

Abstract
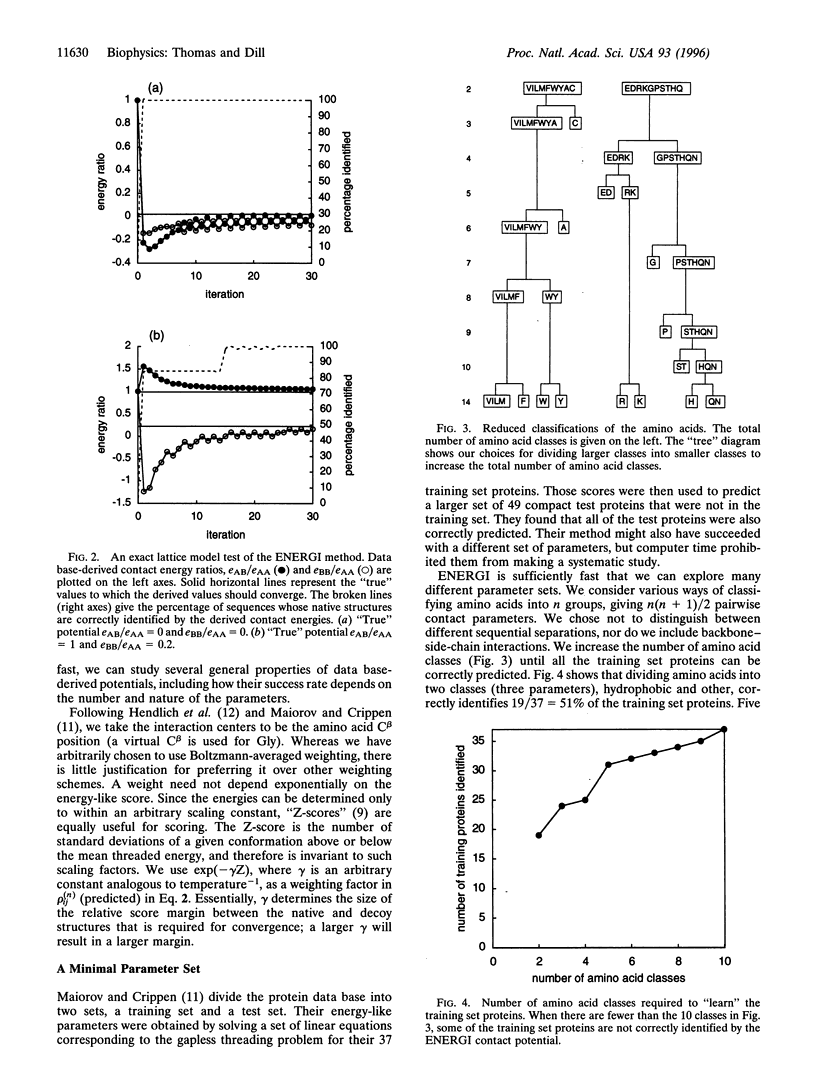
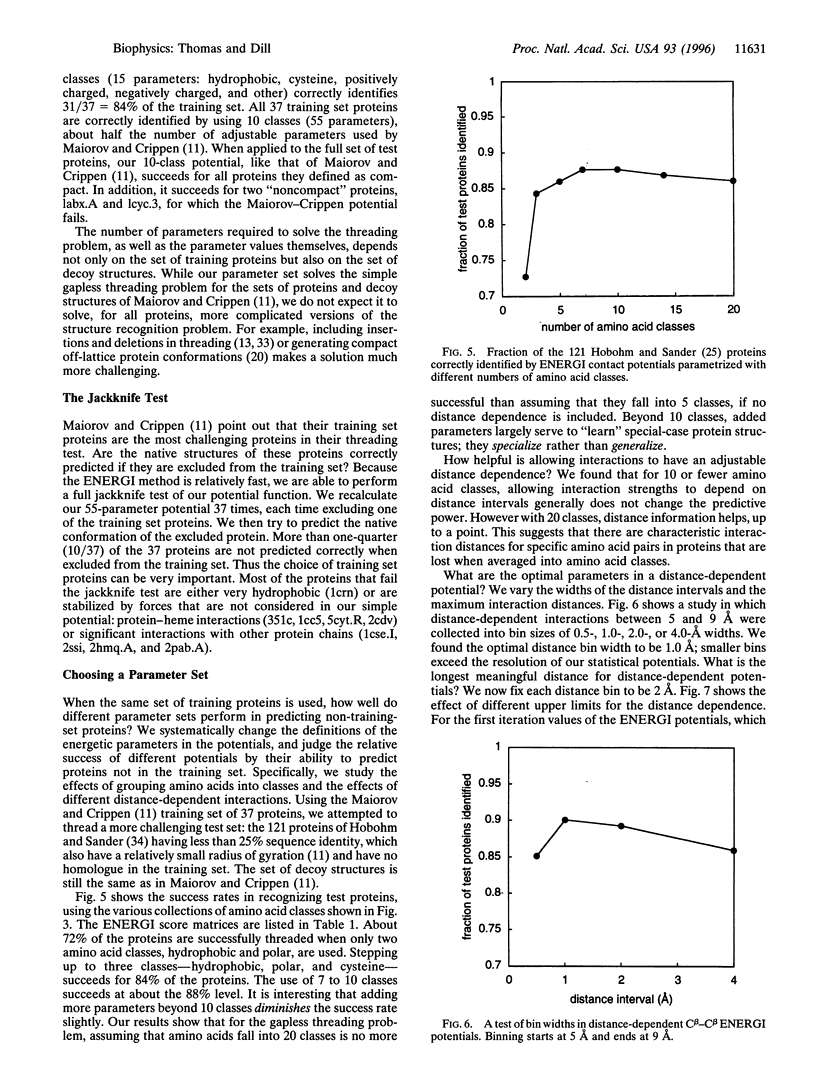
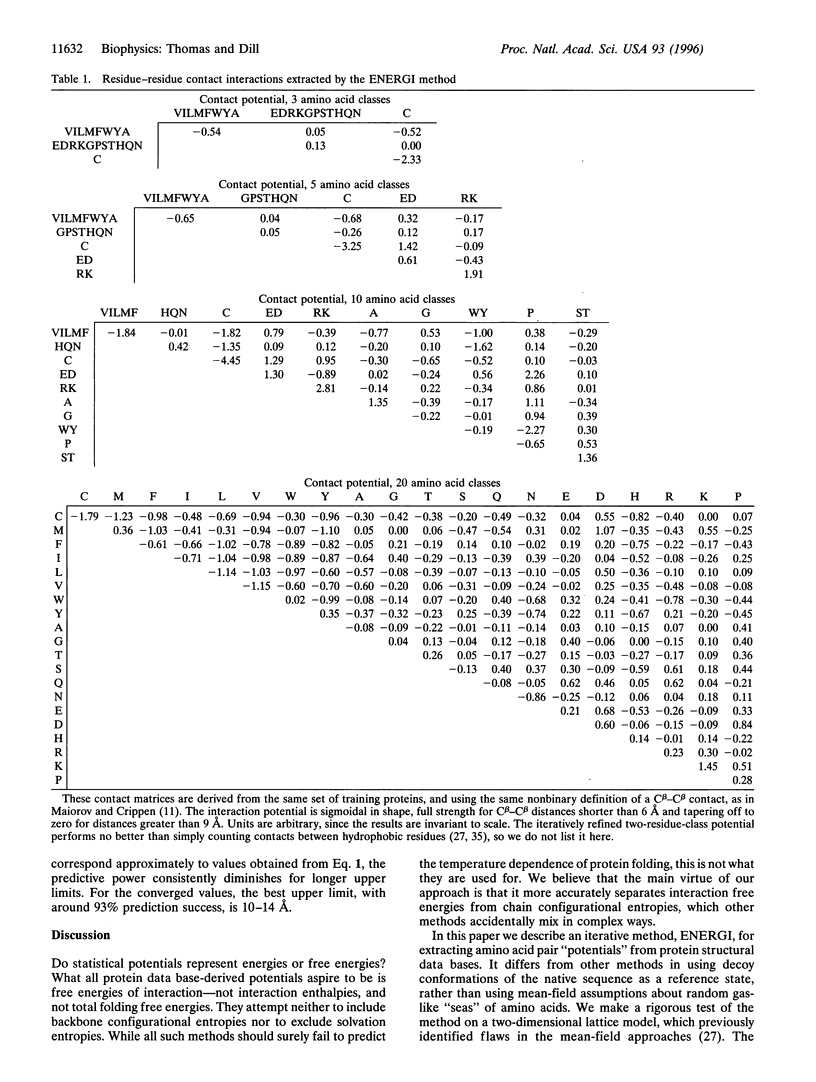
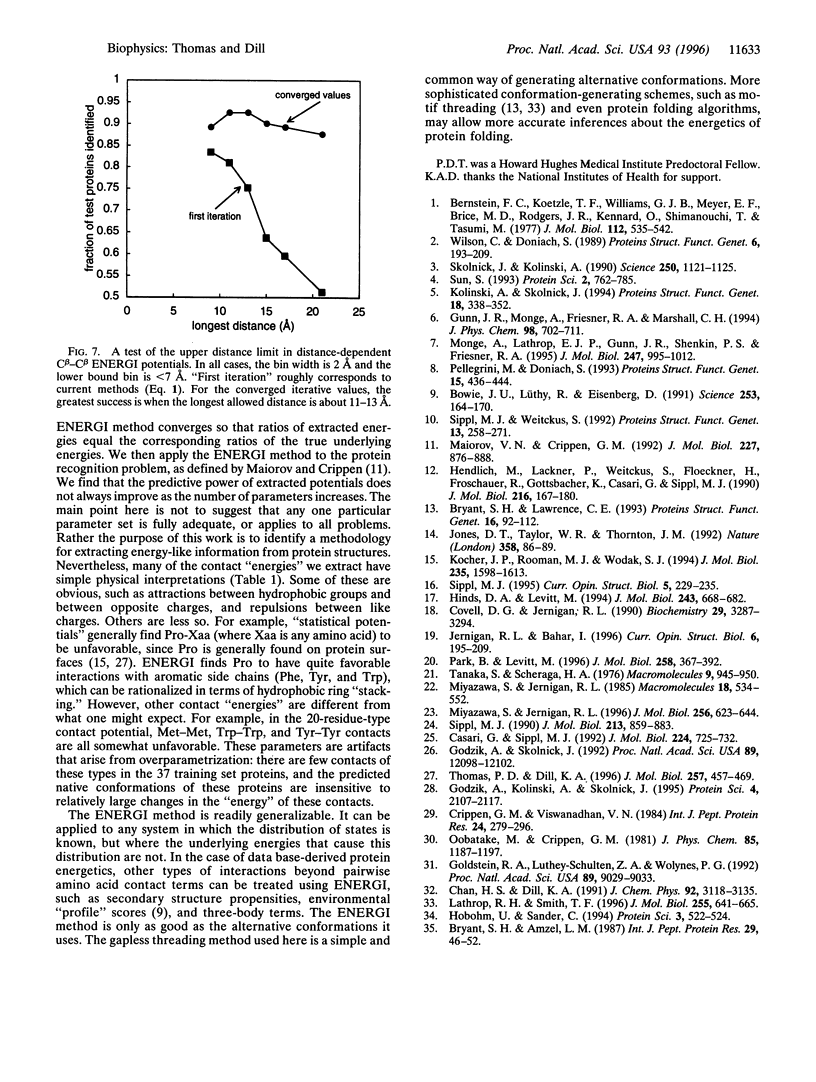
We present a method (ENERGI) for extracting energy-like quantities from a data base of protein structures. In this paper, we use the method to generate pairwise additive amino acid "energy" scores. These scores are obtained by iteration until they correctly discriminate a set of known protein folds from decoy conformations. The method succeeds in lattice model tests and in the gapless threading problem as defined by Maiorov and Crippen [Maiorov, V. N. & Crippen, G. M. (1992) J. Mol. Biol. 227, 876-888]. A more challenging test of threading a larger set of test proteins derived from the representative set of Hobohm and Sander [Hobohm, U. & Sander, C. (1994) Protein Sci. 3, 522-524] is used as a "workbench" for exploring how the ENERGI scores depend on their parameter sets.
Full text
PDF


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bernstein F. C., Koetzle T. F., Williams G. J., Meyer E. F., Jr, Brice M. D., Rodgers J. R., Kennard O., Shimanouchi T., Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977 May 25;112(3):535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Bowie J. U., Lüthy R., Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991 Jul 12;253(5016):164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Bryant S. H., Amzel L. M. Correctly folded proteins make twice as many hydrophobic contacts. Int J Pept Protein Res. 1987 Jan;29(1):46–52. doi: 10.1111/j.1399-3011.1987.tb02228.x. [DOI] [PubMed] [Google Scholar]
- Bryant S. H., Lawrence C. E. An empirical energy function for threading protein sequence through the folding motif. Proteins. 1993 May;16(1):92–112. doi: 10.1002/prot.340160110. [DOI] [PubMed] [Google Scholar]
- Casari G., Sippl M. J. Structure-derived hydrophobic potential. Hydrophobic potential derived from X-ray structures of globular proteins is able to identify native folds. J Mol Biol. 1992 Apr 5;224(3):725–732. doi: 10.1016/0022-2836(92)90556-y. [DOI] [PubMed] [Google Scholar]
- Covell D. G., Jernigan R. L. Conformations of folded proteins in restricted spaces. Biochemistry. 1990 Apr 3;29(13):3287–3294. doi: 10.1021/bi00465a020. [DOI] [PubMed] [Google Scholar]
- Crippen G. M., Viswanadhan V. N. A potential function for conformational analysis of proteins. Int J Pept Protein Res. 1984 Sep;24(3):279–296. doi: 10.1111/j.1399-3011.1984.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Godzik A., Koliński A., Skolnick J. Are proteins ideal mixtures of amino acids? Analysis of energy parameter sets. Protein Sci. 1995 Oct;4(10):2107–2117. doi: 10.1002/pro.5560041016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godzik A., Skolnick J. Sequence-structure matching in globular proteins: application to supersecondary and tertiary structure determination. Proc Natl Acad Sci U S A. 1992 Dec 15;89(24):12098–12102. doi: 10.1073/pnas.89.24.12098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein R. A., Luthey-Schulten Z. A., Wolynes P. G. Protein tertiary structure recognition using optimized Hamiltonians with local interactions. Proc Natl Acad Sci U S A. 1992 Oct 1;89(19):9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendlich M., Lackner P., Weitckus S., Floeckner H., Froschauer R., Gottsbacher K., Casari G., Sippl M. J. Identification of native protein folds amongst a large number of incorrect models. The calculation of low energy conformations from potentials of mean force. J Mol Biol. 1990 Nov 5;216(1):167–180. doi: 10.1016/S0022-2836(05)80068-3. [DOI] [PubMed] [Google Scholar]
- Hinds D. A., Levitt M. Exploring conformational space with a simple lattice model for protein structure. J Mol Biol. 1994 Nov 4;243(4):668–682. doi: 10.1016/0022-2836(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Hobohm U., Sander C. Enlarged representative set of protein structures. Protein Sci. 1994 Mar;3(3):522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan R. L., Bahar I. Structure-derived potentials and protein simulations. Curr Opin Struct Biol. 1996 Apr;6(2):195–209. doi: 10.1016/s0959-440x(96)80075-3. [DOI] [PubMed] [Google Scholar]
- Jones D. T., Taylor W. R., Thornton J. M. A new approach to protein fold recognition. Nature. 1992 Jul 2;358(6381):86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- Kocher J. P., Rooman M. J., Wodak S. J. Factors influencing the ability of knowledge-based potentials to identify native sequence-structure matches. J Mol Biol. 1994 Feb 4;235(5):1598–1613. doi: 10.1006/jmbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- Kolinski A., Skolnick J. Monte Carlo simulations of protein folding. I. Lattice model and interaction scheme. Proteins. 1994 Apr;18(4):338–352. doi: 10.1002/prot.340180405. [DOI] [PubMed] [Google Scholar]
- Lathrop R. H., Smith T. F. Global optimum protein threading with gapped alignment and empirical pair score functions. J Mol Biol. 1996 Feb 2;255(4):641–665. doi: 10.1006/jmbi.1996.0053. [DOI] [PubMed] [Google Scholar]
- Maiorov V. N., Crippen G. M. Contact potential that recognizes the correct folding of globular proteins. J Mol Biol. 1992 Oct 5;227(3):876–888. doi: 10.1016/0022-2836(92)90228-c. [DOI] [PubMed] [Google Scholar]
- Miyazawa S., Jernigan R. L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol. 1996 Mar 1;256(3):623–644. doi: 10.1006/jmbi.1996.0114. [DOI] [PubMed] [Google Scholar]
- Monge A., Lathrop E. J., Gunn J. R., Shenkin P. S., Friesner R. A. Computer modeling of protein folding: conformational and energetic analysis of reduced and detailed protein models. J Mol Biol. 1995 Apr 14;247(5):995–1012. doi: 10.1006/jmbi.1995.0195. [DOI] [PubMed] [Google Scholar]
- Park B., Levitt M. Energy functions that discriminate X-ray and near native folds from well-constructed decoys. J Mol Biol. 1996 May 3;258(2):367–392. doi: 10.1006/jmbi.1996.0256. [DOI] [PubMed] [Google Scholar]
- Pellegrini M., Doniach S. Computer simulation of antibody binding specificity. Proteins. 1993 Apr;15(4):436–444. doi: 10.1002/prot.340150410. [DOI] [PubMed] [Google Scholar]
- Sippl M. J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol. 1990 Jun 20;213(4):859–883. doi: 10.1016/s0022-2836(05)80269-4. [DOI] [PubMed] [Google Scholar]
- Sippl M. J. Knowledge-based potentials for proteins. Curr Opin Struct Biol. 1995 Apr;5(2):229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- Sippl M. J., Weitckus S. Detection of native-like models for amino acid sequences of unknown three-dimensional structure in a data base of known protein conformations. Proteins. 1992 Jul;13(3):258–271. doi: 10.1002/prot.340130308. [DOI] [PubMed] [Google Scholar]
- Skolnick J., Kolinski A. Simulations of the folding of a globular protein. Science. 1990 Nov 23;250(4984):1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- Sun S. Reduced representation model of protein structure prediction: statistical potential and genetic algorithms. Protein Sci. 1993 May;2(5):762–785. doi: 10.1002/pro.5560020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S., Scheraga H. A. Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules. 1976 Nov-Dec;9(6):945–950. doi: 10.1021/ma60054a013. [DOI] [PubMed] [Google Scholar]
- Thomas P. D., Dill K. A. Statistical potentials extracted from protein structures: how accurate are they? J Mol Biol. 1996 Mar 29;257(2):457–469. doi: 10.1006/jmbi.1996.0175. [DOI] [PubMed] [Google Scholar]
- Wilson C., Doniach S. A computer model to dynamically simulate protein folding: studies with crambin. Proteins. 1989;6(2):193–209. doi: 10.1002/prot.340060208. [DOI] [PubMed] [Google Scholar]