

Abstract
As there are more than 50 Adenovirus serotypes, the likelihood of developing an effective vaccine is low. Here we describe inhibitors of the adenovirus proteinase (AVP) with the ultimate objective of developing anti-adenovirus agents. Inhibitors were identified via structure-based drug design using as druggable sites the active site and a conserved cofactor pocket in the crystal structures of AVP. A lead compound was identified that had an IC50 of 18 μM. One of eight structural derivatives of the lead compound had an IC50 of 140 nM against AVP and an IC50 of 490 nM against the AVP with its cofactor bound.
Keywords: Adenovirus proteinase, Inhibitors, Structure-based drug design
1. Introduction
Adenoviruses are major human pathogens that infect a wide range of vertebrates, from hamsters to humans. They are the cause of many, different acute infections, mostly in the respiratory and gastrointestinal tracts, and conjunctivitis. (http://www.vmri.hu/~harrach/AdVtaxlong.htm). Late in adenovirus infection, young virions are assembled in part from precursor proteins. Of the 12 major virion proteins, 6 are precursor proteins. The penultimate step before the appearance of infectious virus is the activation of the adenovirus proteinase (AVP), a 23 kDa cysteine proteinase [1,2], followed by the processing of the virion precursor proteins. AVP is activated by two cofactors, pVIc (GVQSLKRRRCF) [3,4], the 11-amino acid peptide from the C-terminus of the precursor to protein VI, pVI, and the viral DNA genome [3]. We determined crystal structures of the AVP-pVIc complex at 1.6 Å resolution [2] and of AVP in the absence of cofactors at 0.98 Å resolution [5]. Because AVP is essential for synthesis of infectious virus, it is an attractive target for antiviral therapy [6–9].
Our approach toward development of antiviral agents for adenovirus, more specifically inhibitors of AVP, was to use the crystal structures and docking programs against the chemicals in a portion of the Open Chemical Repository of the NCI Developmental Therapeutics Program to identify compounds predicted to bind to sites likely to be critical for AVP activity. In this report, we identify and analyze a lead compound and two derivatives of it that inhibited the enzymes.
2. Materials and Methods
2.1 Structure-based drug design
The X-ray crystal structures of the 1.6 Å structure of the activated covalent AVP-pVIc complex (PDB ID: 1NLN) and the 0.98 Å structure of AVP alone (PDB ID: 4EKF) were used for in silico screening. To prepare the structures for site identification, all waters and heteroatoms were removed from the coordinate files. Then, using Chimera (UCSF), mol2 files were generated utilizing the highest occupancy alternate conformation side chains with all hydrogens assigned based on default charge parameters. Then, the hydrogens were stripped from the structures, and the DMS module of Chimera was used to generate the molecular surface for each protein. In order to identify potential sites on the surface, the SPHGEN module of DOCK (version 5.0, UCSF) was run using a 1.4 Å probe on the molecular surface. The resultant sphere files were converted to PDB format utilizing the DOCK module SHOWSPHERE, and then visual inspection of the spheres for complementarity in regions of the structures involved in cofactor binding or activation was carried out. Sphere clusters residing within an 8 Å radius of those specific regions of interest were compiled, one cluster near the active site residues His54 and Cys122, another cluster in a surface pocket that in the AVP-pVIc structure contains the Gly1′, Val2′ and Gln3′ of pVIc (termed NT-pocket). As an independent validation of the site choices, Fpocket, an open source pocket detection software package, was used to detect cavities from the PDB files, with water and heteroatoms of AVP or AVP-pVIc complexes removed. Visual inspection of the results of the query structures with embedded centers of pocket α-spheres revealed that in the AVP structure, the NT-pocket was the second ranked among the 12 identified in the Fpocket analysis. For the AVP-pVIc structure, the results list the active site pocket was the highest rank of the 12 pockets that were identified in the Fpocket analysis. The Fpocket results confirmed our target choices.
The SHOWBOX module of DOCK was used to generate 4 Å boxes in any direction around each sphere cluster file that was then used as input for the GRID program, which calculates and saves the information concerning the steric and electrostatic environment within the box areas as mol2 files. DOCK 5.0 was then used to screen approximately 140,000 small molecules from the National Cancer Institute/Developmental Therapeutics Program (NCI/DTP) Open Chemical Repository (http://dtp.cancer.gov) within the grids, using the selected spheres as theoretical binding sites. The small molecule output was ranked based on intermolecular AMBER energy scoring (van der Waals plus coulombic), contact scoring and bump filtering. The resultant ligand poses were then ranked by energy score with those in the entire virtual library on a relative basis, and the top ranked ligands were visually inspected for binding poses for each target. The top 10 compounds from each target were obtained from the NCI/DTP for testing the efficacy as an inhibitor in biochemical assays.
2.2 Materials
The gene for adenovirus serotype 2 proteinase, AVP, was expressed in Escherichia coli and the resultant protein purified as described previously[3,10]. pVIc (GVQSLKRRRCF) was purchased from Invitrogen (Carlsbad, CA). pVIc concentrations were determined by titration of the cysteine residue with Ellman’s reagent [11,12] using an extinction coefficient of 14,150 M−1cm−1 at 412 nm for released thionitrobenzoate. TPCK-treated trypsin was obtained from Worthington Biochemical Corporation, and papain was obtained from Sigma Chemical Company. The NCI compounds were obtained upon request to (http://dtp.cancer.gov). DDM (n-Dodecyl-β-D-Maltopyranoside) was obtained from Anatrace. The fluorogenic substrates (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine [3,13], (Pro-Arg-NH)2-Rhodamine [14,15], and (Phe-Arg-NH)2-Rhodamine were synthesized as described.
2.3 AVP-pVIc Complex Formation
Disulfide-linked AVP-pVIc complexes were prepared by overnight incubation at 4°C of 75 μM AVP and 75 μM pVIc in 20 mM Tris-HCl (pH 8.0), 250 mM NaCl, 0.1 mM EDTA and 20 mM β-mercaptoethanol. Under these conditions, Cys104 of AVP and Cys10′ of pVIc undergo oxidative condensation [16,17].
2.4 Enzyme Activity Assays
All enzymatic assays were performed at 21°C in a Corning 96-well half-area black flat bottom plate. The reaction volume was 100 uL and Rhodamine-based fluorogenic substrates were used. The excitation and emission wavelengths were 496 nm and 523 nm, respectively. Substrate hydrolysis was monitored every 10 seconds by measuring the time-dependent increase in fluorescence intensity using a Tecan Safire 2 plate reader. The initial rate (RFI s−1) was determined from a linear fit of the data and converted to nM s−1 as described previously [18].
2.5 Inhibition of AVP-pVIc complexes by lead compound NSC 36806
AVP-pVIc complexes, 0.3 μM, were incubated with the indicated concentrations of NSC 36806 in 5 mM NaCl, 20 mM Tris-HCl (pH 8.0), .025% DDM and 0.01 mM DTT. After 9 min., 5 μM substrate, (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine, was added and the increase in fluorescence monitored as a function of time.
2.6 Coarse screening of structural derivatives for enzyme inhibitors
The NCI compounds were dissolved in neat DMSO to a concentration of 10 mM. A coarse screening of eight NCI compounds, Table 2, was performed: AVP-pVIc complexes, 0.3 μM, were incubated in 150 mM NaCl, 20 mM Tris-HCl (pH 8), 0.025% DDM, and 0.1 mM DTT with 40 μM NCI compound. After 9 min., enzyme activity was assayed with the fluorogenic substrate (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine, 5 μM. Inhibition of AVP was assayed the same way except that after the 9 min. incubation, pVIc was added to 40 μM followed by an additional 9 min. of incubation before measuring enzyme activity.
Table 2.
Potential second generation inhibitors
| NSC No. | Chemical Structure | IUPAC Name |
|---|---|---|
| 36806 |
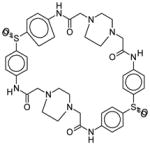
|
2,21-dithia-7,10,13,16,26,29,32,35-octaazaheptacyclo[34.2.2.23,6.210,13.217, 20.222,25.229,32]pentaconta-1(38),3,5,17,19,22,24,36,39,43,45,49-dodecaene-8,15,27,34-tetrone 2,2,21,21-tetraoxide |
| 106721 |
|
N-[4-[4-[(2,2-difluoro-2-morpholin-4-ylacetyl)amino]phenyl]sulfonylphenyl]-2,2-difluoro-2-morpholin-4-ylacetamide |
| 37249 |
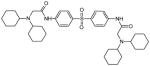
|
2-(dicyclohexylamino)-N-[4-[4-[[2-(dicyclohexylamino)acetyl]amino]phenyl] sulfonylphenyl]acetamide |
| 37248 |
|
2-[bis(3-methylbutyl)amino]-N-[4-[4-[[2-[bis(3-methylbutyl)amino]acetyl]amino]phenyl]s ulfonylphenyl]acetamide |
| 36319 |
|
2-morpholin-4-yl-N-[4-[4-[(2-morpholin-4-ylacetyl)amino]phenyl]sulfonylphenyl] acetamide |
| 36321 |
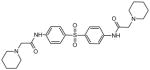
|
2-piperidin-1-yl-N-[4-[4-[(2-piperidin-1-ylacetyl)amino]phenyl]sulfonylphenyl] acetamide |
| 106702 |
|
2-(3,5-dimethylmorpholin-4-yl)-N-[4-[4-[[2-(3,5-dimethylmorpholin-4-yl)acetyl]amino]phenyl]sulfonylphenyl] acetamide |
| 106716 |
|
2-(dihexylamino)-N-[4-[4-[[2-(dihexylamino)acetyl]amino]phenyl] sulfonylphenyl]acetamide |
| 106720 |
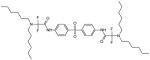
|
2-(dihexylamino)-N-[4-[4-[[2-(dihexylamino)-2,2-difluoroacetyl]amino]phenyl] sulfonylphenyl]-2,2-difluoroacetamide |
2.7 Determination of IC50
The IC50 values of compounds NSC37248 and NSC37249 with AVP and with AVP-pVIc complexes were determined as described in “Coarse screening for enzyme inhibitors” except that: the NaCl concentration in the buffer was 5 mM instead of 150 mM, and the pVIc concentration was 20 μM instead of 40 μM. The IC50 value was determined by fitting the dose-response curve to a four-parameter logistic equation. The equation is:
Where ν is the observed initial rate, Vmin is the minimum initial rate, Vmax is the maximum initial rate, [I] is the inhibitor concentration, and n is the Hill exponent.
2.8 Competitive inhibition of AVP-pVIc complexes by NSC 37249
AVP-pVIc complexes, 0.3 μM, were incubated with the indicated concentrations of NSC 37249 in 5 mM NaCl, 20 mM Tris-HCl, pH 8, 0.025% DDM and 0.1 mM DTT, and, after 9 min, four, different concentrations of substrate(Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine, were added and enzyme activity assayed. The initial rate data were entered into SigmaPlot Kinetic Module (Jandel Scientific) and analyzed for various inhibition models. The best fitting model was competitive inhibition with a Ki of 0.43 μM. The concentrations of (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine were 2.5 μM, 5 μM, 10 μM, and 20 μM.
2.9 Mixed inhibition of AVP by NSC 37249
AVP, 0.15 μM, was incubated with different concentrations of NSC 37249 and pVIc for 10 minutes at 21°C in 5 mM NaCl, 20 mM Tris-HCl (pH 8), 0.025% DDM and 0.1 mM DTT. Substrate, (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine, was then added to the reaction mixture to 5 μM, and the rate of substrate hydrolysis measured as described above. The concentrations of NSC 37249 were 0, 0.05, 0.075, 0.1, 0.2, and 0.4 μM. The concentrations of pVIc were 1.25, 2.5, 5, 10, and 20 μM.. The initial rate data were entered into the SigmaPlot Kinetic Module and analyzed for various inhibition models. The best fitting model was mixed inhibition with a Ki of 0.15 μM.
2.10 Selectivity and specificity experiments with Trypsin and Papain
Trypsin, 20 pM, was incubated with increasing amounts of compound NSC 37249 for ten minutes at 21 °C in 25 mM HEPES (pH 7.5), 1 mM CaCl2. Enzyme activity was measured by monitoring the rate of substrate hydrolysis, 10 μM (Ile-Pro-Arg-NH)2-Rhodamine, as a function of time. The rates were then normalized to the rate in the absence of inhibitor and reported as % activity. A similar assay was performed with papain with the following exceptions: the concentration of papain was 0.18 μM, the assay buffer was 25 mM sodium acetate, pH 5, 1 mM DTT, and the substrate was 10 μM (Phe-Arg-NH)2-Rhodamine.
3. Results
3.1 The lead compound
A comparison of the structures of the inactive AVP structure and active AVP-pVIc complex structure revealed a number of differences which could be considered as targets for drug interactions, Fig 1. Inspection of the surface of the AVP-pVIc complex indicates that the N- and C-termini of pVIc are bound in pockets, Fig. 2B. When comparing these sites to those regions on the surface of the AVP structure, only the N-terminal binding pocket for pVIc, abbreviated NT-pocket, is fully formed, Fig. 2A. The C-terminal binding pocket for pVIc, abbreviated CT-pocket, therefore must be induced upon the binding of pVIc. We predicted that a drug binding in the NT-pocket would prevent the binding of pVIc to AVP and hence prevent the activation of AVP. To see if the NT-pocket is a legitimate druggable site, we mutated the N-terminal amino acid of pVIc to an Ala. The Kd for the binding of wild-type pVIc to AVP is 4.4 μM [19]; the Kd for the mutant is at least an order of magnitude higher. Therefore, this pocket is a legitimate site.
Figure 1.

Surface map of AVP reveals potential drug targets. More than 45% of the surface area of AVP contains potential targets for proteinase inhibitors. The molecular surface of AVP (white) with the amino acid residues involved in regulating enzyme activity shown in colors: Active site region (green), pVIc binding region (pink), DNA binding region (blue), residues common to pVIc or DNA binding (yellow), and remainder of the activation pathway (red).
Figure 2.

From target to inhibitor: A. Surface of AVP (turquoise) with NT-pocket colored in dark gray and the active site in red. B. Surface of the AVP-pVIc complex (yellow) with the CT-pocket colored in mauve. pVIc is colored beige. C. Surface of AVP with energetically favorable ligand atoms (spheres) colored in orange. D. Surface of the AVP-pVIc complex with energetically favorable ligand atoms (spheres) colored in green. E. Surface of AVP with the inhibitor NSC 36806 bound in the NT-pocket. F. Surface of the AVP-pVIc complex with the inhibitor NSC 36806 bound in the active site. G. The structure of the lead inhibitor NSC 36806, in ball and stick form with carbon colored green, nitrogen colored blue, oxygen colored red, sulfur colored yellow and hydrogens colored white. (C40H44N8O8S2): 2,21-dithia-7,10,13,16,26,29,32,35-octaazaheptacyclo-[34.2.2.2~3,6~.2~10,13~.2~17,20~.2~22,25~.2 ~29,32~] pentaconta-1(38),3,5,17,19,22,24,36,39,43,45,49-dodecaene-8,15,27,34-tetrone 2,2,21,21-tetraoxide.
Structure-based drug design on the NT-pocket in AVP yielded several hundred potential inhibitors. The highest energy score ranked hit identified by DOCK was the compound NSC 36806, Fig 2G. The favorable binding pose predicted in the NT-pocket is shown in Fig 2E and Fig 3A. At the same time, we also used DOCK to find ligands predicted to bind to the active site region of the AVP-pVIc complex, Fig. 2D. Here, using identical parameters for DOCKing, we noticed that NSC 36806 was among the top 90 hits. The predicted binding pose of NSC 36806 in the active site of the AVP-pVIc complex is shown in Fig. 2F and Fig. 3B.
Figure 3.

Predicted binding pose of NSC 36806 in the NT-pocket of AVP and in the active site of the AVP-pVIc complex. A. Binding of NSC 36806 in the NT-pocket of AVP. For NSC 36806, carbon is magenta, nitrogen is blue, sulfur is orange and oxygen is red. The surface in red contains amino acid residues that are within 5 Å of NSC 36806. B. Binding of NSC 36806 in the active site of the AVP-pVIc complex. For NSC 36806, carbon is yellow, nitrogen is blue, sulfur is orange and oxygen is red. Two amino acids involved in catalysis, C122 and H54, are colored green. The surface in white contains amino acid residues that are within 5 Å of NSC 36806.
We tested NSC 36806 as an inhibitor of AVP. AVP was incubated with different concentrations of the compound and then pVIc and (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine [3,13] were added and the rate of substrate hydrolysis determined. The data were complex and difficult to interpret, possibly because the compound was binding to more than one site on AVP (data not shown). We also tested NSC 36806 as an inhibitor of AVP-pVIc complexes. Here, we expected a more predictable inhibitor profile as the compound should not be able to bind to the NT-pocket We incubated different concentrations of the compound with the enzyme and then added the fluorogenic substrate (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine [3,13] and measured the rate of substrate hydrolysis. Clearly, the compound inhibited AVP-pVIc complexes, Fig. 4. The IC50 was 18 μM, Table 1. We tested NSC 36806 as a promiscuous inhibitor [20–23] and found it did not inhibit the activity of trypsin (data not shown).
Figure 4.
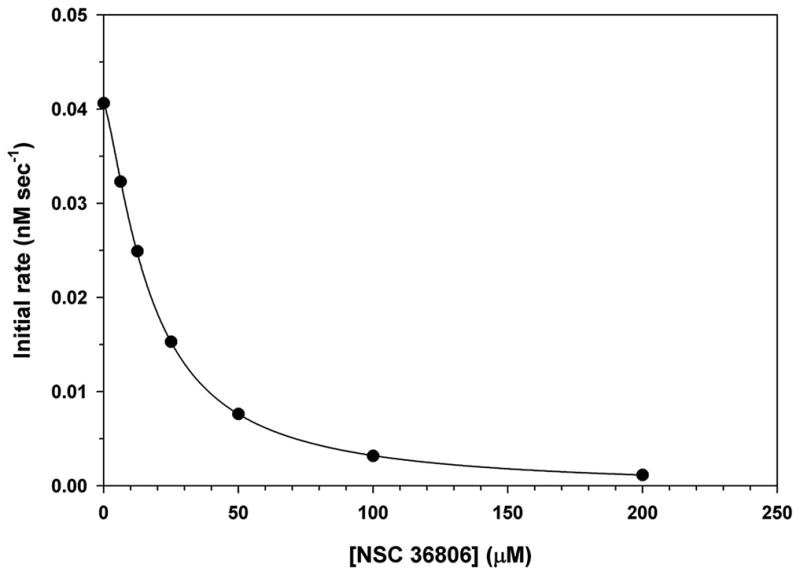
Inhibition of AVP-pVIc complexes by NSC 36806. AVP-pVIc complexes, 0.3 μM, were pre-incubated with increasing amounts of NCI 36806 for 9 minutes at 21°C in 5 mM NaCl, 20 mM Tris-HCl (pH 8), 0.025% DDM, and 0.1 mM DTT. Enzyme activity was measured by monitoring the rate of substrate hydrolysis, 5 μM (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine, as a function of time. The IC50,18 μM, was obtained by fitting the experimental initial rate data to a four-parameter logistic equation.
Table 1.
Testing of first and second generation inhibitors
| NSC 36806 | NSC 37248 | NSC 37249 | |
|---|---|---|---|
| AVP | IC50 = 0.88 μM | IC50 = 0.14 ± 0.06 μM | |
| AVP-pVIc | IC50 = 18 μM | IC50 = 2.1 μM | IC50 = 0.49 ± 0.076 μM |
| Trypsin | No Inhibition | No Inhibition | |
| Papain | No Inhibition | No Inhibition | |
| AVP:pVIc | Competitive Inhibition Ki = 403 nM |
||
| AVP | Mixed Inhibition Ki = 0.15 μM. |
The assays are described in Experimental Procedures
3.2 Structural derivative compounds
Because the DOCK pose of NSC 36806 indicated a significant portion of the compound was not interacting with AVP and, also, its molecular weight was slightly high, we initiated a substructure search of the NCI repository and found 8 compounds structurally related to NSC 36806. These compounds were not among the compounds used in the DOCKing analysis. Their structures are shown in Table 2; essentially all these compounds are derivatives of acetamidophenyl sulfonylphenylacetamides. We tested all 8 compounds for inhibition of AVP and of AVP-pVIc complexes. Four of the compounds were found to inhibit either the activation of AVP or the activity of AVP-pVIc complexes- NSC 37248, NSC 37249, NSC 106702, and NSC 106721. Two of the compounds, NSC 37248 and NSC 37249 were effective inhibitors.
Structurally, NSC 37249 is similar to NSC 36806. In NSC 36806, there are piperazine-based cyclic joinings of the two acetamidophenyl sulfonylphenyl acetamides, while in NSC 37249, each acetamide moiety is derivatized with cyclohexylcyclohexamine. Since NSC 36806 was predicted to bind to the NT-pocket in AVP and to the active site in AVP-pVIc complexes, one might expect NSC 37249 would bind to those same sites. The data, shown in Fig. 5B, are consistent with this prediction of two sites. The IC50 value with AVP (0.14 μM) is different from the IC50 value with AVP-pVIc complexes (0.49 μM), Table 1. Since NSC 37249 was predicted to bind only to the active site in AVP-pVIc complexes, one might predict NSC 37249 would be a competitive inhibitor. The data, presented in the form of a Dixon plot in Fig. 5C, indicate that NSC 37249 is indeed a competitive inhibitor of AVP-pVIc complexes with a Ki of 0.40 μM, Table 1. Similar sets of experiments were done with AVP, and the result indicated mixed inhibition with a Ki of 0.15 μM, Fig. 5D, Table 1.
Figure 5.

Assays of potential second generation proteinase inhibitors. A. Coarse screen of 8 NCI compounds whose structures are shown in Table 2. The inhibition assays are described in Experimental Procedures. All the initial rates of enzyme activity were normalized to the initial rates in the absence of inhibitor and plotted on the same graph for comparison. The control was the activity of AVP (gray) or AVP-pVIc complexes (black) in the absence of inhibitor. B. IC50 values for the inhibition of AVP and AVP-pVIc complexes by NSC 37249. Assays were performed as described in Experimental Procedures. The IC50 value for AVP was 0.14 μM, and for AVP-pVIc complexes it was 0.49 μM. AVP (▫) and AVP-pVIc complexes (○). C. Competitive inhibition of AVP-pVIc complexes by NSC 37249. AVP-pVIc complexes were incubated with the indicated concentrations of NSC 37249 and, after 9 min, four, different concentrations of substrate were added and enzyme activity assayed, as described in Experimental Procedures. The data are presented in a Dixon plot yielding a Ki of 0.403 μM. The concentrations of (Cbz-Leu-Arg-Gly-Gly-NH)2-Rhodamine were 2.5 μM (∙), 5 μM (▽), 10 μM (■), and 20 μM (◇). D. Dixon plot of NSC 37249 inhibition. Data used for graph are from assays where AVP was incubated simultaneously with NSC 37249 and cofactor pVIc. pVIc concentrations were 1.25 (•), 2.5 (△), 5 (■), 10 (◇), and 20 (⬢) μM. AVP activity assays were performed as described in Experimental Procedures. The experimental initial rates were entered into SigmaPlot Kinetic Module and analyzed for various inhibition models. The best model was mixed inhibition with a Ki of 0.15 μM.
How specific is NSC 37249 for AVP and AVP-pVIc complexes? We first determined whether NSC 37249 inhibited trypsin, the canonical serine proteinase. At concentrations of NSC 37249 up to 18 μM, concentrations that would have inhibited AVP and AVP-pVIc complexes, no inhibition was observed, Fig. 6. This result also indicates that NSC 37249 is not a promiscuous inhibitor, a compound that binds and aggregates most anything thereby generating false positives [20–23]. Next, we determined whether NSC 37249 inhibited the canonical cysteine proteinase papain. Again, at concentrations of NSC 37249 up to 50-fold the Ki for AVP, no inhibition was observed, Fig. 6.
Figure 6.

Selectivity of NSC 37249, assays with trypsin, the canonical serine proteinase and papain, the canonical cysteine proteinase. The inhibition assays are described in Experimental Procedures. The rates of substrate hydrolysis were normalized to the rate in the absence of inhibitor and are reported as % Activity. Trypsin (∙) and papain (□).
4. Discussion
During an adenovirus infection, two forms of AVP are present, one early and one late. AVP is initially synthesized in an inactive form. The NT-pocket, where the N-terminus of pVIc binds, is a druggable target. A compound binding there should prevent pVIc from binding and thus inhibit the activation of the enzyme. Parenthetically, the structure of the NT-pocket in AVP is the same in the AVP-pVIc complex. One reason AVP is inactive is that its active site is structurally different from that in AVP-pVIc, the active form of the enzyme. Thus, the active site of AVP was not considered for drugging. However, it is a druggable site in the AVP-pVIc complex. A drug binding in the active site of the AVP-pVIc complex may interfere with substrate binding thereby affecting the activity of the enzyme. Our docking studies revealed a compound predicted to bind in the NT-pocket in AVP and in the active site of the AVP-pVIc complex, and this compound inhibited the enzyme. Searches of structural variants of this lead inhibitor identified two other, even more effective, compounds. Each of these compounds is predicted to bind to two different sites, one in AVP and the other in the AV-pVIc complex. It may be that we have identified a compound that both inhibits a crucial step early in infection and a crucial step late in infection.
Compounds like NSC 36806 or derivatives of it are potentially unique antiviral agents in that the virus may have difficulty in becoming resistant to it. The two sites on the proteinase predicted to bind to NSC 36806, the NT-pocket and the active site, are at the ends of a 62 amino acid long activation path [5]. Because these two binding sites are quite far from each other, a minimum of two mutations may be required for a virus to become resistant. The probability of two independent mutations occurring at specific sites within 615 bases on the same DNA molecule is quite low. Secondly, because the two binding sites are connected by an activation path, a mutation leading to resistance at one site may affect the functioning of the activation path leading to the other site, thereby decreasing the probability of resistance arising even more [24,25]. This is not unlike combination therapy, but, instead of two compounds interacting with two, different sites, it is one compound interacting with two, different sites.
The apparent specificity of NSC 37249 was surprising. At concentrations 50-fold higher than that which would inhibit AVP or AVP-pVIc complexes by more than 90%, the compound did not inhibit trypsin or papain. This result was surprising as the crystal structure of AVP-pVIc complexes indicates the four amino acids involved in catalysis are in identical positions to those in papain [1,2]. However, in examining how NSC 36806 docked to the active site of the AVP-pVIc complex structure aligned to a papain structure active site, several regions within the active site groove predicted to interact with NSC 36806 in the adenovirus proteinase are absent in the papain active site, and this may be the origin of the apparent selectivity of NSC 37249.
Much more needs to be done in characterizing these inhibitors. Especially important would be the structures of co-crystals of inhibitors and enzymes to see if the binding poses are those predicted by DOCK and as an aide in the design of additional compounds for testing. At the very least, however, these molecules should provide a scaffold from which to build more specific second generation inhibitors.
Highlights.
Numerous surface regions of AVP were identified as potential druggable sites
A lead compound was identified and predicted to interact at two different druggable sites
Structural derivatives of the lead compound were 100-fold more effective against AVP
Structural derivatives exhibited selectivity in not inhibiting trypsin and papain
Acknowledgments
We thank Dr. R. Rizzo at Stony Brook University for access to his NCI database formatted for use with DOCK and for helpful discussions. We thank Jeff Aube at the University of Kansas for useful suggestions and discussions. †Research supported by the office of Biological and Environmental Research of the U.S. Department of Energy under Prime Contract DE-AC0298CH10886 with Brookhaven National Laboratory and by National Institutes of Health Grant AI R0141599 to W.F.M. K.Z. was supported by the U.S. Department of Energy, Office of Science, Office of Workforce Development for Teachers and Scientists (WDTS) under the Science Undergraduate Laboratory Internship (SULI) program.
Abbreviations
- AVP
adenovirus proteinase
- AVP-pVIc
covalent complex between AVP and pVIc
- pVI
the precursor to protein VI
- pVIc
11-amino acid peptide from the C-terminus of adenovirus precursor protein pVI
- NT-pocket
pocket on surface of AVP where N-terminal residues of pVIc interact with AVP
- CT-pocket
induced pocket on surface of AVP-pVIc complex where C-terminal residues of pVIc interact with AVP
- RFI
relative fluorescence intensity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ding J, McGrath WJ, Sweet RM, Mangel WF. Crystal structure of the human adenovirus proteinase with its 11 amino acid cofactor. EMBO J. 1996;15:1778–1783. [PMC free article] [PubMed] [Google Scholar]
- 2.McGrath WJ, Ding J, Sweet RM, Mangel WF. Crystallographic structure at 1.6-Å resolution of the human adenovirus proteinase in a covalent complex with its 11-amino-acid peptide cofactor: insights on a new fold. Biochem Biophys Acta. 2003;1648:1–11. doi: 10.1016/s1570-9639(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 3.Mangel WF, McGrath WJ, Toledo DL, Anderson CW. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature. 1993;361:274–275. doi: 10.1038/361274a0. [DOI] [PubMed] [Google Scholar]
- 4.Webster A, Hay RT, Kemp G. The adenovirus protease is activated by a virus-coded disulphide-linked peptide. Cell. 1993;72:97–104. doi: 10.1016/0092-8674(93)90053-s. [DOI] [PubMed] [Google Scholar]
- 5.Baniecki ML, McGrath WJ, Mangel WF. Regulation of a viral proteinase by a peptide and DNA in one-dimensional space. III Atomic Resolution Structure of the Nascent Form of the Adenovirus Proteinase. J Biol Chem. 2013;288:2081–2091. doi: 10.1074/jbc.M112.407429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krausslich HG, Wimmer E. Viral proteinases. Annu Rev Biochem. 1988;57:701–754. doi: 10.1146/annurev.bi.57.070188.003413. [DOI] [PubMed] [Google Scholar]
- 7.Dougherty WG, Semler BL. Microbiological Reviews. 1993. Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes; pp. 781–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kay J, Dunn BM. Viral proteinases: weakness in strength. Biochim Biophys Acta. 1990;1048:1–18. doi: 10.1016/0167-4781(90)90015-t. [DOI] [PubMed] [Google Scholar]
- 9.Patick AK, Potts KE. Protease inhibitors as antiviral agents. Clin Microbiol Rev. 1998;11:614–627. doi: 10.1128/cmr.11.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangel WF, Toledo DL, Brown MT, Martin JH, McGrath WJ. Characterization of three components of human adenovirus proteinase activity in vitro. J Biol Chem. 1996;271:536–543. doi: 10.1074/jbc.271.1.536. [DOI] [PubMed] [Google Scholar]
- 11.Riddles PS, Blakeley RL, Zerner B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)-a reexamination. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 12.Riddles PW, Blakeley RL, Zerner B. Reassessment of Ellman’s reagent. Methods Enzymol. 1983;91:49–60. doi: 10.1016/s0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 13.McGrath WJ, Abola AP, Toledo DL, Brown MT, Mangel WF. Characterization of human adenovirus proteinase activity in disrupted virus particles. Virology. 1996;217:131–138. doi: 10.1006/viro.1996.0100. [DOI] [PubMed] [Google Scholar]
- 14.Leytus SP, Melhado LL, Mangel WF. Rhodamine-based compounds as fluorogenic substrates for serine proteases. Biochem J. 1983;209:299–307. doi: 10.1042/bj2090299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leytus SP, Patterson WL, Mangel WF. New class of sensitive, specific, and selective substrates for serine proteinases: fluorogenic, amino acid peptide derivatives of Rhodamine. Biochem J. 1983;215:253–260. doi: 10.1042/bj2150253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McGrath WJ, Aherne KS, Mangel WF. In the virion, the 11-amino acid peptide cofactor pVIc is covalently linked to the adenovirus proteinase. Virology. 2002;296:234–240. doi: 10.1006/viro.2002.1394. [DOI] [PubMed] [Google Scholar]
- 17.McGrath WJ, Baniecki ML, Peters E, Green DT, Mangel WF. Roles of two conserved cysteine residues in the activation of human adenovirus proteinase. Biochemistry. 2001;40:14468–14474. doi: 10.1021/bi011562d. [DOI] [PubMed] [Google Scholar]
- 18.Graziano V, McGrath WJ, DeGruccio AM, Dunn JJ, Mangel WF. Enzymatic activity of the SARS coronavirus main proteinase dimer. FEBS Letters. 2006;580:2577–2583. doi: 10.1016/j.febslet.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baniecki ML, et al. Interaction of the human adenovirus proteinase with its 11- amino-acid cofactor pVIc. Biochemistry. 2001;40:12349–12356. doi: 10.1021/bi0109008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng BY, Shelat A, Doman TN, Guy RK, Shoichet BK. High-throughput assays for promiscuous inhibitors. Nature Chem Biol. 2005;1:146–148. doi: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- 21.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 22.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46:4265–72. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 23.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–22. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 24.Mangel WF, Brown MT, Baniecki ML, Barnard DL, McGrath WJ. Prevention of viral drug resistance by novel combination therapy. Current Opinion in Investigational Drugs. 2001;2:613–616. [PubMed] [Google Scholar]
- 25.Mangel WF, McGrath WJ, Brown MT, Baniecki ML, Barnard DL, Pang YP. A new form of antiviral combination therapy predicted to prevent resistance from arising, and a model system to test it. Curr Med Chem. 2001;8:933–939. doi: 10.2174/0929867013372742. [DOI] [PubMed] [Google Scholar]