (genotype by time point by tissue). Statistical significance was set at P < 0.05.
Figure 2. Three weeks of one-legged knee extensor exercise training in humans increases Nampt protein in trained, but not untrained, skeletal muscle.
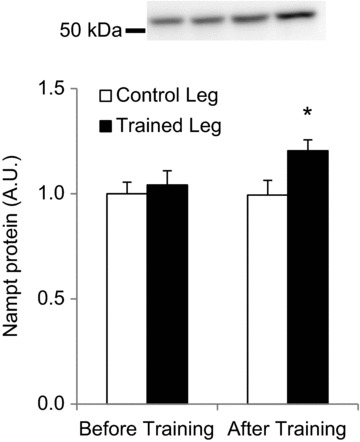
Human volunteers performed 15 sessions of one-legged knee extensor endurance training over the course of 3 weeks. Skeletal muscle biopsies were obtained from both vastus lateralis muscles before and after training (n= 8). * Indicates treatment × time interaction effect (P < 0.05).
Figure 3. Nampt protein levels are related to AMPK activity in mouse skeletal muscle.
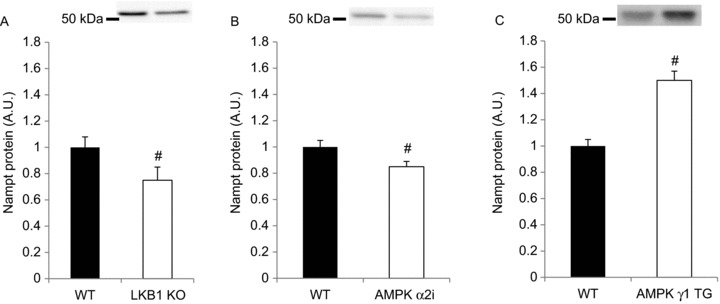
Mouse skeletal muscle Nampt protein was measured in tibialis anterior muscle of mouse models with reduced AMPK activity, such as (A) LKB1 KO (n= 9–11), (B) AMPK α2i (n= 6) or (C) AMPK γ1 transgenic mice, which show chronically elevated AMPK activity in skeletal muscle (n= 5–6). # Indicates vs. WT (P < 0.05).
Figure 4. Acute exercise increases mouse skeletal muscle Nampt mRNA in an AMPK α2-independent manner.
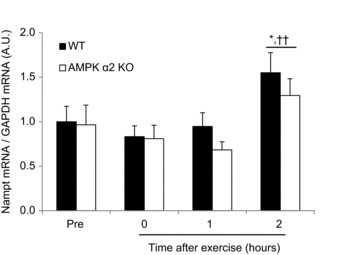
Nampt mRNA was measured in AMPK α2 WT and KO mouse muscle 3 h after completion of a 90 min treadmill exercise bout (n= 6–13). * Indicates vs. Pre (P < 0.05); †† indicates vs. 0, 1 h (P < 0.01).
Figure 5. Combined wheel-cage and treadmill training increases Nampt protein in mouse skeletal muscle in an AMPK α2- and PGC-1α-independent manner.
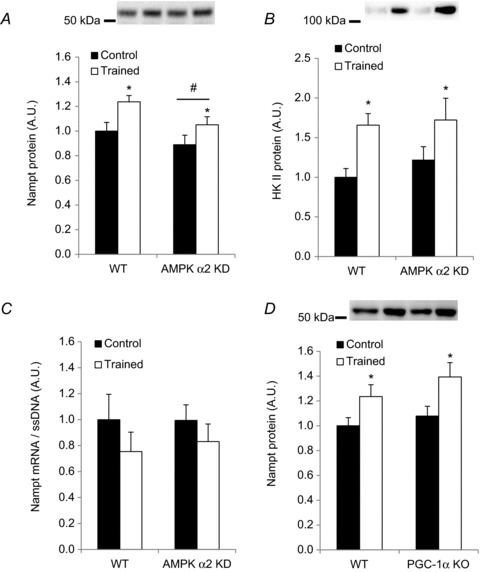
Quadriceps muscles of sedentary or trained (6.5 weeks of combined voluntary wheel-cage and forced exercise training) WT and AMPK α2 KD mice (n= 12–16) were removed the morning following the final exercise bout, and (A) Nampt protein, (B) hexokinase II protein and (C) Nampt mRNA levels were measured. D, Nampt protein abundance was measured in WT and PGC-1α KO mice that underwent 5 weeks of combined voluntary wheel-cage and forced endurance training, or served as sedentary controls (n= 16). * Indicates vs. control (P < 0.05); ** indicates vs. control (P < 0.01); # indicates vs. WT (P < 0.05).
Figure 6. Acute AICAR treatment increases Nampt mRNA independent of AMPK α2.
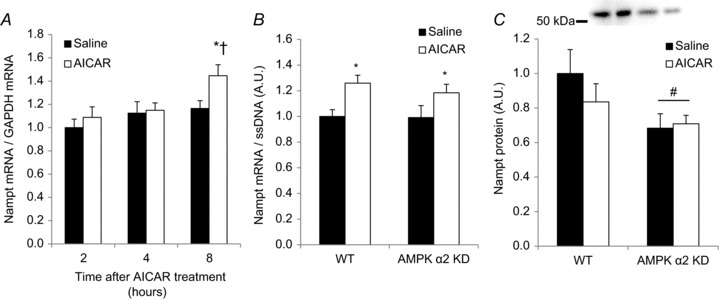
A, Nampt mRNA was measured in C57BL/6J mouse quadriceps muscle 2, 4 and 8 h after AICAR injection (500 mg kg−1 body weight; n= 6). B, Nampt mRNA concentrations and C) Nampt protein abundance were assessed 8 h after AICAR treatment (500 mg kg−1 body weight; n= 10–13). * Indicates vs. saline (P < 0.05); † indicates vs. 2 and 4 h (P < 0.05); # indicates vs. WT (P < 0.05).
Figure 7. Repeated AICAR administration increases skeletal muscle Nampt in an AMPK-dependent but PGC1α-independent manner.
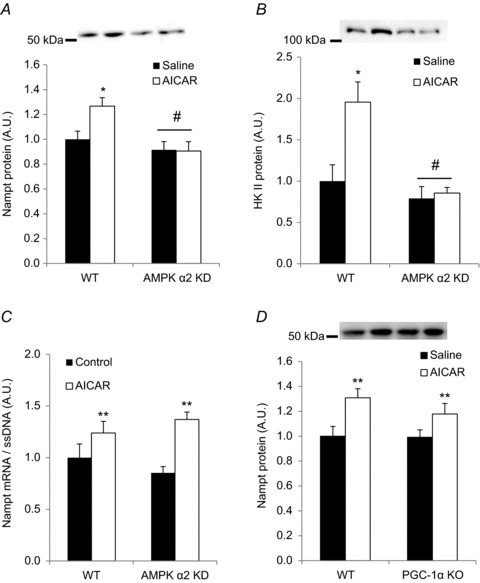
A, Nampt protein; B, hexokinase II protein and C, Nampt mRNA levels were measured in quadriceps of WT or AMPK α2 KD animals following 4 weeks of daily treatment with AICAR (500 mg kg−1 body weight) or saline (n= 7–8). D, Nampt protein levels were measured in both WT and PGC-1α KO mice following 4 weeks of daily treatment with AICAR or saline (n= 8). * Indicates vs. saline (P < 0.05); # indicates vs. WT (P < 0.05); ** indicates vs. saline (P < 0.01).
Figure 8. Effect of repeated metformin treatment on skeletal muscle Nampt concentrations.
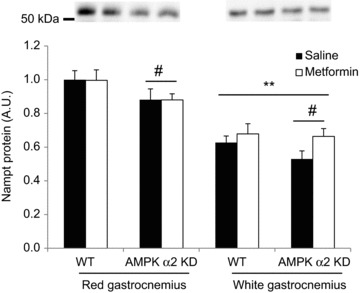
Nampt concentrations were measured in white and red gastrocnemius muscle of WT and AMPK α2 KD that were treated with 2 weeks of oral metformin treatment (300 mg kg−1 body weight) or saline. # Indicates vs. WT (P < 0.05); ** indicates vs. red gastrocnemius (P < 0.01); n= 10–12. Metformin treatment increased Nampt protein nearly significantly in white gastrocnemius (two-way ANOVA; main metformin treatment effect, P= 0.06; observed power = 0.39).
Results
Test of antibody specificity
The validity of the Nampt antibodies (Bethyl, A300–372A (mouse) and A300–779A (human)) used throughout this study was tested in C2C12 myoblast cells after silencing or overexpressing Nampt protein. Nampt was silenced using a shRNA lentiviral approach and transiently overexpressed using FLAG-tagged Nampt in mouse C2C12 myoblast cells. qPCR experiments showed a consistent ≍ 90% reduction in Nampt mRNA levels (Fig. 1A). To confirm the specificity of the Nampt signal, lysates from cells overexpressing Nampt-FLAG were resolved using SDS–PAGE together with control C2C12 and Nampt knockdown cells (Fig. 1B). A ‘split blot’ analysis was performed where the same sample from a FLAG-Nampt-overexpressing cell was resolved in three adjacent wells. After transfer to polyvinylidene difluoride, the membrane was cut through the centre well and the membrane halves were probed with anti-FLAG and the A300–372A anti-Nampt antibodies, respectively. Complete alignment of the bands was confirmed (Fig. 1C). Finally, another split blot analysis was performed using lysates from mouse and human skeletal muscle and mouse liver to compare the signal from the A300–372A antibody with the signal from the A300–779A antibody (Fig. 1D). The bands detected in human skeletal muscle using the two different antibodies ran at the expected molecular mass (≍52 kDa). Collectively, these experiments confirm that the Nampt antibody A300–372A specifically detects Nampt in mouse C2C12 cultured cells and is suitable for the detection of Nampt in mouse skeletal muscle. The antibody A300–779A also detects Nampt in human skeletal muscle, but it does not appear to cross-react with mouse Nampt protein.
Figure 1. Nampt antibody specificity and validity.
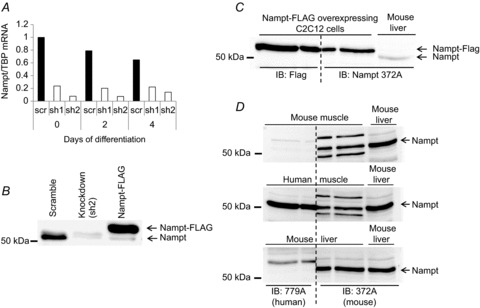
Nampt knockdown reduces (A) Nampt mRNA levels and (B) Nampt protein expression in C2C12 myoblasts. C, three identical C2C12 lysate aliquots from cells overexpressing FLAG-tagged Nampt were resolved alongside a mouse liver sample (right-hand lane). After protein transfer to polyvinylidene difluoride, the membrane was cut in two pieces as indicated by the dashed line, incubated with antibodies specific to FLAG (left side) and Nampt (right side), and re-assembled before visualisation. D, antibody specificity in human and mouse tissues. In separate gels, mouse muscle, human muscle and mouse liver lysates were resolved alongside a mouse liver sample (right-hand lane). After protein transfer to polyvinylidene difluoride, the membrane was then cut in two pieces as indicated by the dashed line, incubated with human Nampt (left side) or mouse Nampt antibody (right side), and re-assembled before visualisation. sh1, sh2, short hairpin clone 1, 2.
Endurance exercise training in humans increases skeletal muscle Nampt protein
A longitudinal study reveals that exercise training increases Nampt expression in human vastus lateralis muscle (Costford et al. 2010). We employed a 3 week one-legged knee extensor training programme to determine whether exercise training increases Nampt protein directly in the exercised muscle or secondarily to improvements or alterations in whole-body metabolism. This is a well-controlled exercise modality that results in specific activation of the quadriceps femoris (Andersen et al. 1985). One-legged endurance exercise training increased Nampt protein abundance in the trained, but not the untrained, leg (Fig. 2; P < 0.05).
AMPK affects basal Nampt protein abundance
AMPK is an important mediator of muscular adaptations to exercise (Jørgensen et al. 2006; Egan & Zierath, 2013). The relationship between AMPK and Nampt, as well as effects on SIRT biology have been documented (Fulco et al. 2008). We hypothesised that Nampt concentrations in sedentary muscle depend on functional AMPK α2 signalling. Our findings show Nampt protein level was reduced in skeletal muscles in which LKB1 or AMPK activity is ablated (Fig. 3A and B). Conversely, Nampt protein abundance was increased in skeletal muscle from a mouse model of increased AMPK activity (AMPK γ1 R70Q transgenic mice; Barréet al. 2007), compared with WT controls (Fig. 3C).
Acute exercise increases Nampt mRNA independent of AMPK α2
Swimming exercise increases Nampt mRNA in mouse skeletal muscle in an AMPK γ3-dependent manner (Cantóet al. 2010). To test whether AMPK α2-containing heterotrimers are involved in exercise-mediated Nampt mRNA induction, we assessed the effect of a single acute bout of treadmill running on Nampt mRNA in WT and AMPK α2 KO mice. Interestingly, Nampt mRNA expression was increased 3 h after exercise cessation in an AMPK-independent manner (Fig. 4).
Endurance training increases skeletal muscle Nampt protein in an AMPK- and PGC-1α-independent manner
Given that: (1) basal Nampt protein abundance partly depends on AMPK α2; and (2) endurance exercise training increases Nampt concentrations, we asked whether exercise training-induced increases in muscle Nampt protein depend on AMPK α2. WT and α2 AMPK KD mice completed an endurance exercise training protocol in which voluntary wheel running was combined with forced exercise on a motorised treadmill for 6.5 weeks. Endurance training elevated Nampt protein levels relative to sedentary controls (P < 0.05; Fig. 5A). This training effect was independent of AMPK activity, as the training response in Nampt protein abundance was not significantly different between WT and AMPK α2 KD mice. WT and KD mice performed similar amounts of voluntary running (WT: 150 ± 20.1 km vs. KD: 150 ± 21.1 km). A similar training-induced adaptation in hexokinase II protein abundance between WT and AMPK α2 KD mice further reinforced a comparable response to exercise training (Fig. 5B). Moreover, Nampt mRNA levels in these samples were similar between WT and AMPK α2 KD muscle, and did not differ between control and trained animals (Fig. 5C).
AMPK phosphorylates PGC-1α on at least two residues that appear to be necessary for SIRT1-mediated deacetylation/activation of PGC-1α (Jäger et al. 2007; Cantóet al. 2009). Based on our evidence that AMPK is necessary for full Nampt expression in skeletal muscle (Figs 3 and 5A), Nampt expression is possibly regulated by the AMPK-PGC-1α signalling axis. To test this hypothesis, we measured Nampt protein abundance in PGC-1α KO and WT mice in the untrained state and in response to endurance exercise training (Leick et al. 2008). Nampt protein abundance was similar between both genotypes in the untrained and trained state (Fig. 5D). These findings suggest either that regulation of Nampt protein levels is independent of PGC-1α, or that redundant signalling mechanisms may compensate for a lack of PGC-1α.
Pharmacological activation of AMPK by AICAR, but not metformin, increases Nampt
Our results from the exercise studies suggest that a functional AMPK α2 subunit is not required for the exercise-induced increases in muscle Nampt. Because exercise causes metabolic adaptations in skeletal muscle via AMPK and several other complementary mechanisms (Jørgensen et al. 2006; Egan & Zierath, 2013), we treated mice with the AMPK activators, AICAR and metformin, to assess the contribution of AMPK in the regulation of Nampt more directly.
AICAR is a cell-permeable nucleotide that can be converted to 5-aminoimidazole-4-carboxamide ribotide (ZMP) in the cell. ZMP shares some structural homologies with AMP and mimics the activating effects of AMP on AMPK (Corton et al. 1995). We tested the hypothesis that a single subcutaneous injection of AICAR would increase Nampt mRNA expression in skeletal muscle in an AMPK-dependent manner. A time course experiment to optimise the timing of the AICAR treatment indicated Nampt mRNA induction 8 h after AICAR treatment in C57BL/6J mice relative to saline-treated animals (P < 0.05; Fig. 6A). Subsequently, WT and AMPK α2 KD mice were injected with AICAR, and Nampt mRNA was evaluated after 8 h. Basal Nampt mRNA levels and AICAR-induced increases in Nampt mRNA were similar in AMPK α2 KD mice and control mice (Fig. 6B). Acute AICAR treatment did not alter Nampt protein abundance (Fig. 6C).
Although AICAR-induced Nampt mRNA induction occurred via an AMPK-independent mechanism, Nampt protein abundance was reduced in mice lacking a functional AMPK α2 subunit (Figs 3B, 5A and 6C). This may indicate that AMPK regulates Nampt protein by a post-transcriptional or -translational mechanism. We therefore determined whether repeated AICAR treatment increases Nampt protein in an AMPK-dependent manner. Four weeks of daily subcutaneous AICAR injections increased Nampt abundance in WT, but not AMPK α2 KD, mice (P < 0.05; Fig. 7A). Similarly, repeated AICAR treatment increased hexokinase II abundance in skeletal muscle of WT but not AMPK α2 KD mice (Fig. 7B). Supporting our finding that AICAR increases Nampt mRNA independent of AMPK (Fig. 6B), we found that Nampt mRNA levels after repeated AICAR treatment were significantly elevated independent of AMPK α2 (P < 0.01; Fig. 7C). Finally, AICAR increased Nampt protein abundance in the quadriceps muscle by a PGC-1α-independent mechanism (P < 0.01; Fig. 7D). These data indicate that pharmacological activation of AMPK can increase Nampt protein abundance in an AMPK α2-dependent manner that does not require the transcriptional co-activator PGC-1α.
Metformin is a potent anti-diabetic drug that has a major effect on the suppression of hepatic glucose production. However, metformin activates AMPK in skeletal muscle (Musi et al. 2002) and increases glucose uptake (Zhou et al. 2001) by both AMPK-dependent and -independent mechanisms (Turban et al. 2012). Therefore, we tested the hypothesis that metformin would increase Nampt protein levels in an AMPK-dependent manner. Although we have found that a single oral dose of metformin significantly increases AMPK phosphorylation in skeletal muscle in the hours after administration (J. M. Kristensen, J. T. Treebak and J. F. P. Wojtaszewski, unpublished observation), Nampt protein levels were unaltered overall in the gastrocnemius muscle of WT or AMPK α2 KD mice after 2 weeks of oral metformin administration (Fig. 8). However, Nampt protein levels were consistently lower in white relative to red gastrocnemius muscle (P < 0.01). When white gastrocnemius samples were analysed separately, we detected a borderline significant increase in Nampt following metformin treatment (main effect, P= 0.06; observed power = 0.39), with a greater relative response to metformin in KD muscle (25%) than WT muscle (8%).
Discussion
Activation of AMPK raises intracellular NAD concentrations and activates SIRT1, whereas AMPK deficiency compromises SIRT1-dependent responses to exercise and fasting (Cantóet al. 2009). A putative adaptive response to an accelerated NAM turnover caused by augmentations in SIRT activity may involve an increase in Nampt expression or activity. Several lines of evidence suggest that Nampt gene expression is dependent on a functional AMPK signalling cascade (Fulco et al. 2008). However, direct evidence to suggest that AMPK is necessary for maintaining Nampt protein abundance is lacking.
Here we demonstrate that skeletal muscle Nampt expression is partly dependent on AMPK heterotrimers containing a functional α2 catalytic subunit. Nampt protein abundance is consistently reduced in skeletal muscle of mouse models with ablated AMPK activity, and increased in a model of chronically increased AMPK activity. Moreover, repeated AICAR injections increased skeletal muscle Nampt protein abundance in WT mice, but not in AMPK α2 KD mice, implicating AMPK signalling in regulating Nampt protein levels. Together, these results suggest that Nampt protein abundance is partly determined by cellular energy status via AMPK α2-containing complexes in skeletal muscle, where deficiency or sustained activation of AMPK results in reduced or increased protein levels of Nampt, respectively.
We provide evidence that acute exercise increases Nampt mRNA induction in both WT and AMPK α2 KO mice. How these data agree with previous findings of a blunted Nampt mRNA induction in the quadriceps muscle of AMPK γ3 KO mice following 2 h of acute swimming is not immediately apparent (Cantóet al. 2010). The difference between these studies may be related to the alternative modes of exercise studied (90 min of treadmill running vs. four 30 min bouts of swimming separated by 5 min recovery). These exercise modalities may differentially affect muscle bioenergetics, and consequently influence the role of AMPK in the exercise-induced upregulation of Nampt mRNA. Skeletal muscle from AMPK γ3 KO mice rapidly fatigues during acute intensive exercise (Barnes et al. 2005) and shows reduced glycogen re-synthesis during recovery (Barnes et al. 2004), indicating a key role of the AMPK γ3 subunit in supporting muscle bioenergetics in response to exercise. Our treadmill exercise experiments were performed in fed mice, whereas the AMPK γ3 KO mice were fasted prior to swimming exercise (Cantóet al. 2010). Considering the impaired glycogen re-synthesis in AMPK γ3 KO mice and a compromised effect of caloric restriction on skeletal muscle Nampt protein abundance in AMPK α2 KO mice (Wang et al. 2012), nutritional status or cellular energy charge before the start of exercise may influence the role of AMPK in determining an exercise-induced increase in Nampt mRNA. Alternatively, other AMPK subunits, such as the α1 subunit that is upregulated in the AMPK α2 KO mice (Jørgensen et al. 2007), may play yet unidentified specialised roles in mediating the acute effects of exercise on Nampt mRNA induction.
Increases in Nampt protein abundance following exercise training, but not repeated AICAR administration, are preserved in AMPK α2 KD mice. These data are consistent with earlier evidence suggesting exercise-induced protein synthesis in skeletal muscle does not depend solely on AMPK signalling, while AICAR treatment requires functional AMPK signalling. For example, exercise training but not AICAR-induced metabolic adaptations in skeletal muscle are maintained in AMPK α2 KO mice (Jørgensen et al. 2005, 2007). Thus, redundant pathways such as calcium-calmodulin signalling may mediate the synthesis of specific proteins in response to exercise (Rose et al. 2009).
Our data on mRNA levels following exercise training and repeated AICAR are consistent with mRNA data from our acute exercise and AICAR treatment studies in that an effect of AMPK α2 on Nampt mRNA was not detected. Nampt mRNA was significantly elevated in the quadriceps muscle following 4 weeks of AICAR treatment, similar to the response observed after acute AICAR treatment. In contrast, Nampt mRNA was not increased after exercise training. Thus, we speculate that the metabolic effects of exercise on Nampt mRNA induction may be more transient than the effect of AICAR. Exercise-induced increases in AMP levels are relatively transient, and while skeletal muscle ZMP levels return to near baseline values within an hour after AICAR infusion (Sabina et al. 1982), a single dose of AICAR, comparable to the dose given in this study, elevates intracellular ZMP for hours in skeletal muscle as well as other tissues (Holmes et al. 1999; Bumpus & Johnson, 2011). This prolonged perturbation of cellular energy charge in response to AICAR treatment may account for the differential effect of exercise training and repeated AICAR treatment on Nampt mRNA expression and protein abundance.
A pool of AMPK α2 is thought to translocate to the nucleus upon activation (McGee et al. 2003), where it phosphorylates PGC-1α that is subsequently deacetylated by SIRT1 (Jäger et al. 2007; Cantóet al. 2009). However, PGC-1α KO was without effect on Nampt protein abundance in sedentary or trained skeletal muscle. In AMPK α2 KD mice, Nampt mRNA expression was similar between WT and AMPKα2 KD mice in basal, as well as AICAR-stimulated muscle, although Nampt protein abundance partly depends on AMPK. Collectively, these data are consistent with a post-transcriptional or -translational regulation of Nampt by AMPK. Interestingly, AMPK activation suppresses endothelial cell expression of angiotensin-converting enzyme post-translationally via phosphorylation of p53 and upregulation of miR 143/145 (Kohlstedt et al. 2013). These data suggest that AMPK can regulate protein abundance via post-translational mechanisms. Whether a similar mechanism can account for the ability of AMPK to regulate Nampt protein abundance remains to be determined.
Metformin is a biguanide that primarily acts by activating hepatic AMPK, with modest effects on skeletal muscle AMPK (Zhou et al. 2001; Musi et al. 2002). We are aware of only one other report concerning the effects of repeated metformin treatment on Nampt protein abundance (Caton et al. 2011). However, Nampt abundance was evaluated in adipose tissue, rather than skeletal muscle as studied here. Using a similar dose of metformin (250 mg kg−1 day−1 for 7 days vs. 300 mg kg−1 day−1 in this study), metformin treatment increased Nampt protein abundance in adipose tissue of db/db mice. Here we find that metformin did not consistently alter skeletal muscle Nampt protein content, despite the fact that we chose a metformin dosage that was intended to mimic pharmacologically active circulating metformin concentrations in humans (Bailey & Puah, 1986; Cusi & Defronzo, 1998). Metformin treatment was shown to ameliorate defects in mitochondrial respiration in predominantly glycolytic skeletal muscle from AMPK α2 KD mice (Kristensen et al. 2013). We detected borderline significant increases of Nampt protein in white (also predominantly glycolytic) gastrocnemius muscle with metformin, and we speculate that the effects of metformin on mitochondrial function and Nampt abundance may be particularly evident in glycolytic muscle fibres.
In conclusion, endurance exercise training increases Nampt protein abundance directly in exercise-trained muscle in humans. Thus, intrinsic changes in skeletal muscle, rather than systemic factors, contribute to the regulation of Nampt protein in response to exercise training. Moreover, AICAR- but not exercise-induced increases in Nampt protein abundance in mouse skeletal muscle depend on AMPK α2. In contrast, AMPK α2-containing heterotrimers are not required for regulating Nampt mRNA expression in response to either AICAR or treadmill exercise. Thus, AMPK-independent mechanisms may control Nampt-mediated gene transcription. Our study establishes a clear connection between AMPK activation and recycling of NAD by Nampt. Future studies are warranted to identify the exact mechanism by which AMPK regulates Nampt protein abundance, as well as other regulatory signals that determine Nampt expression.
Acknowledgments
The authors wish to acknowledge Dr. Ho-Jin Koh, Dr. Haiyan Yu, Dr. Peter Schjerling, Christine B. Janssens, Rasmus Kjøbsted, Radhika Vadalasetty and Camilla Stavnsbjerg for their efforts and technical expertise. Prof. Morris J. Birnbaum (Howard Hughes Medical Institute and University of Pennsylvania School of Medicine), Dr Benoit Viollet (Inserm, U1016, Institut Cochin, Paris) and Prof. Bruce Spiegelman (Dana-Farber Cancer Institute, Harvard Medical School) are acknowledged for providing founder AMPK α2 KD, AMPK α2 KO and PGC-1α KO mice, respectively.
Glossary
- α2i
catalytically inactive alpha 2 subunit
- γ1 TG
transgenic γ1 subunit
- AICAR
5-amino-1-β-d-ribofuranosyl-imidazole-4-carboxamide
- AMPK
AMP-activated protein kinase
- A.U.
arbitrary units
- DMEM
Dulbecco's modified Eagle's medium
- FBS
foetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- KD
kinase dead
- KO
knockout
- NAM
nicotinamide
- Nampt
nicotinamide phosphoribosyl transferase
- PGC-1α
peroxisome proliferator-activated receptor γ-coactivator-1α
- P/S
penicillin streptomycin
- qPCR
quantitative polymerase chain reaction
- sh
short hairpin
- SIRT
sirtuin
- TBP
tata box-binding protein
- TG
transgenic
- WT
wild-type
- ZMP
5-aminoimidazole-4-carboxamide ribotide
Additional information
Competing interests
The authors report no conflict of interest.
Author contributions
The experiments in the present manuscript were conducted in Section of Integrated Physiology at the Novo Nordisk Foundation Center for Basic Metabolic Research and at Joslin Diabetes Center. Conception and design of the experiments: J.B., J.T.T., L.J.G; collection, analysis and interpretation of data: J.B., J.T.T., S.G.V., M.A.A., S.R., S.R., P.S.L., J.M.K., C.F., L.L., J.F., S.J., B.K., J.F.P.W., E.A.R., L.J.G., H.P; drafting the article or revising it critically for important intellectual content: J.B., J.T.T., S.G.V., M.A.A., J.F.P.W., E.A.R., J.R.Z., L.J.G., H.P.
Funding
Support for this project was provided by the Novo Nordisk Foundation Center for Basic Metabolic Research and the Novo Nordisk Foundation to Jonas T. Treebak. Jonas T. Treebak was supported by a postdoctoral fellowship from The Danish Agency for Science, Technology and Innovation. Josef Brandauer was supported by a Research and Professional Development Grant by Gettysburg College. J. F. P. Wojtaszewski was supported by a Hallas Møller fellowship from the Novo Nordisk Foundation.
The human training study was supported by grants from the Danish National Research Foundation (no. 504–14), a Research and Technological Development Projects (QLG1-CT-2001-01488) grant funded by the European Commission, by the Media and Grants Secretariat of the Danish Ministry of Culture, by the Danish Diabetes Association, by the Novo Nordisk Foundation, and by the Danish Medical Research Council.
Experiments in PGC-1α KO mice were supported by the Lundbeck Foundation, the Novo Nordisk Foundation and a grant from the European Commission FP6 Integrated Project Exgenesis (Ref. LSHM-CT-2004-005272). Support was also provided by the Danish National Research Foundation (grant no. 02-512-555) to the Centre of Inflammation and Metabolism.
Studies in AMPK α2 KO mice were funded by the Danish National Research Foundation (#504-14), the Danish Diabetes Association, the Danish Medical Research Council, the Danish Natural Science Research Council, the Novo Nordisk Foundation and by an integrated project (contract LSHM-CT-2004-005272) funded by the European Commission.
Author's present address
J. M. Kristensen: Department of Endocrinology, Odense University Hospital, and Section of Molecular Diabetes & Metabolism, Institute of Clinical Research, University of Southern Denmark, Odense, Denmark.
References
- Andersen P, Adams RP, Sjøgaard G, Thorboe A, Saltin B. Dynamic knee extension as model for study of isolated exercising muscle in humans. J Appl Physiol. 1985;59:1647–1653. doi: 10.1152/jappl.1985.59.5.1647. [DOI] [PubMed] [Google Scholar]
- Bailey CJ, Puah JA. Effect of metformin on glucose metabolism in mouse soleus muscle. Diabete Metab. 1986;12:212–218. [PubMed] [Google Scholar]
- Barnes BR, Glund S, Long YC, Hjälm G, Andersson L, Zierath JR. 5′-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. FASEB J. 2005;19:773–779. doi: 10.1096/fj.04-3221com. [DOI] [PubMed] [Google Scholar]
- Barnes BR, Marklund S, Steiler TL, Walter M, Hjälm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. J Biol Chem. 2004;279:38441–38447. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- Barré L, Richardson C, Hirshman MF, Brozinick J, Fiering S, Kemp BE, Goodyear LJ, Witters LA. Genetic model for the chronic activation of skeletal muscle AMP-activated protein kinase leads to glycogen accumulation. Am J Physiol Endocrinol Metab. 2007;292:E802–E811. doi: 10.1152/ajpendo.00369.2006. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- Bumpus NN, Johnson EF. 5-Aminoimidazole-4-carboxyamide-ribonucleoside (AICAR)-stimulated hepatic expression of Cyp4a10, Cyp4a14, Cyp4a31, and other peroxisome proliferator-activated receptor α-responsive mouse genes is AICAR 5′-monophosphate-dependent and AMP-activated protein kinase-independent. J Pharmacol Exp Ther. 2011;339:886–895. doi: 10.1124/jpet.111.184242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton PW, Kieswich J, Yaqoob MM, Holness MJ, Sugden MC. Metformin opposes impaired AMPK and SIRT1 function and deleterious changes in core clock protein expression in white adipose tissue of genetically-obese db/db mice. Diabetes Obes Metab. 2011;13:1097–1104. doi: 10.1111/j.1463-1326.2011.01466.x. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells. Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, Smith SR. Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab. 2010;298:E117–E126. doi: 10.1152/ajpendo.00318.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusi K, Defronzo RA. Metformin: a review of its metabolic effects. Diabetes Reviews. 1998;6:89–131. [Google Scholar]
- Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the “magnificent seven”, function, metabolism and longevity. Ann Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Frøsig C, Jørgensen SB, Hardie DG, Richter EA, Wojtaszewski JFP. 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286:E411–E417. doi: 10.1152/ajpendo.00317.2003. [DOI] [PubMed] [Google Scholar]
- Fujii N, Ho RC, Manabe Y, Jessen N, Toyoda T, Holland WL, Summers SA, Hirshman MF, Goodyear LJ. Ablation of AMP-activated protein kinase alpha2 activity exacerbates insulin resistance induced by high-fat feeding of mice. Diabetes. 2008;57:2958–2966. doi: 10.2337/db07-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garten A, Petzold S, Körner A, Imai S-I, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen SB, Richter EA, Wojtaszewski JFP. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. J Physiol. 2006;574:17–31. doi: 10.1113/jphysiol.2006.109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JFP, Richter EA. Role of AMPKα2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab. 2007;292:E331–E339. doi: 10.1152/ajpendo.00243.2006. [DOI] [PubMed] [Google Scholar]
- Jørgensen SB, Wojtaszewski JFP, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of α-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005;19:1146–1148. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- Koh H-J, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh H-J, Brandauer J, Goodyear LJ. LKB1 and AMPK and the regulation of skeletal muscle metabolism. Curr Opin Clin Nutr Metab Care. 2008;11:227–232. doi: 10.1097/MCO.0b013e3282fb7b76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlstedt K, Trouvain C, Boettger T, Shi L, Fisslthaler B, Fleming I. AMP-activated protein kinase regulates endothelial cell angiotensin-converting enzyme expression via p53 and the post-transcriptional regulation of microRNA-143/145. Circ Res. 2013;112:1150–1158. doi: 10.1161/CIRCRESAHA.113.301282. [DOI] [PubMed] [Google Scholar]
- Kristensen JM, Larsen S, Helge JW, Dela F, Wojtaszewski JFP. Two weeks of metformin treatment enhances mitochondrial respiration in skeletal muscle of AMPK kinase dead but not wild type mice. PLoS ONE. 2013;8:e53533. doi: 10.1371/journal.pone.0053533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leick L, Fentz J, Biensø RS, Knudsen JG, Jeppesen J, Kiens B, Wojtaszewski JFP, Pilegaard H. PGC-1α is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2010;299:E456–E465. doi: 10.1152/ajpendo.00648.2009. [DOI] [PubMed] [Google Scholar]
- Leick L, Wojtaszewski JFP, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H. PGC-1alpha is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E463–E474. doi: 10.1152/ajpendo.00666.2007. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jäger S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Kaeberlein M, Andalis AA, Sturtz LA, Defossez P-A, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes. 2003;52:926–928. doi: 10.2337/diabetes.52.4.926. [DOI] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rechsteiner M, Hillyard D, Olivera BM. Magnitude and significance of NAD turnover in human cell line D98/AH2. Nature. 1976;259:695–696. doi: 10.1038/259695a0. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Alsted TJ, Jensen TE, Kobberø JB, Maarbjerg SJ, Jensen J, Richter EA. A Ca(2+)-calmodulin-eEF2K-eEF2 signalling cascade, but not AMPK, contributes to the suppression of skeletal muscle protein synthesis during contractions. J Physiol. 2009;587:1547–1563. doi: 10.1113/jphysiol.2008.167528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabina RL, Kernstine KH, Boyd RL, Holmes EW, Swain JL. Metabolism of 5-amino-4-imidazolecarboxamide riboside in cardiac and skeletal muscle. Effects on purine nucleotide synthesis. J Biol Chem. 1982;257:10178–10183. [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JFP. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol. 2009;297:C1041–C1052. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- Turban S, Stretton C, Drouin O, Green CJ, Watson ML, Gray A, Ross F, Lantier L, Viollet B, Hardie DG, Marette A, Hundal HS. Defining the contribution of AMP-activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J Biol Chem. 2012;287:20088–20099. doi: 10.1074/jbc.M111.330746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jørgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhang R-Y, Song J, Guan Y-F, Xu T-Y, Du H, Viollet B, Miao C-Y. Loss of AMP-activated protein kinase-α2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61:1051–1061. doi: 10.2337/db11-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JFP, Birk JB, Frøsig C, Holten M, Pilegaard H, Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J Physiol. 2005;564:563–573. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]