

Abstract
Igf2 mRNA binding protein 1 (IMP1, CRD-BP, ZBP-1) is a messenger RNA binding protein that we have shown previously to regulate colorectal cancer (CRC) cell growth in vitro. Furthermore, increased IMP1 expression correlates with enhanced metastasis and poor prognosis in CRC patients. In the current study, we sought to elucidate IMP1-mediated functions in CRC pathogenesis in vivo. Using CRC cell xenografts, we demonstrate that IMP1 overexpression promotes xenograft tumor growth and dissemination into the blood. Furthermore, intestine-specific knockdown of Imp1 dramatically reduces tumor number in the Apc Min/+ mouse model of intestinal tumorigenesis. In addition, IMP1 knockdown xenografts exhibit a reduced number of tumor cells entering the circulation, suggesting that IMP1 may directly modulate this early metastatic event. We further demonstrate that IMP1 overexpression decreases E-cadherin expression, promotes survival of single tumor cell-derived colonospheres and promotes enrichment and maintenance of a population of CD24+CD44+ cells, signifying that IMP1 overexpressing cells display evidence of loss of epithelial identity and enhancement of a tumor-initiating cell phenotype. Taken together, these findings implicate IMP1 as a modulator of tumor growth and provide evidence for a novel role of IMP1 in early events in CRC metastasis.
Introduction
Igf2 mRNA binding protein 1 (IMP1, CRD-BP, ZBP-1) is a messenger RNA (mRNA) binding protein originally identified as a translational stabilizer of c-Myc, β-actin and Igf2 mRNAs (1–4). IMP1 is expressed ubiquitously during development in mice, yet has low expression in adult tissues, and its function during adulthood remains unclear (5). Global Imp1 deletion in mice leads to dwarfism and a lethal intestinal phenotype at or near birth, suggesting a particularly critical role for Imp1 in fetal development of the intestine (5). In humans, IMP1’s aberrant reexpression has been implicated in various cancers, including colon, lung, skin and breast (5–10). Thus, IMP1 has been termed an ‘oncofetal’ factor for its role in both development and carcinogenesis.
Mechanistically, IMP1 has been described in a variety of roles in vitro, including mRNA trafficking and translational stabilization of various genes. In addition, studies of IMP1 and its orthologs have demonstrated specific roles for IMP1 in cytoskeletal organization and cellular movement (2,11–13). Imp1 has been shown to bind to and stabilize β-TrCP1 mRNA upstream of Skp-1-Cullin1-F-box E3 ubiquitin ligase activation, which leads to increased turnover of IκB and β-catenin (14). Most recently, studies have reported that IMP1 may regulate cellular migration and polarization via regulation of MAPK4 and PTEN in osteosarcoma and ovarian tumor cell lines and may promote wound repair in colonic mesenchymal stem cells via stabilization of Ptgs2 mRNA (15,16). Collectively, these studies suggest a multitude of roles for IMP1 that may explain clinical observations in the context of cancer development and progression. However, these functional roles remain largely untested in vivo.
Despite its potential for diagnostic or therapeutic importance in human cancers, only one study has addressed the direct role of IMP1 in tumorigenesis in vivo to date. Transgenic expression of Imp1 driven by the whey acidic protein promoter, which is expressed in mammary epithelium of pregnant and lactating females, led to breast tumors in 95% of transgenic mice compared with controls, with a subset of mice exhibiting metastatic disease (7). Although compelling, these findings are limited to breast neoplasia of relatively long tumor latency. Importantly, tumors from whey acidic protein Imp1 mice did not exhibit upregulation of c-Myc or Igf2 protein levels, indicating a role for other pathways either directly or indirectly influenced by IMP1 in tumor initiation and/or progression.
IMP1 is expressed in rare populations of cells in the normal adult intestine (10 and unpublished observations). Furthermore, human CRC tumors that exhibit high IMP1 expression correlate with enhanced metastasis and recurrence, and IMP1 positive tumors correlate with shorter survival time (10). We have shown previously that IMP1 loss decreases proliferation and colony formation in CRC cell lines and that high IMP1 expression is correlated positively with β-catenin and K-ras activation in primary colorectal tumors and predicts poor clinical outcome in CRC patients (17). Here, we present data indicating that IMP1 overexpression promotes tumor-cell growth in CRC xenografts and that intestine-specific Imp1 knockdown reduces tumor number in a genetic mouse model of intestinal tumorigenesis. In addition, xenograft mice with IMP1 overexpressing CRC cells exhibited enhanced dissemination into the blood, whereas knockdown of IMP1 decreased the number of circulating tumor cells. Furthermore, IMP1 promotes a tumor-initiating cell phenotype in vitro. Taken together, these data support a role for IMP1 in modulating CRC tumor growth and indicate IMP1 as a potential initiator of metastasis by promoting tumor-cell dissemination into the blood.
Materials and methods
Generation of IMP1 overexpression and knockdown cell lines
SW480 and LoVo cell stocks were obtained commercially from American Type Culture Collection (Manassas, VA) and have been previously authenticated in the Rustgi laboratory via cytogenetic analyses. Cells were maintained according to American Type Culture Collection recommendations. The IMP1/pMSCV-PIG retroviral vector (plasmid 21659) and control vector were purchased from Addgene (Cambridge, MA) and used to stably infect SW480 or LoVo cells (described previously) (17,18). As we have shown previously that IMP1 knockdown decreases cell proliferation using multiple small interfering RNAs (17), we opted to generate an inducible IMP1 short hairpin RNA cell line. This was achieved by transducing SW480 cells with the doxycycline-inducible short hairpin RNA pTripz lentiviral vector (Open Biosystems, Huntsville, AL, clone ID V3THS_342778). For both overexpression and knockdown experiments, transduced cells were selected using puromycin and if necessary sorted in the Penn Flow Cytometry and Cell Sorting Core to obtain greater than 99% efficiency. IMP1 overexpression or knockdown was confirmed using quantitative reverse transcription–polymerase chain reaction (qRT–PCR), western blotting and fluorescent imaging of either overexpression [green fluorescent protein (GFP)] or knockdown [red fluorescent protein (RFP)].
Gene expression analysis
Cell lines were monitored for IMP1 overexpression or knockdown via qPCR for the probe-specific complementary DNA using the following proprietary primer/probe sets: IGF2BP1 Hs00198023_m1; GAPDH Hs99999905_m1 (Applied Biosystems, Carlsbad, CA) using ABI Prism 7000 Sequence Detection System. Additional primer/probe sets include the following: CDH1 Hs01023894_m1; VIM Hs00185584_m1; SNAI1 Hs00195591_m1; SNAI2 Hs00161904_m1; ZEB1 Hs00232783_m1; ZEB2 Hs00207691_m1. For western blotting, cells were lysed in radioimmunoprecipitation assay buffer [phosphate-buffered saline (pH 7.4), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% (v/v) IGEPAL, 100mM sodium orthovanadate and proteinase inhibitor cocktail (Sigma, St Louis, MO)]. Protein was quantified with the BCA Protein Assay Kit (Thermo Scientific, Rockford, IL) and loaded on 4–12% gradient gels for electrophoresis using Invitrogen (Carlsbad, CA) western blotting apparatus. Antibodies used were as follows: rabbit anti-IMP1 (Cell Signaling, Danvers, MA); mouse anti-GAPDH (Millipore, Billerica, MA).
Xenograft tumors
Athymic nude mice (6-weeks-old, at least eight mice per group, Taconic, Cranbury, NJ) were injected in the rear flank with 1 × 106 IMP1 overexpressing or knockdown cells or empty vector cells suspended in BD Matrigel™ Basement Membrane Mix (San Jose, CA) as described previously (18). All mice were cared for in accordance with University Laboratory Animal Resources requirements and under an Institutional Animal Care and Use Committee approved protocol. Mice were evaluated weekly for 8 weeks postinjection and tumors measured manually using digital calipers. Mice were euthanized 8 weeks postinjection or sooner in accordance with Institutional Animal Care and Use Committee guidelines for tumor size and animal welfare. Mice with IMP1 knockdown were given doxycycline (2 g/l in 5% sucrose water) 1 week postinjection for the duration of the study. Live animal imaging of IMP1 knockdown xenograft mice was performed using the IVIS imaging system (Xenogen, Alameda, CA) through the Small Animal Imaging Core Facility at the University of Pennsylvania. This imaging served as an additional measure of tumor volume for knockdown studies, as well as confirmation that knockdown was maintained (via visualization of RFP) for the duration of the study. Portions of all tumors were harvested in 10% formalin for histology, and hematoxylin and eosin staining was performed on all tumors, as well as staining for collagen using Masson’s trichrome. Slides were evaluated by a pathologist for tumor morphology (A.J.K.-S.).
Generation of Imp1-loxP mice
Mice heterozygous for Imp1-loxP were generated by Ozgene (www.Ozgene.com), from which homozygous Imp1-loxP mice were obtained through in-house breeding. Genotype of the mice was confirmed by PCR using primers for the loxP arm with the following sequences; loxP arm 1F: 5′TGTGTGTGTGTGAGGGAGGT3′; loxP arm 1R: 5′TAGGTGACGTTGACCACAGC3′. Native gels of 15% were used to differentiate between mice homozygous for Imp1-loxP, which showed a PCR product size around 270 bp, wild-type mice (235 bp) and mice heterozygous for Imp1-loxP, which showed both PCR product sizes.
Generation and characterization of mouse embryonic fibroblasts
A homozygous female Imp1-loxP mouse was mated with a homozygous male Imp1-loxP mouse. At 14 days of gestation, the female was euthanized using isoflurane for anesthesia followed by cervical dislocation. Embryos were removed from the uterus and used to generate mouse embryonic fibroblasts (MEFs) according to the WiCell protocol (WiCell, Madison, WI, http://www.wicell.org). Briefly, in aseptic conditions, the embryos were cleaned of their visceral tissue, washed several times with phosphate-buffered saline, then minced into grain-sized pieces using dissecting scissors, mixed with trypsin/ethylenediaminetetraacetic acid (0.25% trypsin/2.21 mM ethylenediaminetetraacetic acid; Mediatech, Herndon, VA) and placed in a 37°C tissue culture incubator for 20 min. The derivation culture media was then added to the minced tissue and the mixture was transferred into different T75 flasks and placed into the 37°C tissue culture incubator overnight. Attached MEFs were observed the next day and the cells were propagated. Whole cell lysates from MEFs transfected with a Cre expression vector or an empty vector were lysed using radioimmunoprecipitation assay buffer. Immunoblotting procedures were performed as described previously (19). Western blot antibody against β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) was purchased, as well as the secondary antibody conjugated with horseradish peroxidase (Chemicon, Billerica, MA). Mouse monoclonal antibody against Imp1 was generated in the Spiegelman Laboratory.
Immunostaining
Eight-week old Imp1-loxP +/+ ;Villin-Cre +/− (Imp1-loxP homozygous; Villin-Cre heterozygous) mice and littermate wild-type mice were euthanized by CO2 asphyxiation followed by cervical dislocation, and their intestines were collected, preserved in 10% formalin for 48 h and then in 70% ethanol. Cross-tissue sections were generated from formalin-fixed paraffin-embedded intestine. The sections were subsequently deparaffinized in xylene (Fisher, NJ) and rehydrated in grades of alcohol. The sections were subjected to antigen retrieval by incubating in 10mM citrate buffer for 20 min in a microwave oven. The sections were blocked with normal goat serum, probed with specific primary antibodies to Imp1, followed by appropriate fluorescent-labeled secondary antibody (Invitrogen, Carlsbad, CA). Tissue sections were mounted with Prolong Gold Antifade with 4′,6-diamidino-2-phenylindole mounting media (Invitrogen).
Tumor count
Imp1-loxP +/+ mice were crossed sequentially with Villin-Cre and Apc Min+/− mice to obtain Imp1-loxP +/− ; Villin-Cre +/− ;Apc Min/+ and Villin-Cre +/− ;Apc Min/+ animals. At 90 days of age, the mice were euthanized by CO2 asphyxiation followed by cervical dislocation, and the different parts of their intestine were collected, opened and preserved in 10% formalin for 48 h and then in 70% ethanol. The tumors were counted under the microscope. Mann–Whitney U-test was used for statistical analysis.
Analysis of circulating tumor cells
Analysis of circulating tumor cells was performed as described previously (20). Briefly, immediately posteuthanasia, blood was collected from mice using 1 ml syringes containing 0.1 ml chilled lithium heparin via cardiac puncture. Samples were enriched via negative selection of erythrocytes using FACS Lyse buffer (BD Biosciences) and analyzed in 10% fetal calf serum/Dulbecco’s modified Eagle’s medium/F12 using the Accuri C6 Flow Cytometer. Pulse height and forward scatter/side scatter gating were used to gate out dead cells, and high expression of GFP or RFP was correlated with live cells. The number of GFP or RFP positive cells per milliliter of blood was then calculated. We analyzed blood from at least three mice per group.
Colonosphere assay
Upon being euthanized, one half of each xenograft tumor was used to sort single, live tumor cells into 96-well utlralow attachment plates to assay for colonosphere growth as described previously (21). Briefly, tumors were minced into 1 mm3 pieces and then subjected to sequential collagenase (Sigma), trypsin/ethylenediaminetetraacetic acid (Invitrogen) and DNase/Dispase (Roche, Indianapolis, IN) digestions. Single-cell suspensions were obtained by then filtering cell suspensions over a 40 μM filter. Single GFP+ tumor cells were sorted using the BD FACS Aria with assistance from the UPenn Flow Cytometry and Cell Sorting Facility. Dead cells were excluded using 4′,6-diamidino-2-phenylindole staining. One tumor cell was sorted into each well of a 96-well ultralow attachment plates (Corning Inc, Lowell, MA) containing 100 μl of prewarmed serum-free ‘stem cell media’ (Dulbecco’s modified Eagle’s medium/F12, B27 (Life Technologies, Gaithersburg, MD), 20 ng/ml epidermal growth factor (Sigma), 10 ng/ml fibroblast growth factor (Sigma). Tumors from three mice per condition were analyzed for number of spheres per 96-well plate, and sphere size was analyzed by photographing individual spheres and measuring radii using NIH ImageJ. Data are represented as mean ± standard error. For passaging, colonospheres were mechanically dissociated by removing single spheres to a new 96-well ultralow attachment plate containing stem cell media and vigorous pipetting (25 times) to break apart individual spheres. Successful dissociation was determined immediately following passaging by visualization via light microscopy. Fourteen days postpassaging, total growth of new spheres and spheres with ‘spreading’ or ‘budding’ were evaluated together by blind scoring (P.S.K.) of total growth within individual wells, with a score of 0 being zero living, GFP+ cells remaining and a score of 10 being 100% coverage of the well with GFP+ spheres or budding spheres.
Fluorescence-activated cell-sorting analysis for CD24+CD44+ cells
Single-cell suspensions were prepared from stable IMP1 overexpression cells (SW480). Cells were trypsinized from cell culture plates, resuspended in complete media, passed through a 70 μm cell strainer, rinsed with fluorescence-activated cell-sorting buffer (phosphate-buffered saline/1% bovine serum albumin) and stained with the following antibodies: PE/Cy7 anti-human CD24 (BioLegend, San Diego, CA); APC anti-human CD44 (BD Biosciences, San Jose, CA). Flow cytometry was done using a BD FACS Calibur (BD Biosciences), and data were analyzed using FlowJo Flow Cytometry Analysis Software. For functional analyses of CD24+CD44+ cells, the top 25% were collected as CD24+CD44+high, with remaining cells (CD24+CD44+ neg/low expression) using the BD FACS Aria with assistance from the UPenn Flow Cytometry and Cell Sorting Facility. Cells were immediately plated onto polystyrene tissue culture plates and passaged via trypsinization at 70–90% confluence. Passaged cells were subsequently characterized for CD24+CD44+ expression on the FACS Calibur, and proportions of CD24+CD44+high cells were analyzed using FlowJo Flow Cytometry Analysis Software. Unsorted MSCV-IMP1 cells were used to create a standardized gate for each experimental passage, wherein 20% of the cells present were gated as CD24+CD44+. This normalized gating to an unsorted population allowed us to compare relative proportion of CD24+CD44+ cells in control versus IMP1 overexpressing cells. Cells were analyzed at passage 0–3, and data were represented as relative difference between control and IMP1 overexpressing CD24+CD44+high cells over the course of these passages.
Statistical analyses
For xenografts and circulating tumor cells, statistical significance of comparisons between empty vector and IMP1 tumors was determined by applying student’s t-test, with P < 0.05 as statistically significant. For all other analyses, data from at least triplicate experiments were presented as mean ± standard error and analyzed by applying student’s t-test, with P < 0.05 as statistically significant.
Results
IMP1 modulates tumor volume in vivo
IMP1 expression is enhanced in CRC tumors and its overexpression correlates with advanced disease and poor prognosis in humans. We therefore hypothesized that overexpressing IMP1 may drive an increase in tumor volume and potentially metastatic growth, and conversely, that IMP1 knockdown would reduce tumor burden. To assess the effect of IMP1 overexpression or knockdown on CRC xenograft tumor growth, we initially chose SW480 cells because they express relatively moderate levels of endogenous IMP1 and have been shown previously to grow robust but non-metastatic xenograft tumors (22). IMP1 overexpression was confirmed by qRT–PCR and western blotting and consistently showed a minimum of 2-fold increase in IMP-1 (Figure 1A). We performed subcutaneous injections of 1 × 106 cells into the rear flanks of nude mice and monitored tumor growth weekly over the course of 8 weeks. We found a significant increase in SW480 xenograft tumor volume in mice with IMP1 overexpression at 8 weeks postinjection (Figure 1B). There was no significant difference in total body weights between control and IMP1 overexpression groups (data not shown). Both control and IMP1 overexpressing tumors were poorly differentiated adenocarcinoma with no significant differences in tumor morphology upon histopathological analysis (Figure 1C), and there was no significant difference in collagen deposition between control and overexpression tumors (data not shown). In a second, independent overexpression CRC cell line, LoVo cells, we observed an increased tumor burden with IMP1 overexpression by 2.5 weeks postinjection; however, the primary tumors grew too rapidly, resulting in lesions that required early euthanasia (Supplementary Figure S1, available at Carcinogenesis Online).
Fig. 1.

IMP1 overexpression increases tumor volume. (A) SW480 cells stably expressing MSCV or MSCV-IMP1 were assayed for IMP1 expression. Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control. IMP1 expression was significantly increased in SW480 MSCV-IMP1 cells. *P < 0.05, N = 3. (B) Xenograft mice were injected with control or IMP1 overexpressing cells as described in Materials and methods. Mean tumor volume was significantly increased in mice injected with IMP1 overexpressing cells compared with controls. *P < 0.05, N = 8 mice per group. (C) Upon being euthanized, xenograft tumors were analyzed histologically. There was no significant difference in tumor histology in IMP1 overexpression tumors compared with control tumors.
In order to address IMP1 knockdown effects on tumorigenesis, we generated Imp1-loxP mice. In these mice, the exons 5 and 6 are flanked by loxP sequences (design of the targeting construct in Figure 2A). The effectiveness of inducible knockout of Imp1 in these animals was first verified in MEFs isolated from Imp1-loxP homozygous mouse embryos. A dramatic reduction in Imp1 expression is achieved in these MEFs when transfected with a Cre recombinase expression plasmid (Figure 2B). In addition, we have shown that the expression of Cre recombinase under the Villin promoter (Imp1-loxP +/+;Villin-Cre +/−) results in significant inhibition of Imp1 expression in the intestinal epithelium of these mice (Figure 2C).
Fig. 2.

Intestinal-specific knockout of Imp1 suppresses Apc Min/+ -driven intestinal tumorigenesis. (A) Design of targeting construct for generation of Imp1-LoxP mice. In these mice, the exons 5 and 6 of Imp1 are flanked by loxP sequences. (B) Immunoblot analysis of protein extract isolated from Imp1-loxP homozygous MEFs (transfected as indicated) 48 h after transfection. (C) Imp1 expression in small intestine and colon of wild-type and Imp1-loxP +/+ ; Villin-Cre +/− mice. Arrows indicate high expression of Imp1. (D) Tumor number in the mouse intestine in Apc Min/+-driven intestinal tumorigenesis. **P < 0.001 in Mann–Whitney U-test, N ≥ 5 mice per group.
To analyze the contribution of Imp1 to Apc Min/+ -driven intestinal tumorigenesis, which we have shown previously to express high Imp1 in both small intestine and colon tumors, we crossed Imp1-loxP mice sequentially with Villin-Cre, and Apc Min/+mice to obtain Imp1-loxP +/− ;Villin-Cre +/− ;Apc Min/+animals (and Villin-Cre +/− Apc Min/+ littermate controls) (23). We observed a robust 66% reduction in the total number of intestinal tumors in the mice deficient for the expression of Imp1 in the intestinal epithelium (Figure 2D) at 90 days of age, signifying that Apc Min/+-driven tumorigenesis is inhibited by intestinal- specific Imp1 knockdown. These data, together with xenograft studies, lead us to conclude that IMP1 modulates primary tumor growth.
Surprisingly, we did not see a decrease in tumor burden in mice inje wcted with SW480 IMP1 knockdown cells (Supplementary Figure S2, available at Carcinogenesis Online). This was not due to ineffective knockdown, which was confirmed initially by qRT–PCR and western blot and monitored throughout the experiment using live animal imaging of the RFP-expressing IMP1 short hairpin RNA-transduced cells. Viability analysis via 4′,6-diamidino-2- phenylindole staining and pathological analysis for necrosis revealed that there was a greater degree of cell death in IMP1 knockdown tumors (Supplementary Figure S2, available at Carcinogenesis Online), suggesting that perhaps an unknown factor may be, in part, compensating for IMP1 loss-associated cell death in this cell line. We analyzed expression of the IMP1 paralog IMP3, which is also increased in CRC in humans, in our IMP1 knockdown cell lines and saw no change in IMP3 levels in vitro (data not shown) (24,25). This suggests that IMP3 is probably not the factor compensating for IMP1 loss in these cells.
IMP1 modulates tumor dissemination into the blood
We did not observe gross metastases in our IMP1 overexpression xenograft models, which may be a consequence of the short time course of the studies performed. To circumvent this limitation, we evaluated the burden of tumor cells circulating within the blood, which represents a critical first step in the metastatic cascade and a newly emerging approach for the detection of premalignant disease (20,26). We collected whole blood via cardiac puncture immediately posteuthanasia and utilized GFP or RFP expressed by tumor cell lines to quantify tumor cells per volume of blood. In SW480 cells, IMP1 overexpression enhanced the number of tumor cells in the blood (Figure 3A). We did not observe tumor cells in the blood of LoVo cells, which we attribute to having euthanized the mice at 2.5 weeks rather than 8 weeks postinjection. Despite inconclusive data for primary tumor formation in IMP1 knockdown xenografts, we saw a significant decrease in the number of cells circulating in the blood (Figure 3B). This observation supports the concept that primary tumor size does not necessarily correlate with metastatic potential and suggests a novel role for IMP1 in modulating tumor dissemination into the blood.
Fig. 3.
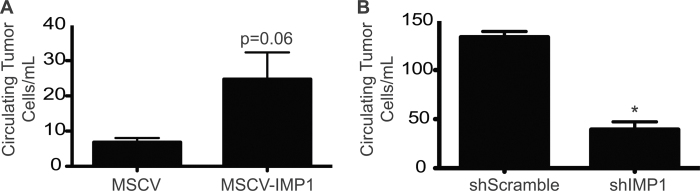
IMP1 modulates tumor dissemination into the blood. Blood was collected from xenograft mice after euthanasia and number of circulating tumor cells quantified using fluorescence-activated cell sorting for GFP (overexpressing cells) or RFP (knockdown cells). (A) There was an increase in the number of circulating tumor cells in mice harboring IMP1 overexpressing tumors. N ≥ 3 mice per group. (B) In mice with IMP1 knockdown tumors, there was a significant decrease in the number of circulating tumor cells detected in the blood upon euthanasia. *P < 0.05, N ≥ 3 mice per group.
IMP1 overexpression promotes colonosphere growth from single tumor cells
To evaluate the presence of tumor cells with enhanced clonogenic growth in control and IMP1 overexpressing cells, we dissociated xenograft tumors into single cells and grew individual cells in serum-free, ultralow attachment conditions as described previously (Figure 4A) (20,21,27). Individual spheres were counted after 10 days, and photographs were taken. Mean sphere growth was determined by counting the number of wells with spheres per 96-well plate for each of three individual tumors per condition. Mean sphere size was evaluated by measuring average radii using ImageJ. We did not initially observe an increase in colonosphere size or number with IMP1 overexpression (Figure 4B and C); however, when spheres were passaged via mechanical dissociation, there was a significant increase in sphere growth in IMP1 overexpressing cells compared with controls (Figure 4D). This suggests that IMP1 overexpression may promote survival and clonogenic growth of a subpopulation of cells exhibiting characteristics of tumor-initiating cells (TICs).
Fig. 4.

IMP1 promotes colonosphere growth after passaging. Xenograft tumors were dissociated into single cells and grown in serum-free, ultralow attachment conditions. (A) Individual tumor cells were evaluated after 10 days for sphere growth, followed by mechanical disruption, passaging and evaluation of sphere growth and spreading after 14 days. Scale bars = 50 μM. (B) There was no difference in sphere number or (C) size in IMP1 overexpressing tumor cells compared with controls. (D) We observed a significant increase in the outgrowth of mechanically passaged colonospheres in IMP1 overexpressing cells compared with controls. *P < 0.05, N = 3 individual tumors per condition.
IMP1 overexpression promotes a TIC phenotype
Our in vivo and in vitro findings suggest that IMP1 may promote clonogenic growth in xenograft tumors and enhancement of a TIC phenotype. To confirm these findings, we chose to evaluate further this phenotype using fluorescence-activated cell sorting. Cell surface markers CD24 and CD44 have been shown previously to identify a TIC phenotype in CRC cells (28–31) and were thus used to evaluate IMP1 overexpressing cells. In particular, CRC cells that exhibit enhanced CD24+CD44+ phenotype are postulated to be TICs (31). We observed a significant enhancement in the proportion of CD24+CD44+ populations in both SW480 (Figure 5A and B) and LoVo (data not shown) IMP1 overexpression cell lines, suggesting that IMP1 does indeed promote a TIC phenotype, which may confer a prosurvival advantage for cells that disseminate into the blood.
Fig. 5.

IMP1 overexpression promotes a tumor-initiating phenotype. Based on findings in colonospheres, we sought to further characterize the effect of IMP1 overexpression on TIC phenotype in vitro. (A and B) Control and IMP1 overexpressing cells were evaluated for cell surface expression of CD24 and CD44, markers of TICs. IMP1 overexpression dramatically enhanced the proportion of cells expressing high levels of both CD24 and CD44 compared with controls. *P < 0.05, N = 3. (C) CD24+CD44+high cells were sorted and grown in two-dimensional culture conditions. Cells were passaged three times, with analysis for CD24+CD44+high cells at each passage in both control and IMP1 overexpressing cell lines. There was a significant increase in the ability of IMP1 overexpressing cells to maintain the CD24+CD44+high phenotype compared with controls. *P < 0.05, N = 3 passages.
In order to test if IMP1 promotes stabilization of a TIC phenotype over the course of multiple passages, we sorted and passaged CD24+CD44+high cells from both control and IMP1 overexpressing cell lines (Figure 5C). We consistently observed that IMP1 overexpressing cells maintained a greater proportion of CD24+CD44+high cells with passaging, whereas control cells appeared to lose this phenotype over time. This supports our finding that IMP1 enhances the longevity of this cell population in colonospheres (Figure 4D), as well as in cells grown in two-dimensional culture conditions.
Tumor dissemination is often associated with a TIC phenotype and/or an epithelial to mesenchymal transition (EMT) phenotype (20). We therefore used qRT–PCR to examine EMT in IMP1 cell lines and saw a significant downregulation of E-cadherin when IMP1 is overexpressed, indicating that these cells may be losing their ‘epithelial’ phenotype (Figure 6A). We observed a trend for concomitant upregulation of EMT genes vimentin, Snail, Slug and Zeb1, although these were not statistically significant (Figure 6B–E). Further, we were not able to detect Zeb2 in these cells (data not shown). Taken together, the upregulation of CD24+CD44+ markers and downregulation of E-cadherin suggest that IMP1 overexpression promotes both an acquisition of a ‘stem-like’ phenotype and a loss of epithelial characteristics, both of which support the novel role of high-IMP1-expressing tumor cells in tumor progression.
Fig. 6.

IMP1 overexpression decreases E-cadherin expression. We evaluated whether IMP1 overexpression promotes EMT in vitro. SW480 cells overexpressing IMP1 were analyzed for E-cadherin, vimentin, snail, slug and zeb1 gene expression in order to assay for loss of an epithelial phenotype or gain of mesenchymal phenotype. Glyceraldehyde 3-phosphate dehydrogenase was used as the internal control. (A) E-cadherin was significantly decreased with IMP1 overexpression. *P < 0.05, N = 3. (B–E) We observed a trend for upregulation of EMT gene in IMP1 overexpressing cells.
Discussion
The studies herein suggest an oncogenic role for IMP1 in vivo, providing additional functional insight into its role in CRC progression beyond proapoptotic functions associated previously with IMP1 knockdown in vitro (17,32). Using xenograft and genetic models of CRC, we demonstrate that IMP1 overexpression promotes increased tumor growth, and Imp1 knockdown limits tumor growth. In addition, IMP1 overexpression enhances tumor dissemination into the blood, increases survival and outgrowth of individual tumor clones and enriches a population of CD24+CD44+ TICs. Together, these findings may in part provide a rationale as to why IMP1 has been correlated with poor survival and prognosis in CRC patients (10,17) and suggest IMP1 as a potential driver of robust, therapy-resistant TICs.
Our previous studies indicated that IMP1 loss decreased proliferation and colony formation in CRC cells in vitro, and we thus hypothesized that xenograft studies of IMP1 overexpression may promote increased tumor growth and potentially enhanced tumor progression. In SW480 cells, we observed an increase in tumor load with IMP1 overexpression over a time course of 8 weeks, with a trend for increased tumor load as early as 2.5 weeks in LoVo cell tumors (Figure 1B; Supplementary Figure S1B, available at Carcinogenesis Online). However, in SW480 cells with inducible IMP1 knockdown, we observed inconclusive results (Supplementary Figure S2B, available at Carcinogenesis Online). In these knockdown tumors, we observed enhanced necrosis and decreased viability with IMP1 loss (Supplementary Figure S2C –E, available at Carcinogenesis Online), which is not surprising considering our previous in vitro work in which we observed that IMP1 loss increased apoptosis (17). Increased cell death associated with IMP1 loss does not, however, appear to lead to an overall decrease in xenograft tumor size, suggesting potential compensatory mechanisms for IMP1 loss in these cells. We therefore turned to a genetic mouse model of Imp1 depletion during intestinal tumorigenesis: conditional Imp1 knockdown mice crossed with APC Min/+ mice (Imp1-loxP +/− ;Villin-Cre +/− ;Apc Min/+ mice). These mice exhibited a dramatic reduction in tumor number compared with Villin-Cre +/− Apc Min/+ littermates. Taken together, our combined xenograft and genetic mouse models provide novel evidence that IMP1 modulates CRC tumor growth in vivo.
We found that IMP1 overexpression increased and that IMP1 knockdown decreased the number of tumor cells circulating in the blood, independent of IMP1’s effect on primary xenograft tumor size (Figure 3). Several recent studies have reported that tumor cells may enter the circulation prior to or concurrent with malignant growth, challenging the current dogma of tumor progression from localized disease followed by distal metastasis (20,33,34). In the current xenograft studies, we did not observe gross metastases in our 8 week time course, but we cannot rule out the existence of micrometastases or that eventual metastases could arise from ‘dormant’ TICs. A second possibility for lack of observation of gross metastases in our model could be that the circulating tumor cells lacked the appropriate distal metastatic niche, which several studies suggest is a critical component for the establishment of metastatic lesions (35–37). Future studies in genetic mouse models will be required to determine if IMP1-mediated tumor dissemination alone can lead to metastases or if additional ‘hits’ may be required.
Several mechanisms may act in concert to promote tumor dissemination into the blood and subsequent metastatic growth. Early tumor dissemination has been linked with tumor cells adopting a mesenchymal phenotype (20); however, other factors such as the existence of a subpopulation of TICs (31,38) have also been proposed as critical components of cancer metastasis. We evaluated clonogenic growth by dissociating xenograft tumors and evaluating growth of individual clones and their subsequent progeny. Although we did not observe a significant difference in the number of clones giving rise to colonospheres with IMP1 overexpression (Figure 4B), there was a significant increase in survival and outgrowth of IMP1 overexpressing clones postpassage (Figure 4D), suggesting that IMP1 may favor long-term growth and/or expansion of individual clones.
To further substantiate our in vivo findings, we explored whether IMP1 overexpression promotes a TIC phenotype and/or EMT in vitro. Gene expression of E-cadherin was downregulated with IMP1 overexpression (Figure 6); however, we did not observe an increase in mesenchymal genes in these cells in vitro. E-cadherin loss signifies a loss of intercellular contacts, an event that on its own can promote a metastatic phenotype (39). More strikingly, we observed that IMP1 overexpression promoted a dramatic increase in the percentage of CD24+CD44+ cells (Figure 5A and B), indicating that IMP1 may favor or enhance for a population of TICs. Furthermore, IMP1 overexpression promotes maintenance of this phenotype over the course of several passages (Figure 5C). This is intriguing because IMP1 has been shown previously to bind to and stabilize CD44 mRNA in vitro, and in the same study, IMP1 depletion was associated with decreased invadopodia formation (40). It is therefore possible that IMP1 overexpression promotes a TIC phenotype in our studies via the stabilization of CD44 mRNA.
Taken together, these studies provide novel in vivo data suggesting that IMP1 modulates tumor growth and dissemination of tumor cells into the blood. Our finding that IMP1 overexpression promotes a shift toward a TIC phenotype may explain clinical findings correlating high IMP1 with poor prognosis or clinical outcome in CRC patients and underscores a potential role for IMP1 in early metastatic events or as a marker for a more aggressive or therapeutic-resistant cell population.
Supplementary material
Supplementary Figures 1 and 2 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health (T32 DK007066, F32 DK093207-01 to K.E.H.; K08 DK088945 to A.D.R.; R01 DK056645-12, U01 DK085551, P30 DK050306 to K.E.H., P.S.K., C.M.H., S.R.D., E.T.L., A.J.K.-S., A.D.R., A.K.R.; CA153102 to F.K.N.; AR063361 to V.S.S.); Hansen Foundation and National Colon Colon Research Alliance to A.K.R.
Conflict of Interest Statement: None declared.
Supplementary Material
Acknowledgements
We thank A.Bedenbaugh, D.Budo and R.Hasan (Penn Molecular Pathology and Imaging Facility) for IHC; P.Hallberg (Penn Flow Cytometry and Sorting Facility); D. Heitjan (Penn Biostatistics); S.Keilbaugh (Penn Molecular Biology Core) and E.Roberston (Penn Cell Culture Core).
Glossary
Abbreviations:
- CRC
colorectal cancer
- EMT
epithelial to mesenchymal transition
- GFP
green fluorescent protein
- IMP1
Igf2 mRNA binding protein 1
- MEF
mouse embryonic fibroblast
- mRNA
messenger RNA
- qRT–PCR
quantitative reverse transcription–polymerase chain reaction
- RFP
red fluorescent protein
- TIC
tumor-initiating cell.
References
- 1. Leeds P., et al. (1997). Developmental regulation of CRD-BP, an RNA-binding protein that stabilizes c-myc mRNA in vitro . Oncogene, 14, 1279–1286 [DOI] [PubMed] [Google Scholar]
- 2. Ross A.F., et al. (1997). Characterization of a beta-actin mRNA zipcode-binding protein. Mol. Cell. Biol., 17, 2158–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doyle G.A., et al. (1998). The c-myc coding region determinant-binding protein: a member of a family of KH domain RNA-binding proteins. Nucleic Acids Res., 26, 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nielsen J., et al. (1999). A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol., 19, 1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hansen T.V., et al. (2004). Dwarfism and impaired gut development in insulin-like growth factor II mRNA-binding protein 1-deficient mice. Mol. Cell. Biol., 24, 4448–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ross J., et al. (2001). Overexpression of an mRNA-binding protein in human colorectal cancer. Oncogene, 20, 6544–6550 [DOI] [PubMed] [Google Scholar]
- 7. Tessier C.R., et al. (2004). Mammary tumor induction in transgenic mice expressing an RNA-binding protein. Cancer Res., 64, 209–214 [DOI] [PubMed] [Google Scholar]
- 8. Elcheva I., et al. (2008). Overexpression of mRNA-binding protein CRD-BP in malignant melanomas. Oncogene, 27, 5069–5074 [DOI] [PubMed] [Google Scholar]
- 9. Kato T., et al. (2007). Increased expression of insulin-like growth factor-II messenger RNA-binding protein 1 is associated with tumor progression in patients with lung cancer. Clin. Cancer Res., 13(Pt 1)434–442 [DOI] [PubMed] [Google Scholar]
- 10. Dimitriadis E., et al. (2007). Expression of oncofetal RNA-binding protein CRD-BP/IMP1 predicts clinical outcome in colon cancer. Int. J. Cancer, 121, 486–494 [DOI] [PubMed] [Google Scholar]
- 11. Lapidus K., et al. (2007). ZBP1 enhances cell polarity and reduces chemotaxis. J. Cell Sci., 120(Pt 18)3173–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oberman F., et al. (2007). VICKZ proteins mediate cell migration via their RNA binding activity. RNA, 13, 1558–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vainer G., et al. (2008). A role for VICKZ proteins in the progression of colorectal carcinomas: regulating lamellipodia formation. J. Pathol., 215, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Noubissi F.K., et al. (2006). CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature, 441, 898–901 [DOI] [PubMed] [Google Scholar]
- 15. Stöhr N., et al. (2012). IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev., 26, 176–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manieri N.A., et al. (2012). Igf2bp1 is required for full induction of Ptgs2 mRNA in colonic mesenchymal stem cells in mice. Gastroenterology, 143, 110–21.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mongroo P.S., et al. (2011). IMP-1 displays cross-talk with K-Ras and modulates colon cancer cell survival through the novel proapoptotic protein CYFIP2. Cancer Res., 71, 2172–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. King C.E., et al. (2011). LIN28B promotes colon cancer progression and metastasis. Cancer Res., 71, 4260–4268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spiegelman V.S., et al. (2000). Wnt/beta-catenin signaling induces the expression and activity of betaTrCP ubiquitin ligase receptor. Mol. Cell, 5, 877–882 [DOI] [PubMed] [Google Scholar]
- 20. Rhim A.D., et al. (2012). EMT and dissemination precede pancreatic tumor formation. Cell, 148, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kanwar S.S., et al. (2010). The Wnt/beta-catenin pathway regulates growth and maintenance of colonospheres. Mol. Cancer, 9, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leibovitz A., et al. (1976). Classification of human colorectal adenocarcinoma cell lines. Cancer Res., 36, 4562–4569 [PubMed] [Google Scholar]
- 23. Noubissi F.K., et al. (2009). Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res., 69, 8572–8578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li D., et al. (2009). IMP3 is a novel prognostic marker that correlates with colon cancer progression and pathogenesis. Ann. Surg. Oncol., 16, 3499–3506 [DOI] [PubMed] [Google Scholar]
- 25. Lochhead P., et al. (2012). Insulin-like growth factor 2 messenger RNA binding protein 3 (IGF2BP3) is a marker of unfavourable prognosis in colorectal cancer. Eur. J. Cancer, 48, 3405–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faltas B. (2012). Cornering metastases: therapeutic targeting of circulating tumor cells and stem cells. Front. Oncol., 2, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu Y., et al. (2009). Elimination of colon cancer stem-like cells by the combination of curcumin and FOLFOX. Transl. Oncol., 2, 321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weichert W., et al. (2005). Cytoplasmic CD24 expression in colorectal cancer independently correlates with shortened patient survival. Clin. Cancer Res., 11, 6574–6581 [DOI] [PubMed] [Google Scholar]
- 29. Choi D., et al. (2009). Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J. Gastroenterol., 15, 2258–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herrmann I., et al. (2010). Highly efficient elimination of colorectal tumor-initiating cells by an EpCAM/CD3-bispecific antibody engaging human T cells. PLoS One, 5, e13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yeung T.M., et al. (2010). Cancer stem cells from colorectal cancer-derived cell lines. Proc. Natl Acad. Sci. USA, 107, 3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boyerinas B., et al. (2012). Let-7 modulates acquired resistance of ovarian cancer to Taxanes via IMP-1-mediated stabilization of multidrug resistance 1. Int. J. Cancer, 130, 1787–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hüsemann Y., et al. (2008). Systemic spread is an early step in breast cancer. Cancer Cell, 13, 58–68 [DOI] [PubMed] [Google Scholar]
- 34. Eyles J., et al. (2010). Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Invest., 120, 2030–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cameron M.D., et al. (2000). Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res., 60, 2541–2546 [PubMed] [Google Scholar]
- 36. Psaila B., et al. (2009). The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer, 9, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oskarsson T., et al. (2011). Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med., 17, 867–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dieter S.M., et al. (2011). Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell, 9, 357–365 [DOI] [PubMed] [Google Scholar]
- 39. Onder T.T., et al. (2008). Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res., 68, 3645–3654 [DOI] [PubMed] [Google Scholar]
- 40. Vikesaa J., et al. (2006). RNA-binding IMPs promote cell adhesion and invadopodia formation. EMBO J., 25, 1456–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.