

Abstract
Summary: Here we introduce catRAPID omics, a server for large-scale calculations of protein–RNA interactions. Our web server allows (i) predictions at proteomic and transcriptomic level; (ii) use of protein and RNA sequences without size restriction; (iii) analysis of nucleic acid binding regions in proteins; and (iv) detection of RNA motifs involved in protein recognition.
Results: We developed a web server to allow fast calculation of ribonucleoprotein associations in Caenorhabditis elegans, Danio rerio, Drosophila melanogaster, Homo sapiens, Mus musculus, Rattus norvegicus, Saccharomyces cerevisiae and Xenopus tropicalis (custom libraries can be also generated). The catRAPID omics was benchmarked on the recently published RNA interactomes of Serine/arginine-rich splicing factor 1 (SRSF1), Histone-lysine N-methyltransferase EZH2 (EZH2), TAR DNA-binding protein 43 (TDP43) and RNA-binding protein FUS (FUS) as well as on the protein interactomes of U1/U2 small nucleolar RNAs, X inactive specific transcript (Xist) repeat A region (RepA) and Crumbs homolog 3 (CRB3) 3′-untranslated region RNAs. Our predictions are highly significant (P < 0.05) and will help the experimentalist to identify candidates for further validation.
Availability: catRAPID omics can be freely accessed on the Web at http://s.tartaglialab.com/catrapid/omics. Documentation, tutorial and FAQs are available at http://s.tartaglialab.com/page/catrapid_group.
Contact: gian.tartaglia@crg.eu
1 INTRODUCTION
Increasing evidence indicates that ribonucleoprotein interactions are fundamental for cellular regulation (Khalil and Rinn, 2011). Moreover, several studies highlighted the involvement of RNA molecules in the onset and progression of human diseases including neurological disorders (Johnson et al., 2012). To our knowledge, there are two sequence-based methods for prediction of protein–RNA interactions: catRAPID (Bellucci et al., 2011) and RPISeq (Muppirala et al., 2011). The catRAPID algorithm exploits predictions of secondary structure, hydrogen bonding and van der Waals’ contributions to estimate the binding propensity of protein and RNA molecules. RPISeq is based on support vector machine (SVM) and random forest (RF) models predicting protein–RNA interactions from primary structure alone (Muppirala et al., 2011). Both methods show remarkable performances, but catRAPID discriminates positive and negative cases with higher accuracy (Cirillo et al., 2013b) and has been tested on long non-coding RNAs (Agostini et al., 2013).
Here we introduce catRAPID omics to perform high-throughput predictions of protein–RNA interactions using the information on protein and RNA domains involved in macromolecular recognition.
2 WORKFLOW AND IMPLEMENTATION
The catRAPID omics server provides two main services to explore the interaction potential of (i) a protein of interest with respect to a target transcriptome or (ii) a given RNA with respect to the nucleic acid binding proteome. Several options are available to refine the type of analysis in eight model organisms or custom libraries (see online documentation):
In the case of a protein query, catRAPID omics takes as input the protein sequence (FASTA format): full-length or, alternatively, nucleic acid binding regions.
For a transcript query (FASTA format), the server uses the full-length sequence if below 1200 nt, or, alternatively, uses fragments with predicted stable secondary structure (Agostini et al., 2013). Full-length proteins and nucleic acid binding regions can be searched.
The server automatically detects disordered proteins lacking canonical RNA binding domains. Indeed, it has been observed that disordered regions are enriched in RNA binding proteins (Castello et al., 2012).
As RNA motifs are important for protein recognition (Kazan et al., 2010), a search for these elements is carried out. The motifs were taken from RNA-Binding Protein DataBase (RBPDB) (Cook et al., 2011), SpliceAid-F (Giulietti et al., 2013) and a recent motif compendium (Ray et al., 2013).
Using the interaction propensities distribution, catRAPID omics predicts the RNA binding ability of the input protein (86% accuracy) and ranks RNA interactions (downloadable by the user).
In the output page (Fig. 1A), we report all the variables used to estimate protein–RNA associations: interaction propensity (Bellucci et al., 2011), discriminative power (Bellucci et al., 2011), interaction strength (Agostini et al., 2013) and presence of protein RNA binding domains as well as RNA motifs. A ‘star rating system’ ranks the binding propensities (http://service.tartaglialab.com/static_files/shared/faqs.html). As for the reference sets, ENSEMBL (version 68) is used for retrieval and classification of coding and non-coding RNAs, whereas protein sequences are gathered from the UniProtKB database (release 2012_11). Finally, catRAPID omics uses hmmscan, a Hidden Markov Model-based algorithm from the HMMER3 package (Finn et al., 2011), to identify known PfamA domains (Finn et al., 2009) and recognize protein regions involved in binding nucleic acid molecules. Algorithm hit significance is determined according to the PfamA ‘gathering thresholds’.
Fig. 1.
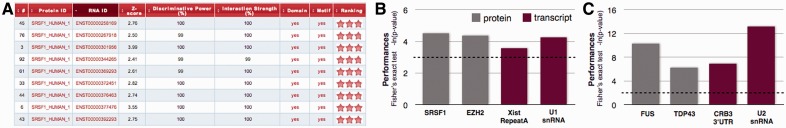
catRAPID omics features and performances. (A) Example of the output table showing Z-score (interaction propensity normalized with respect to experimental cases), discriminative power (with respect to training sets), interaction strength (enrichment with respect to random interactions) and presence of RNA binding domains as well as RNA motifs. Interaction scores are ranked according to a ‘star rating system’ ranging from 0 to 3 (http://service.tartaglialab.com/static_files/shared/faqs.html). A click on the text redirects to reference pages. Performances on (B) full-length proteins and (C) RNA binding protein domains. Gray is used to highlight transcriptomic studies (i.e. RNA sequencing) and red indicates proteomic analyses (i.e. mass spectrometry). The significance of our predictions was assessed using Fisher’s exact test (the dashed line corresponds to P = 0.05)
3 PERFORMANCES
The catRAPID algorithm has been previously validated on a number of protein–RNA associations (Agostini et al., 2013; Bellucci et al., 2011; Cirillo, et al., 2013a; Johnson et al., 2012). To evaluate large-scale performances of catRAPID omics, we used data from recent large-scale experiments. To compare predicted and experimental interactions, we used Fisher’s exact test. As shown in Figure 1B, performances on the human splicing factor serine/arginine-rich splicing factor 1 (SRSF1) (Sanford et al., 2009) and murine nucleic acid binding protein Histone-lysine N-methyltransferase EZH2 (EZH2) (Zhao et al., 2010) are highly significant (P-values: 0.01 and 0.01, respectively). Good performances are found for low-throughput experiments on murine non-coding X inactive specific transcript (Xist) repeat A region (RepA) (Maenner et al., 2010; Royce-Tolland et al., 2010) and yeast small nuclear RNA U1 (Cvitkovic and Jurica, 2012) (P-values: 0.03 and 0.015) (Fig. 1B). To illustrate the ability of catRAPID omics to predict interactions with nucleic acid binding domains (Fig. 1C), we used murine FUS (Han et al., 2012) and rat TAR DNA-binding protein 43 (TDP43) (Sephton et al., 2011) (P-values: 3e-05 and 0.002) as well as human Crumbs homolog 3 (CRB3) 3′-untranslated region (Iioka et al., 2011) and yeast small nuclear U2 (Cvitkovic and Jurica, 2012) (P-values: 0.001 and 2e-0.6). To evaluate catRAPID’s performances on high-throughput data, we collected positive interactions (TDP43: 568, FUS: 99, SRSF1: 358, EZH2: 1141) as well as negative controls (same numbers as positives and generated in four random extractions). Comparing the interaction scores of positives and negatives, we found enrichment (calculated as discriminative power) in 72% (TDP43), 88% (FUS), 74% (SRSF1) and 56% (EZH2) of cases. On the same datasets, SVM RPIseq showed enrichment in 58% (TDP43; RF has enrichment in 53%), 83% (FUS; RF has enrichment in 68%), 47% (SRSF1; RF has enrichment in 59%) and 41% (EZH2; RF has enrichment in 48%) of cases.
4 CONCLUSIONS
Despite recent technical developments, detection of protein–RNA associations remains a challenging task. For this reason, we developed an algorithm that can be used to complement experimental efforts (Zanzoni et al., 2013). The catRAPID omics server offers unique features such as organism-specific proteomic and transcriptomic libraries, possibility to generate custom datasets, analysis of long sequences and calculation of interaction specificities. Moreover, we implemented an algorithm for the detection of RNA motifs as well as protein RNA binding domains, which will help to retrieve recognition motifs embedded in sequences. Our server enables fast calculations of ribonucleoprotein associations and predicts RNA binding activity of proteins with high accuracy, thus resulting in a powerful tool for designing new experiments.
ACKNOWLEDGEMENTS
The authors would like to thank Dr J.R. Sanford CLIP-seq data on SFSR1, Prof R. Guigó and Dr G. Bussotti for stimulating discussions.
Funding: Spanish Ministry of Economy and Competitiveness (SAF2011-26211), the European Research Council (ERC Starting Grant to G.G.T) and the RTTIC project (to A.Z.). ‘La Caixa’ fellowship (to P.K.). Programa de Ayudas FPI del Ministerio de Economia y Competitividad—BES-2012-052457 (to D.M.).
Conflict of Interest: none declared.
REFERENCES
- Agostini F, et al. X-inactivation: quantitative predictions of protein interactions in the Xist network. Nucleic Acids Res. 2013;41:e31. doi: 10.1093/nar/gks968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci M, et al. Predicting protein associations with long noncoding RNAs. Nat. Methods. 2011;8:444–445. doi: 10.1038/nmeth.1611. [DOI] [PubMed] [Google Scholar]
- Castello A, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Cirillo D, et al. Neurodegenerative diseases: quantitative predictions of protein–RNA interactions. RNA. 2013a;19:129–140. doi: 10.1261/rna.034777.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo D, et al. Predictions of protein–RNA interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013b;3:161–175. [Google Scholar]
- Cook KB, et al. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvitkovic I, Jurica MS. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2012;41:D132–D141. doi: 10.1093/nar/gks999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. The Pfam protein families database. Nucleic Acids Res. 2009;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulietti M, et al. SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013;41:D125–D131. doi: 10.1093/nar/gks997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Iioka H, et al. Efficient detection of RNA-protein interactions using tethered RNAs. Nucleic Acids Res. 2011;39:e53. doi: 10.1093/nar/gkq1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, et al. Neurodegeneration as an RNA disorder. Prog. Neurobiol. 2012;99:293–315. doi: 10.1016/j.pneurobio.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazan H, et al. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol. 2010;6:e1000832. doi: 10.1371/journal.pcbi.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Rinn JL. RNA-protein interactions in human health and disease. Semin. Cell Dev. Biol. 2011;22:359–365. doi: 10.1016/j.semcdb.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenner S, et al. 2-D structure of the A Region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010;8:e1000276. doi: 10.1371/journal.pbio.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muppirala UK, et al. Predicting RNA-protein interactions using only sequence information. BMC Bioinformatics. 2011;12:489. doi: 10.1186/1471-2105-12-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royce-Tolland ME, et al. The A-repeat links ASF/SF2-dependent Xist RNA processing with random choice during X inactivation. Nat. Struct. Mol. Biol. 2010;17:948–954. doi: 10.1038/nsmb.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, et al. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009;19:381–394. doi: 10.1101/gr.082503.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011;286:1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A, et al. Principles of self-organization in biological pathways: a hypothesis on the autogenous association of alpha-synuclein. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt794. [Epub ahead of print, doi: 10.1093/nar/gkt794, September 3, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]