

Abstract
Gold nanoparticles (AuNPs) were synthesized in situ in a green and rapid method from the reaction of reducing linear and cyclic peptides containing tryptophan and lysine residues, (KW)5 and cyclic [KW]5, with an aqueous solution of HAuCl4 and were evaluated as cellular nanodrug delivery systems. The cyclic or linear nature of the peptide was found to determine the morphology and size of the formed peptide-AuNPs and their in vitro molecular transporting efficiency. While cyclic [KW]5-AuNPs formed sponge-like agglomerates, linear (KW)5-AuNPs demonstrated ball-shaped structures. A comparative flow cytometry study showed that the cellular uptake of fluorescence-labeled anti-HIV drugs (emtricitabine (FTC) and lamivudine (3TC)) in human Leukemia (CCRF-CEM) cells, and a negatively charged cell-impermeable phosphopeptide (GpYEEI) in human ovarian adecarcinoma (SK-OV-3) cells was significantly higher in the presence of cyclic [KW]5-AuNPs than that of linear (KW)5-AuNPs, parent cyclic [KW]5, and linear (KW)5 peptides. For example, the cellular uptake of F′-GpYEEI was enhanced 12.8-fold by c[KW]5-AuNPs. Confocal microscopy revealed the localization of fluorescence-labeled-3TC in the presence of c[KW]5-AuNPs mostly in nucleus in SK-OV-3 cells after 1 h. On the other hand, l(KW)5-AuNPs delivered fluorescence-labeled-3TC in cytoplasm. These data suggest that non-cell penetrating peptides can be converted to efficient molecular transporters through peptide-capped AuNPs formation.
Keywords: Cellular Uptake, Cyclic Peptide, Drug Delivery, Gold Nanoparticles, Lysine, Nuclear Targeting, Tryptophan
Introduction
The cellular delivery of hydrophobic and poorly water-soluble compounds to enhance the efficacy using Drug delivery systems (DDS) is a subject of major interest.1 Among all delivery systems, nuclear targeting nanodrug delivery systems (nano-DDS) have attracted significant attention. Common approaches in gene therapy involve using DNA as a pharmaceutical agent to replace or correct a mutated gene.2 Moreover, the nucleus is a target for a diverse number of anticancer and antiviral drugs,3 due to the presence of the genetic information and transcription systems.
The nuclear membrane (envelope) is composed of a double lipid bilayer that works as a physical barrier between cell nucleus content and the cytoplasm. The nuclear membrane contains many nuclear pore complexes (NPCs) that facilitate and regulate the exchange of proteins and RNA between the cell nucleus and the cytoplasm. Depending on the cell line, NPCs have different sizes from 20 to 60 nm.4 Although NPCs operate as available passages for all exchange between the nucleoplasm and cytoplasm, they can be exploited as pathways for nanoparticle delivery.5,6 Therefore, the delivery of the therapeutic agents to the nucleus by using nano-DDS offers several advantages compared to other systems.
The application of prodrug strategy among researchers is now well established. Prodrugs are chemically modified analogous of the active metabolite that can improve pharmacokinetics and pharmacodynamic (PK/PD) properties of the active drug. However, intracellular chemical transformation needs to be occurred in the presence of different enzymes to convert prodrugs to their corresponding pharmacologically potent compounds in in vivo systems. Prodrug approach offers several advantageous, such as enhancing water solubility, improved chemical stability, decreased toxicity, and insufficient brain penetration.7
Gold nanoparticles (AuNPs) have received an increased attention due to their unique potential use as nano-DDS.8 Most of the known AuNPs as nano-DDS lead to the intracellular localization of nanoparticles mainly in the cytoplasm.9 However, appropriate functionalization of AuNPs with other compounds can be used for their optimization as nano-DDS for a specific application.
Peptide-mediated drug delivery has been widely used for a broad range of cargo molecules including drugs, siRNA, and genes due to their versatility and the presence of a wide range of amino acids.10 Thus, the peptides have the potential to be used for decoration of the surface of metal nanoparticles. Recently, AuNPs functionalized by peptides have been used as biocompatible systems in drug delivery.11 For example, AuNPs capped by linear Tat was reported to be able to get internalized into the cytoplasm of 3T3 and HepG2 cells.11c
After the localization of DDS in the destination, it should release the cargo. Thus, drug loading and release issues are critical for optimized DDS function. Two major strategies for drug loading include covalent and non-covalent binding between AuNPs and drugs.12 Non-covalent loading offers two main advantages, including ease of the drug loading and facilitating drug release in intact form over covalent loading.13 Thus, the entrapment of unmodified drugs into functionalized AuNPs through non-covalent interaction is introduced as one of the prodrug strategies for the delivery of a broad range of drugs. Employing appropriate surface functionalization provides a hydrophobic pocket for the loading of drugs.
We have previously reported cyclic peptides containing alternative arginine (R) and tryptophan (W) as efficient molecular transporters that showed improving the cellular uptake of different cargos.14 Subsequently, it was found that a physical mixture of the cell-penetrating cyclic peptide [WR]4 and HAuCl4 led to the formation of peptide-capped gold nanoparticles (P-AuNPs) and enhanced the cellular uptake of drugs significantly.15 It remains to be determined whether non-cell penetrating peptides can be converted to a nuclear targeting DDS through generation of peptide-AuNPs.
Herein, we report the generation of a novel nuclear-targeting nano-DDS containing AuNPs and a non-cell penetrating peptide containing alternative lysine and tryptophan residues. The presence of these amino acids was found to be appropriate to generate in situ biocompatible AuNPs. To the best of our knowledge, this is the first report of converting a non-cell penetrating peptide to an efficient nuclear targeting nano-DDS through cyclic peptide-capped AuNPs formation.
Experimental Section
General
All reactions were carried out in Bio-Rad polypropylene columns by shaking and mixing using either a Glass-Col small tube rotator or PS3 automated peptide synthesizer in dry conditions at room temperature unless otherwise stated. In general, all cyclic and linear peptides were synthesized by the solid-phase synthesis strategy employing N-(9-fluorenyl)methoxycarbonyl (Fmoc)-based chemistry and Fmoc-L-amino acid building blocks. 2-(1H–Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) were used as coupling and activating reagents, respectively. 2-Chlorotrityl chloride resin, Fmoc-amino acid trityl chloride resins, coupling reagents, and Fmoc-amino acid building blocks were purchased from Chempep (Miami, FL). Other chemicals and reagents were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI). Fmoc deprotection at each step was carried out using piperidine in DMF (20%). Side chain protected peptides were cleaved from the resins by shaking the resins with a mixture of trifluoroethanol (TFE)/acetic acid/dichloromethane (DCM) (1:2:7 v/v/v, 15 ml) for 2 h. The resins were filtered off, and the liquid was evaporated to dryness to get side-chain protected linear peptide. Generally, the cyclization of the peptides was carried out in the presence of a mixture of 1-hydroxy-7-azabenzotriazole (HOAt) and N,N′-diisopropylcarbodiimide (DIC) in dry DMF and dry dichloromethane (DCM) for 24 h. DMF and DCM were evaporated. The side chain deprotection was carried with TFA/thioanisole/anisole/1,2-ethanedithiol (EDT) (90:5:2:3 v/v/v/v) for 2 h. The crude peptides were precipitated by the addition of cold diethyl ether (Et2O) and were purified by using a reversed-phase Hitachi HPLC (L-2455) on a Phenomenex Prodigy 10 μm ODS reversed-phase column (2.1 cm × 25 cm) and a gradient system. The peptides were separated by eluting the crude peptides at 10.0 mL/min using a gradient of 0-100% acetonitrile (0.1% TFA) and water (0.1% TFA) over 60 min, and then were lyophilized to yield cyclic peptides (Figure 1). The purity of final products (≥95%) was confirmed by analytical HPLC. The analytical HPLC was performed on a Hitachi analytical HPLC system using a C18 Shimadzu Premier 3 μm column (150 cm × 4.6 mm) and a gradient system (water/CH3CN), and a flow rate of 1 mL/min with detection at 220 nm. The chemical structures of final products were confirmed by high-resolution MALDI AXIMA performance TOF/TOF mass spectrometer (Shimadzu Biotech) or a high-resolution Biosystems QStar Elite time-of-flight electrospray mass spectrometer. As a representative example, the synthesis of linear (KW)4 is outlined here.
Figure 1.

Chemical structures of synthesized peptides.
Synthesis of Linear (KW)4 and Linear (KW)5
Fmoc-Trp(Boc)-Wang resin (555 mg, 0.54 mmol/g, 0.3 mmol) was swelled in anhydrous DMF for about 15 min under dry nitrogen. The excess of the solvent was filtered off. The swelling and filtration steps were repeated for 2 more times before the coupling reactions. Fmoc-Lys(Boc)-OH (325 mg, 0.75 mmol) was coupled to the N-terminal of tryptophan Wang resin in the presence of HBTU (285 mg, 0.75 mmol) and DIPEA (262 μL, 1.50 mmol) in DMF (7 mL) by mixing for 1.5 h. After the coupling was completed, the reaction solution was filtered off, and the resin was collected by filtration and washed with DMF (7 × 15 mL), followed by N-terminal Fmoc deprotection using piperidine in DMF (20% v/v, 10 mL, 2 times, 5 and 10 min). The resin was washed with DMF (7 × 15 mL). The subsequent amino acids, Fmoc-Trp(Boc)-OH (395 mg, 0.75 mmol) and Fmoc-Lys(Boc)-OH (325 mg, 0.75 mmol), were coupled alternatively three times, respectively, in a similar manner (Scheme S1). Fmoc-deprotection at the N-terminal was carried out in the presence of piperidine in DMF (20% v/v, 10 mL, 2 × 10 min) to afford the linear peptide on solid phase. The resin was washed with DMF and DCM respectively (each 2 × 25 mL). The resins were dried under vacuum for 24 h. Fresh cleavage cocktail, reagent R, TFA/thioanisole/EDT/anisole (90:5:3:2 v/v/v/v, 10 mL), was added to the resins. The mixture was shaken at room temperature for 2 h. The resin was collected by filtration and washed with another 2 mL of cleavage cocktail. Combined filtrates were evaporated to a minimum volume under dry nitrogen. The crude peptide was precipitated by the addition of cold diethyl ether (75 mL, Et2O), lyophilized, and purified by reversed-phase Hitachi HPLC (L-2455) on a Phenomenex Prodigy 10 μm ODS reversed-phase column (2.1 cm × 25 cm) using a gradient system as described above to yield linear (KW)4. MALDI-TOF (m/z) [C68H90N16O9]: calcd, 1274.7077; found, 1275.1780 [M + H]+, 1297.1747 [M + Na]+. A simliar procedure was performed for the synthesis of the linear (KW)5. l(KW)5: MALDI-TOF (m/z) [C85H112N20O11]: calcd, 1588.8819; found, 1589.7442 [M + H]+, 1611.7890 [M + Na]+, 1627.7601 [M + K]+.
Synthesis of Cyclic Peptides
The synthesis of cyclic peptides was carried out according to the previously reported procedure.14,16 As a representative example, the synthesis of cyclic [KW]4 is described here.
Cyclic [KW]4
The linear protected peptide (KWKWKWKW) was assembled on the H-Trp(Boc)-2-chlorotrityl chloride resin (975 mg, 0.41 mmol/g) in 0.40 mmol scale as described above. After the final deprotection of the N-terminal Fmoc group by piperidine in DMF (20% v/v, 15 mL, 2 times, 5 and 10 min), the side chain protected peptide was cleaved from assembled trityl resin in the presence of the cleavage cocktail TFE/acetic acid/DCM (1:2:7 v/v/v, 50 mL) by shaking for 1 h at room temperature to yield the side chain protected linear peptide. The filtrates were evaporated to dryness under reduced pressure. To the residue was added hexane (2 × 25 mL) and DCM (1 × 25 mL) to remove the acetic acid from the mixture and to solidify the crude material as white solid. The crude protected peptide was dried under vacuum overnight. Examining a small portion of the crude by MALDI analysis confirmed the formation of the linear protected peptide. Thus, the crude was used directly for the cyclization reaction. The dried crude linear protected peptide was dissolved in DMF/DCM (2:1 v/v, 150 mL). HOAt (135 mg, 1 mmol) and DIC (137 μL, 0.88 mmol) were added to the mixture, and the solution was stirred for 12 h under the nitrogen atmosphere. After completion of the cyclization as confirmed by the MALDI-TOF, the solvents were removed under reduced pressure on a rotary evaporator. The crude product was dried overnight in vacuum before the final cleavage. Freshly prepared cleavage cocktail, reagent R, TFA/thioanisole/EDT/anisole (90:5:3:2 v/v/v/v, 15 mL) was added to the crude product. The mixture was stirred at room temperature for 2 h. The cleavage cocktail was concentrated to a minimum volume under reduced pressure by a rotary evaporator. After precipitation of crude peptide in cold diethyl ether (75 mL, Et2O) and centrifugation, the crude peptide was lyophilized and purified by reversed-phase Hitachi HPLC (L-2455) on a Phenomenex Prodigy 10 μm ODS reversed-phase column (2.1 cm × 25 cm) using a gradient system to yield cyclic [KW]4. A similar procedure was employed for the synthesis of other cyclic peptides by using appropriate resins and protected amino acids. The mass spectra data for c[KW]5, c[KF]4, c[KA]4, c[RFEF]2, and c[KE]4 have been previously reported by us.16
c[KW]4
MALDI-TOF (m/z) [C68H88N16O8]: calcd, 1256.6971; found, 1257.2083 [M + H]+, 1281.2555 [M + H + Na]+, 1295.2420 [M + K]+; C[EL]4: HR-MS (ESI-TOF) (m/z): C44H72N8O16 calcd, 968.5066 ; found, 969.4220 [M + H]+, 485.2551 [M + 2H]2+; C[RF]4: HR-MS (ESI-TOF) (m/z): C60H84N20O8: calcd, 1212.6781; found, 1212.6052 [M + H]+, 607.3026 [M + 2H]2+, 405.5428 [M+ 3H]3+, 304.1588 [M + 4H]4+; C[RC]4: HR-MS (ESI-TOF) (m/z): C36H68N20O8S4 calcd, 1036.4412; found, 1037.4096 [M + H]+, 345.1323 [M + 3H]3+, 259.1024 [M + 4H]4+.
Synthesis of Fluorescence Labeled-βA-Cyclic[KWKWKWKWKW] (F′- [KW]5)
The linear peptide (Dde-K(Fmoc)-WKWKWKWKW) was assembled on solid phase using Fmoc/tBu solid phase methodology. Appropriately protected amino acids were assembled on H-Trp(Boc)2 chlorotrityl resin (513 mg, 0.40 mmol, 0.78 mmol/g) according to the solid-phase synthesis strategy described above. Dde-Lys(Fmoc)-OH was used at the N-terminal of the peptide. The Fmoc protecting group was removed from the side chain of lysine using piperidine in DMF (20%, 10 mL, 2 times, 5 and 10 min) and a short linker Fmoc-β-alanine (312 mg, 1 mmol) was coupled with the unprotected side chain amine group of lysine using HBTU/DIPEA (380 mg, 1 mmol/349 uL, 2 mmol) in DMF (8 mL). Fmoc of β-alanine was removed in the presence of piperidine in DMF (20%, 10 mL, 2 times, 5 and 10 min) to attach carboxyfluorescein. 5(6)-Carboxyfluorescein diisobutyrate (CFDI, US Biological) (516 mg, 1 mmol) was used for labeling the peptide. This was achieved by adding resin-bound linear peptide (0.4 mmol) into a solution of CFDI (516 mg, 1 mmol), azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 520 mg, 1 mmol), HOAT (135 mg, 1 mmol), and DIPEA (350 μL, 2 mmol) in dry DMF (12.0 mL). The mixture was shaken for 3 h at room temperature and then the solvents were filtered off. The resin was washed with DMF (3 × 10 mL). The ester group of carboxyfluorescein was deprotected with 20% piperidine (20 mL) for 30 min. The N-terminal Dde protection was deprotected by treating the resin-bound peptide with hydrazine hydrate in DMF (2% v/v, 20 mL, 2 × 5 min). Finally, the side chain protected peptide was cleaved from the resin by shaking the resins with a mixture of TFE/acetic acid/DCM (15 mL, 1:2:7 v/v/v) for 1 h. The resin was filtered off, and the solution was evaporated to dryness under reduced pressure to yield side-chain protected linear peptide. The cyclization and purification was carried out according the described general method above used for other cyclic peptides to yield fluorescence-labeled cyclic [KW]5. MALDI-TOF (m/z) [C109H127N21O17]: calcd, 2001.9719; found, 2000.9316 [M]+, 2023.0032 [M + Na]+, 2038.9458[M + K]+.
Synthesis of Fluorescence Labeled-βA-Linear(KWKWKWKWKW) (F′-(KW)5)
The labeled linear sequence was assembled on H-Trp(Boc)-2 chlorotrityl resin (513 mg, 0.40 mmol, 0.78 mmol/g). The synthesis of the fluorescence-labeled side chain-protected linear peptide was carried out according to the similar procedure that described above. The peptide was assembled on resin using Dde-Lys(Fmoc)-OH at N-terminal. The Fmoc was removed in the presence of piperidine (20%, DMF). After the coupling of β-alanine and carboxyfluorscein in the side chain, the Dde protection at the N-terminal was removed by reacting with 2% hydrazine hydrate in DMF followed by washing with DMF. Then, the linear peptide was removed from the resin by using TFA/thioanisole/anisole/EDT (15 mL, 90:5:2:3 v/v/v/v) for 2 h to obtain the crude product. The crude peptide was precipitated by the addition of cold diethyl ether (75 mL, Et2O). Reversed-phase Hitachi HPLC (L-2455) and the gradient system described above were used for the purification of the peptide. MALDI-TOF (m/z) [C109H129N21O18]: calcd, 2019.9824; found, 2019.8717 [M]+, 2042.8717 [M + Na]+, 2058.8717 [M + K]+.
HAuCl4 Reduction By Peptides Dissolved In DMSO/Water Mixtures
The peptides were dissolved in 1 mL of deionized water in order to prepare a stock solution of 1 mM. The stock solution (1 mL, 1 mM) was physically mixed with HAuCl4 solution (1 mL, 1 mM) in deionized water to obtain AuNPs. The final concentrations of gold and peptides were 500 μM. The formation of purple AuNPs was observed by the apparent change in the color of solution and confirmed by UV-Vis spectroscopy.
UV-Vis Spectroscopy Studies
The formation of P-AuNPs was studied in the presence of different peptides. In general, a peptide stock solution (1 mM) was mixed with an aqueous solution of HAuCl4 (1 mM) at room temperature. The color of solution turned red after 1 h due to the formation of peptide capped-AuNPs. UV-Vis spectroscopy study was carried out using HAuCl4 (1 mM) and different cyclic peptides (1 mM) using 96-well plate, and the absorbance was read using SpectraMax M2 spectrophotometer (Molecular Devices, CA) (Figure S1). The visible range was chosen because of the characteristic maxima peak of AuNPs appeared around 520-560 nm. All experiments were performed in triplicate.
Encapsulation of Camptothecin (CPT)
CPT was dissolved in DMSO, and 10 μL of 10-3 M camptothecin solution was mixed with 100 μL of 10-4 M aqueous peptide solution in a 1:1 molar ratio. CPT in DMSO was mixed with water as a negative control. Mixed solution was incubated in a 96 well plate at room temperature for 4 h. Fluorescence of the solution was measured using SpectraMax M2 fluorescence spectrophotometer (Molecular Devices, CA) with excitation at 290 nm and emission at 360 nm.
Drug Loading
Doxorubicin (Dox) aqueous solution (100 μL, 100 μM) was added into [KW]5-AuNPs solution (900 μL, 500 μM) with the concentration ratio of 1:5 for Dox to peptide-capped AuNPs. The mixture was transferred into a 1 mL dialysis membrane (with the cutoff molecular weight 1000 D; Float-A-Lyzer G2, Spectrum Labs). The membrane was sealed and submerged into water (500 mL) as a medium. After stirring in dark for 24 h, unloaded Dox in the mixture was collected and evaporated to reduce the volume. The concentration of Dox was measured using a reverse phase HPLC system installed with Hitachi premier C18 column (3 μm, 4.6 × 150 mm) at UV/Vis 490 nm wavelength. Mobile phases were water (0.1% trifluoroacetic acid) and acetronitrile (0.1% trifluoroacetic acid). The gradient of 10% to 90% acetonitrile was applied to separate the Dox peak from other impurities. The calibration standard solutions were injected to quantify Dox in the sample solution.
The loading efficiency was calculated using the following equation:
To measure the loading capacity, the amount of the AuNPs in the dialysis membrane was measured after 24 h by using ICP-MS. The ICP-MS results showed the amount of AuNPs were responsible for the encapsulation of Dox. Thus, the loading capacity was calculated based on the following equation.
Transmission Electron Microscopy (TEM)
TEM analyses were conducted in JEOL Transmission Electron Microscope (Tokyo, Japan) at an accelerating voltage 80 keV. The stock solution (1 mL, 1 mM) was physically mixed with HAuCl4 solution (1 mL, 1 mM) in deionized water to obtain AuNPs. The final concentrations of gold and peptides were 500 μM. The mixture was incubated for 12 days. TEM samples of c[KW]5-AuNPs and l(KW)5-AuNPs were prepared by depositing a droplet of 5 μL of 0.5 mM solution in H2O on a carbon-coated Cu support grid of mesh size 300 that were dried in air overnight.
Cell Culture
Human leukemia cell line CCRF-CEM (ATCC no. CCL-119), ovarian carcinoma SK-OV-3 (ATCC no HTB-77), and colon myofibroblasts CCD-18Co (ATCC no. CRL-1459) were obtained from American Type Culture Collection. Cells were grown on 75 cm2 cell culture flasks with RPMI-16 medium (for leukemia cells) and EMEM medium (for SK-OV-3 and CCD-18Co cells), supplemented with 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin solution (10,000 units of penicillin and 10 mg of streptomycin in 0.9 % NaCl) in a humidified atmosphere of 5% CO2, 95% air at 37 °C.
Cytotoxicity Assay
SK-OV-3, CCRF-CEM, and CCD-18Co cells were seeded at 5,000 cells, 30,000 cells, and 3,000 cells, respectively, in 0.1 ml per well in 96-well plates 24 h prior to the experiment. The old medium (EMEM containing FBS (10%)) was replaced (not in case of CCRFCEM) by different concentrations of c[KW]5, l(KW)5, c[KW]5-AuNPs or l(KW)5-AuNPs in serum containing medium and incubated for 24 h (or 72 h) at 37 °C in a humidified atmosphere of 5% CO2. Cell viability was then determined by measuring the fluorescence intensity at 490 nm using a SpectraMax M2 microplate spectrophotometer. The percentage of cell survival was calculated as [(OD value of cells treated with the test mixture of compounds) – (OD value of culture medium)]/[(OD value of control cells) – (OD value of culture medium)] × 100% (Figure S2).
Flow Cytometry Studies
Human leukemia and ovarian carcinoma cells (CCRF-CEM and SK-OV-3) were taken in 6-well plates (1 × 107 cells/well (CCERF-CEM) and 3 × 105 cells/well (SK-OV-3)) in opti-MEM. Then the fluorescence-labeled compound (F′-3TC, F′-FTC, or F′-GpYEEI) (5 μM) was added to the different wells containing c[KW]5-AuNPs (25 μM), l(KW)5-AuNPs (25 μM), l(KW)5 (25 μM), or c[KW]5 (25 μM) in serum-free media. The plates were incubated for 1 h at 37 °C. Cells, P-AuNPs, and the fluorescence-labeled compound alone were used as negative controls. After 1 h incubation, the media containing the peptide was removed. The cells were digested with 0.25% trypsin/EDTA (0.53 mM) for 5 min to remove any artificial surface binding. Then the cells were washed twice with PBS. Finally, the cells were resuspended in flow cytometry buffer and analyzed by flow cytometry (FACSCalibur™:Becton Dickinson) using FITC channel and CellQuest software. The data presented were based on the mean fluorescence signal for 10,000 cells collected. All assays were performed in triplicate.
In the case of fluorescence-labeled peptide gold nanoparticles (F′-Peptide-AuNPs) and their corresponding peptides (F′-Peptide), SK-OV-3 cells were seeded in 6-well plates (3 × 105 cells/well) in opti-MEM. The F′-labeled compound (F′-c[KW]5, F′-l(KW)5, F′-c[KW]5-AuNPs, or F′-l(KW)5-AuNPs (5 μM) was added to the various wells. The plates were incubated for 1 h at 37 °C. Wells containing no treatment and 5-(6) carboxyfluorescein (FAM) were used as negative controls. After 1 h incubation, the medium was removed. The cells were digested with 0.25% trypsin/EDTA (0.53 mM) for 5 min to remove any artificial surface association and to detect only intracellular uptake. Then the cells were washed twice with PBS. Finally, the cells were resuspended in flow cytometry buffer and analyzed by flow cytometry (FACSCalibur: Becton Dickinson) using FL2 channel and CellQuest software. The data presented are based on the mean fluorescence signal for 10,000 cells collected. All assays were performed in triplicate.
Cellular Uptake Studies In The Presence of Inhibitors
Human ovarian adenocarcinoma cells (SK-OV-3) were seeded in six well plates (3 × 105 cells/well) in opti-MEM. The cells were pre-incubated by various inhibitors including, nystatin (50 μg/ml), chloroquine (100 μM), chlorpromazine (30 μM), methyl-β-cyclodextrin (2.5 mM), and 5-(N-ethyl-N-isopropyl)amiloride (EIA, 50 μM) for 30 min. The treatment was removed, and the cells were incubated with F′-l(KW)5-AuNPs or F′-c[KW]5-AuNPs (5 μM) a similar concentration of inhibitors for 1 h. Consequently, similar FACS protocol was performed as described above.
Membrane Integrity Test
Ovarian carcinoma cells SK-OV-3 were grown in six well plates (1 × 105 cells/well). Cells were treated with [KW]5-AuNPs (50 μM) and (KW)5-AuNPs (50 μM) in serum-free media for 1 h at 37 °C. After 1 h incubation, the cells were detached with trypsinization for 5 min followed by washing twice with PBS. Then they were resuspended in serum-free media. Trypan blue (2 μL) was added into the cell suspension (18 μL) and incubated for 5 min. Finally, trypan bluestained cells were counted using cellometer vision (Nexcelom Bioscience, Lawrence, MA, USA) (Figure S3).
Confocal Microscopy on Live Cells
Adherent SK-OV-3 cells were seeded with EMEM media overnight on coverslips in six well plates (1×105 cells per well). Then, the media were removed and washed with opti-MEM. The cells were treated with the mixture of F′-3TC (5 μM) and c[KW]5-AuNPs (25 μM), and the mixture of F′-3TC (5 μM) and l(KW)5-AuNPs (25 μM) in opti-MEM for 1 h at 37 °C. After 1 h incubation, the media containing the treatments were removed followed by washing with PBS three times. To stain the nuclei, cells were incubated with 4,6-diamidino-2-phenylindole (DAPI, 0.5 mL) solution in PBS for 5 min. After the incubation, the cells were softly washed twice with PBS to remove the excessive DAPI. The coverslips were mounted on a microscope slide with mounting media with cells-attached side facing down. Laser scanning confocal microscopy was carried out using Carl Zeiss LSM 700 system. The cells were imaged using FITC and phase contrast channels. The fluorescence images were taken under 20 × objective. Blue and green luminescent emissions from DAPI and FITC were excited at the wavelength of 405 nm and 488 nm, respectively. The emission wavelengths were ranged from 425 nm to 475 nm for DAPI and 500 nm to 550 nm for FITC. There was no interference between these two channels. The scanning mode was in sequential frame.
Time-Dependent Antiproliferative Assay
The antiproliferative activity of CPT alone and in the presence of c[KW]5-AuNPs against SK-OV-3 cells was determined by MTS assay. All cells were plated overnight in 96-well plates with a density of 5000 cells per well in 0.1 mL of appropriate growth medium at 37 °C. CPT alone (5 μM) or a combination of CPT (5 μM) and c[KW]5-AuNPs (25 μM) were incubated with the cells for 4 h. Excess of compounds was removed and washed by fresh media. The cells were kept in an incubator for 24–72 h. The cells without compounds were included in each experiment as controls. After 24 h, 48 h, and 72 h incubation, 20 μL of MTS solution was added and incubated for 2 h. The absorbance of the formazan product was measured at 490 nm using microplate reader. The percentage of cell viability was calculated as (OD value of untreated cells - OD value of treated cells)/OD value of untreated cells × 100%.
Intracellular Release of Dox
Intracellular release and accumulation of Dox were determined in CCRF-CEM cells by HPLC analysis. CCRF-CEM cells were grown in 75 cm2 culture flasks with RPMI medium (containing 10% FBS and 1% penicillin-streptomycin) to achieve ∼70–80% confluence (1.2 × 107 cells/mL). The cells were partitioned/transferred to culture plates (six well) having 1.2 × 107 cells per well in 2 mL of medium. The treatment in fresh RPMI medium containing Dox-loaded c[KW]5-AuNPs (7.5/37.5 μM) was added to cells, and they were incubated at 37 °C for different incubation times including 2, 12, 24, and 48 h. After the incubation time, the cells were collected by centrifugation. The medium was removed and cell pellets were washed with ice-cold PBS (5 mL) twice to remove any medium. The cell pellets were thoroughly extracted with an equal volume of methanol, chloroform, and isopropanol mixture (4:3:1 v/v/v) and filtered through 0.2 μm filters. The solvents were evaporated under N2 gas. The released Dox was quantified by reverse phase HPLC system with UV/Vis detector (490 nm) (Figures S4-S9, Supporting Information). The HPLC condition was using mobile phase water (0.1% trifluoroacetic acid) and acetronitrile (0.1% trifluoroacetic acid) and the gradient of 10% to 90% acetonitrile as described above in drug loading section.
Results and Discussion
Chemistry
A number of cyclic octapeptides containing diverse combination of amino acids including c[KW]4, c[KF]4, c[KA]4, c[EL]4, c[RFEF]2, c[EK]4, c[RF]4, and c[RC]4 (Figure S1) were synthesized by employing 9-fluorenylmethyoxycarbonyl (Fmoc)-based chemistry according to the previously reported procedure.14,16 Synthesis of c[KW]4 and l[KW]4 is described in Supporting Information (Schemes S1 and S2).
Fluorescently-labeled conjugates of linear (KW)5 and cyclic [KW]5 were synthesized as described previously.14 For example, F′-l(KW)5 was synthesized by the Fmoc protection group at the N-terminal was removed. Subsequent conjugation reaction with 5(6)-carboxyfluorescein isobutyrate was performed in the presence of HOAt, PyAOP, and N,N-diisopropylethylamine (DIPEA) in DMF:DCM followed by deprotection and cleavage from the resin in the presence of reagent R afforded the crude product that was purified by preparative reversed-phase HPLC (Scheme S1). The structures of all the final compounds were confirmed by high-resolution MALDI TOF/TOF and/or high-resolution electrospray mass spectrometry.
Evaluation of Cyclic and Linear Peptides for Generation of Peptide-AuNPs and Characterization
All peptides were examined for their ability to generate AuNPs by direct addition of the peptide solution (1 mM) into an aqueous solution of HAuCl4 (1 mM) at room temperature. UV-Vis spectra and monitoring of surface plasmon resonance peak at ∼550 nm showed that among all peptides, c[KW]4 and the corresponding linear peptide l(KW)4 were able to produce peptide capped AuNPs (P-AuNPs) (Figure S1).
Mechanistically, c[KW]4 acts as reducing and capping agent, making this method a one-step (one-pot) reaction without any need for chemical modification on the surface of AuNPs. Amino acids have been used as reducing agents. Among them, tryptophan (W) has been shown to be the most efficient reducing agents among 20 amino acids.17 Addition of positively charged residues, such as lysine (K) in the structure of the peptide enhances the reduction through the favorable charge interactions with the chloroaurate anions.18
To optimize the yield of P-AuNPs by increasing the reducing and capping efficiency of the peptide, cyclic c[KW]5 containing ten alternative tryptophan and lysine (Figure 1) was synthesized. The corresponding linear peptide l(KW)5 (Figure 1) was also designed and synthesized to determine the effect of the peptide cyclic nature of the peptide on the shape and size of P-AuNPs. UV-Vis spectra of both c[KW]5-AuNPs and l(KW)5-AuNPs showed also a SPR peak at approximately 550 nm.
Further studies were conducted by Transmission electron microscopy (TEM) to characterize the size and shape of P-AuNPs. TEM images of c[KW]5-AuNPs and l(KW)5-AuNPs showed that these two systems formed entirely different nanosized structures. c[KW]5-AuNPs formed sponge-like agglomerates containing peptide and AuNPs containing peptide and AuNPs with an approximate diameter size of 250-450 nm (Figure 2) along with some isolated AuNPs in mostly spherical and triangular shapes (6-60 nm). Compared to cyclic peptide-capped AuNPs, TEM images of 1(KW)5-AuNPs demonstrated ball-shaped structures with the approximate diameter size of 900-1000 nm composed of a large number of AuNPs (Figure 3). The majority of individual l(KW)5-AuNPs were found to be spherical and approximately in the size range of 4-35 nm. The distribution pattern of AuNPs in the ball-shape structure of l(KW)5-AuNPs showed an organized arrangement (Figure 3). No visible peptide agglomerates were observed. Cavities between AuNPs were found to be tailored in a similar range size (approximately 45 ± 2 nm). The presence of the tryptophan in the structure of the linear peptide induces presumably hydrophobic interactions that could be an efficient driving force for the formation of this specific structure. c[KW]5 and l(KW)5 alone did not form any specific nanostructures under similar conditions, suggesting that the binding between AuNPs and peptides contributes to different morphology in P-AuNPs.
Figure 2.

TEM images of c[KW]5-AuNPs.
Figure 3.
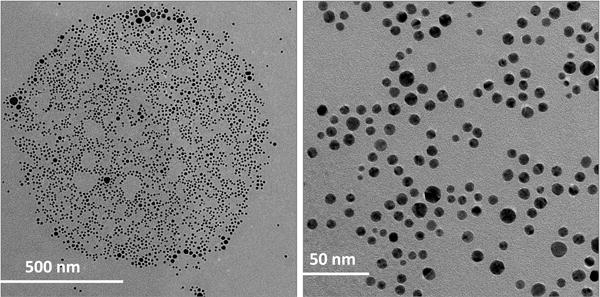
TEM images of l(KW)5-AuNPs.
These data showed that the cyclic nature and rigidity of the peptide in c[KW]5-AuNPs and different orientation of amino acids in the skeleton of peptide generated a different environment surrounding the complex that led to aggregation or agglomeration of the peptides. This orientation altered the number of available tryptophan and/or lysine residues in reduction of Au3+ reaction.18 However, l(KW)5 has more flexible conformation compared to the corresponding conformationally constrained cyclic counterpart c[KW]5 during the AuNPs formation. The presence of additional positively charge amino group and negatively charge carboxylate at N-terminal lysine and C-terminal tryptophan, respectively, could also contribute in intermolecular interactions of the peptide. Thus, the cyclic and linear nature of the peptide and the orientation of amino acids were found to be responsible for the different morphology and size of formed P-AuNPs.
Circular dichroism (CD) was used to obtain a better understanding about the effect of AuNPs formation on the secondary structure of these peptides. CD experiment of aqueous solution of peptides and their corresponding P-AuNPs (100 μM) was performed at room temperature. CD spectra of c[KW]5 (Figure 4) revealed a negative band at approximately 216 nm and a positive band around 230 nm. Compared to c[KW]5, the linear counterpart l(KW)5 showed a different CD spectra pattern with a negative band at 201 nm and a positive band at 227 nm. However, the CD spectra of both c[KW]5-AuNPs and l(KW)5-AuNPs exhibited relatively similar pattern with a minima at 204 ± 2 nm and a maxima at maxima at 229 ± 1 nm. Although two P-AuNPs showed approximately similar pattern and intensity, l(KW)5-AuNPs (206 nm) minima was shifted compared to that of c[KW]5-AuNPs (203 nm). The CD spectra demonstrated a significant decrease in spectral elipticity of both P-AuNPs compared to their corresponding parent peptides, suggesting significant modification of secondary structures upon reducing Au3+ and/or binding to AuNPs. These data show the binding between peptides and AuNPs leads to the formation of c[KW]5-AuNPs and l(KW)5-AuNPs with differential CD spectra pattern compared to the parent peptides.
Figure 4.

Comparative CD of cyclic c[KW]5, linear l(KW)5, and compared to c[KW]5-AuNPs and l(KW)5-AuNPs.
Encapsulation of Camptothecin By Peptide-AuNPs
To evaluate the potential of c[KW]5-AuNPs and l(KW)5-AuNPs to encapsulate drugs, camptothecin (CPT), a hydrophobic potent anticancer drug targeting topoisomerase I19 was selected as a model drug for evaluation of loading using fluorescence spectroscopy. Incubation of CPT with c[KW]5-AuNPs and l(KW)5-AuNPs demonstrated significant shift in emission maxima of CPT. A characteristic maxima band of CPT alone at 458 nm was shifted to 433 nm and 430 nm in the presence of c[KW]5-AuNPs and l(KW)5-AuNPs, respectively (Figure S10). The spectral blue shift in the presence of c[KW]5-AuNPs and l(KW)5-AuNPs was possibly due to partitioning of CPT into a hydrophobic core and hydrophobic interactions. Moreover, the intensity of the maxima band was decreased that could be due to the selfquenching of bound-drug and/or the result of the partitioning and entrapment in the hydrophobic pocket generated by the P-AuNPs.20 These data exhibited that the P-AuNPs were able to encapsulate CPT due to the generated hydrophobic pocket possibly by tryptophan in the structure of the peptide.
Drug Loading
To get a better understanding of the quantitative drug loading efficiency of the system, Dox was selected as a model drug because of its stability and fluorescence property. The loading measurement was performed based on the previously reported method.27 Aqueous Dox solution (100 μL, 100 μM) was mixed with aqueous c[KW]5-AuNPs solution (900 μL, 500 μM) to obtain 1:5 molar ratio. The unloaded Dox was separated by using dialysis to analyze the Dox loading efficiency and loading capacity in the c[KW]5-AuNPs. After 24 h stirring, the loading efficiency was found to be 72 ± 1% when the weight ratio of Dox to c[KW]5-AuNPs in feed is 1:5. The loading capacity was calculated to be 16 ± 1 % based on the weight ratio of Dox to c[WK]5-AuNPs (1:5) when the amount of AuNPs was determined using inductively coupled plasma mass spectrometry (ICP-MS).
Cytotoxicity of Peptide-AuNPs
Generated c[KW]5-AuNPs and l(KW)5-AuNPs did not show any significant toxicity in human ovarian adecarcinoma (SK-OV-3), human leukemia (CCRF-CEM) cancer cells, and normal human colon myofibroblast (CCD-18Co) cells at a concentration of 100 μM after 24 and 72 h incubation times (Figure S2). Thus, concentration of □100 μM was selected for further cell-based studies. It is important to emphasize that the cytotoxicity of c[KW]5 and l(KW)5 (100 μM) was decreased through P-AuNPs formation dramatically. For example, c[KW]5 was found to be toxic in normal colon CCD-18Co cells (61.3% cell viability) compared to c[KW]5-AuNPs (99.3% cell viability) after 24 h at a concentration of 100 μM. These data indicate that the cytotoxicity of the parent peptide significantly reduces upon generation of P-AuNPs.
Cellular Uptake of Peptide-AuNPs
To confirm the internalization of P-AuNPs alone, F′-l(KW)5-AuNPs and F′-c[KW]5-AuNPs were prepared as described above by incubating of fluorescently-labeled conjugates of linear (KW)5 and cyclic [KW]5, F′-l(KW)5 and F′-c[KW]5, with HAuCl4 solution according to the previously reported procedure.15 Incubation of F′-l(KW)5-AuNPs and F′-c[KW]5-AuNPs with SK-OV-3 cells showed 3.3- and 4.3-fold higher cellular uptake compared to the corresponding fluorescently-labeled peptides F′-l(KW)5 and F′-c[KW]5, respectively, thus suggesting the formation of P-AuNPs is crucial for the enhanced cellular permeability (Figure 5). FACS results confirmed that F′-c[KW]5-AuNPs showed approximately 1.6-fold higher cellular uptake than F′-l(KW)5-AuNPs, suggesting that the cyclic nature of the peptide in P-AuNPs contributes to improving the cellular uptake.
Figure 5.

Cellular uptake of F′-l(KW)5-AuNPs and F′-c[KW]5-AuNPs (5 μM) versus the corresponding fluorescently-labeled peptides F′-l(KW)5 and F′-c[KW]5 (5 μM) after 1 h incubation.
To investigate whether c[KW]5-AuNPs and l(KW)5-AuNPs localize in different locations in the cell, F′-l(KW)5-AuNPs (5 μM) and F′-c[KW]5-AuNPs (5 μM) were incubated with SK-OV-3 for 1 h. 4′,6-Diamidino-2-phenylindole (DAPI) was employed as a marker of nucleus. Confocal microscopy showed significantly higher nuclear localization of the cyclic peptide-capped AuNPs (F′-c[KW]5-AuNPs) versus the corresponding linear system (F′-l(KW)5-AuNPs) as shown by overlayed picture with DAPI (Figure 6).
Figure 6.
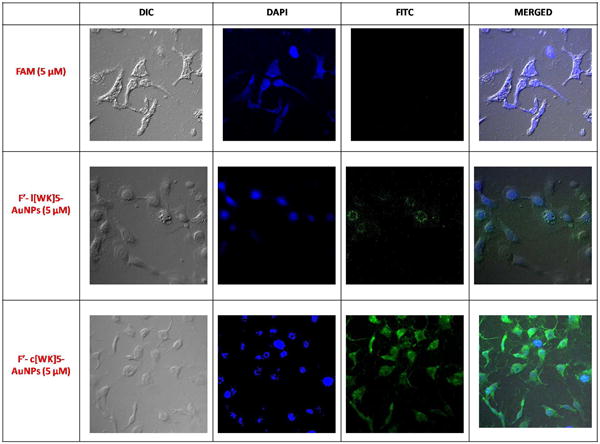
Confocal microscope images of F′-l(KW)5-AuNPs (5 μM) and F′-c[KW]5-AuNPs (5 μM) uptake by SK-OV-3 cells after 1 h incubation.
Cells use different mechanisms to internalize macromolecules and particles, such as phagocytosis, micropinocytosis, and receptor-mediated endocytosis (RME) pathways including clathrin-mediated, caveolae-mediated, and caveolae/clathrin independent endocytosis.21 To get a better understanding of the mechanism of peptide-capped gold nanoparticles uptake by cells, the cellular uptake of fluorescein-labeled peptide capped gold nanoparticles, F′-l(KW)5-AuNPs and F′-c[KW]5-AuNPs (5 μM), were tested quantitatively in the presence of several inhibitors by using FACS including, nystatin, chloroquine, chlorpromazine, methyl-β-cyclodextrin, and 5-(N-ethyl-N-isopropyl)- amiloride (EIA).
As it is shown in Figure 7, the intracellular uptake of F′-l(KW)5-AuNPs and F′-c[KW]5-AuNPs did not significantly change in SK-OV-3 cells in the presence of different endocytic inhibitors after 1 h incubation, suggesting that the mechanism of cellular uptake is not exclusively clathrin-mediated or caveolae-mediated endocytosis, and macropinocytosis. These peptide-AuNPs provide an advantage to known gold nanoparticles and cell-penetrating peptides that their uptake is dependent mainly on endocytotic entry.22
Figure 7.

Cellular uptake of (a) F′-l(KW)5-AuNPs (5 μM) and (b) F′-c[KW]5-AuNPs (5 μM) in the absence or presence of different endocytic inhibitors in SK-OV-3 cells after 1 h.
The surface decoration of AuNPs by amphipathic c[KW]5 and l(KW)5 peptides could improve the interactions of lysine and tryptophan residues with the corresponding negatively charged phospholipids and hydrophobic residues in lipid bilayer. This interaction could be a strong driving force for the initial entry into the cell membrane. Hydrophobic interactions generated by tryptophan residues and the lipids can potentially distort the outer phospholipid monolayer. This process will be followed by peptide internalization and enhanced cellular uptake of the cargo. The nature of the peptide in surface of AuNPs is an important parameter that can alter the mechanism of nanoparticle uptake by cells. Further investigation is required to pinpoint the detailed mechanism of cell entry by these P-AuNPs.
The cellular membrane integrity in SK-OV-3 cells was examined in the presence of c[KW]5-AuNPs and l(KW)5-AuNPs and using trypan blue and showed no significant difference to the control cells (Figure S3), ruling out the damage of the plasma membrane by P-AuNPs at 50 μM and confirming that the highly efficient transport of the complex was not a result of decreased cellular membrane integrity.
Evaluation of Peptide-AuNPs As Molecular Transporters
To evaluate P-AuNPs as molecular transporters, model experiments with anti-HIV drugs 2′,3′-dideoxy-5-fluoro-3′-thiacytidine (emtricitabine, FTC) and 2′,3′-didehydro-2′,3′-dideoxythymidine (lamivudine, 3TC) as cargo drugs were performed. FTC and 3TC are nucleoside reverse transcriptase inhibitors those blocks HIV-1 and hepatitis B virus replication.22 The efficient cellular uptake of FTC and 3TC are critical for anti-HIV activity. To monitor the molecular transporter ability of P-AuNPs, carboxyfluorescein derivatives of FTC (F′-FTC) and 3TC (F′-3TC) were synthesized as described previously, where F′= fluorescein.24,25
CCRF-CEM cells were incubated with F′-FTC and F′-3TC (5 μM) in the presence or absence of diluted carriers c[KW]5-AuNPs and l(KW)5-AuNPs (25 μM) and their parent peptides for 1 h at 37 °C and then treated with trypsin to remove cell surface-bound drugs. Intracellular uptake of F′-FTC and F′-3TC (5 μM) was measured in cells using fluorescence activated cell sorter (FACS). FACS showed significantly higher fluorescence signals in cells treated with F′-Drug-loaded P-AuNPs compared to those with drug alone. The cellular uptake of F′-drug-loaded c[KW]5-AuNPs and F′-drug–loaded l(KW)5-AuNPs were found to be 5.2–5.3- and 2.5–2.9-fold higher for F′-FTC and F′-3TC, respectively, than those of drug alone respectively (Figure 8), suggesting that the uptake of drugs is facilitated by P-AuNPs. The results showed that parent cyclic or linear parent peptides did not improve the cellular uptake of drugs. However, after the formation of P-AuNPs, the cellular uptake of drugs was increased significantly. c[KW]5-AuNPs were found to be more efficient transporter than l(KW)5-AuNPs, while c[KW]5 and l(KW)5 exhibited similar results. These data suggest that a non-cell penetrating cyclic peptide can be converted to cell-penetrating P-AuNPs, presumably due to the new orientation of amino acids and the secondary structure in peptides as shown with CD in the presence of generated AuNPs for drug entrapment.
Figure 8.

Cellular uptake studies for F′-FTC and F′-3TC alone (5 μM) in the presence of cyclic and linear peptides and their corresponding P-AuNPs (25 μM) after 1 h incubation.
To visualize the enhancement of the cellular uptake of drugs, F′-3TC was used as a model for confocal microscopy in SK-OV-3 cells. Confocal microscopy showed nuclear localization of F′-3TC–loaded c[KW]5-AuNPs compared to the corresponding linear system and drug alone. F′-3TC-loaded l(KW)5-AuNPs exhibited a modest fluorescence mostly in the cytoplasm of the cells. However, incubation of cells with F′-3TC alone did not show any fluorescence intensity in cells. These data confirm that the cyclic nature of the peptide is critical for the enhanced cellular permeability (Figure 9) and the nuclear targeting delivery of cargos.
Figure 9.
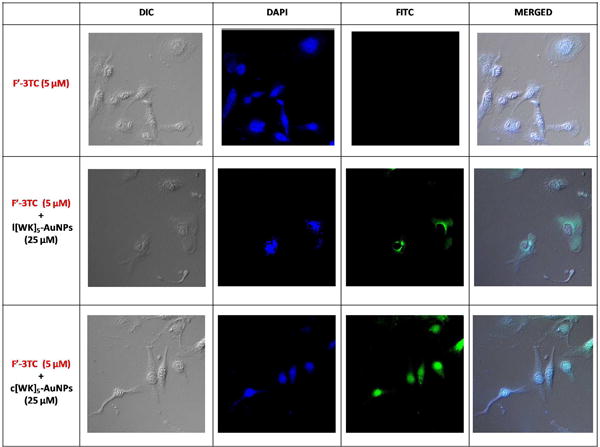
Confocal microscope images of F′-3TC (5 μM) uptake by SK-OV-3 cells in the presence of l(KW)5-AuNPs and c[KW]5-AuNPs (25 μM) after 1 h incubation.
Thus, differential application of linear and cyclic [KW]5-AuNPs was discovered for localization of fluorescence-labeled-3TC. Confocal microscopy revealed the localization of fluorescence-labeled-3TC in the presence of c[KW]5-AuNPs mostly in nucleus in SK-OV-3 cells after 1 h. On the other hand, l(KW)5-AuNPs delivered fluorescence-labeled-3TC in cytoplasm. Furthermore, c[KW]4-AuNPs offered two major advantages over c[RW]4-AuNPs that we previously reported15. First, the reaction time for the formation of c[KW]4-AuNPs (1 h) was significantly shorter compared to that of c[RW]4-AuNPs (4-8 h). The Surface Plasmon Resonance peak for both c[KW]4-AuNPs and c[RW]4-AuNPs15 was compared by using UV-Vis spectroscopy (Figure S11) after 1 h. The results showed that c[KW]4-AuNPs formed faster than c[RW]4-AuNPs. Second, the size and morphology of nanoparticles can be used to control the amount of intracellular uptake of AuNPs by cells. Intracellular accumulation of 233 ng and 288 ng of c[RW]4-AuNPs and c[KW]4-AuNPs was detected, respectively, in SK-OV-3 cells after 24 h incubation15 as measured by ICP-MS.
The molecular transporting efficiency of P-AuNPs for cell-impermeable negatively charges phosphopeptides was also evaluated using flow cytometry. The phosphopeptide do not cross the cell-membrane readily because of the presence of the negatively charged phosphate group. The pTyr-Glu-Glu-Ile (GpYEEI) peptide template has been reported to be an optimal binding sequence for the Src SH2 domain of Src kinase.25 The cellular uptake of F′-GpYEEI was monitored in the presence of P-AuNPs in SK-OV-3 cells. FACS analysis showed that the cellular uptake of F′-GpYEEI was enhanced 3.5- and 12.8-fold by l(KW)5-AuNPs and c[KW]5-AuNPs, respectively (Figure 10), suggesting that these systems may function as a delivery tools for F′-GpYEEI. c[KW]5-AuNPs improved the cellular uptake of F′-GpYEEI 4.2-fold higher when compared to l(KW)5-AuNPs, suggesting the critical role of the cyclic peptide in improving the cellular uptake. These results are consistent with cellular uptake studies of lamivudine and emtricitabine.
Figure 10.

Cellular uptake of F′-GpYEEI (5 μM) in the presence of l(KW)5-AuNPs, c[KW]5-AuNPs, l(KW)5, and c[KW]5 (25 μM) after 1 h incubation.
Considering the significant enhancement of F′-GpYEEI in the presence of P-AuNPs (Figure 10), confocal microscopy was used to confirm the cellular uptake of this negatively charged phosphopeptide in the presence and absence of P-AuNPs. Thus, confocal microscopy was conducted by measuring the fluorescence intensity of F′-GpYEEI-loaded P-AuNPs compared to F′-GpYEEI alone in SK-OV-3 cells. 4′,6-Diamidino-2-phenylindole (DAPI) was employed as a marker of nucleus. The cells were incubated with F′-GpYEEI-loaded c[KW]5-AuNPs, F′-GpYEEI-loaded L[KW]5-AuNPs, and F′-GpYEEI alone. No green fluorescence was observed for the parent fluorescence-labeled phosphopeptide, suggesting that F′-GpYEEI alone did not cross the membrane because of the presence of negative charge phosphate. As shown in Figure 11, the cell nuclei stained by DAPI (blue) were circumvented by the green fluorescence showing the fluorescent-labeled phosphopeptide localized in the cytoplasm in the presence of L[KW]5-AuNPs (Figure 11b). However, the presence of c[KW]5-AuNPs led an overlay of green and blue fluorescence signals revealing the localization of the phosphopeptide mostly in the nuclei of SK-OV-3 cells (Figure 11c). These results indicated that the presence of P-AuNPs is critical to improve the cellular uptake of cell-impermeable phosphopeptide. Furthermore, the linear or cyclic nature of the peptide determines the final destination of the cargo.
Figure 11.
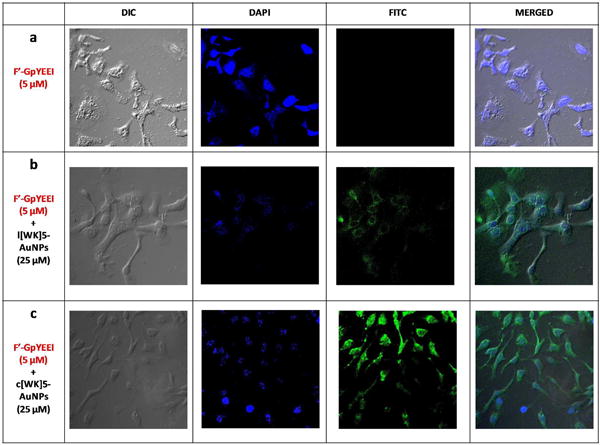
Confocal microscope images of F′-GpYEEI (5 μM) uptake by SK-OV-3 cells in the presence of l(KW)5-AuNPs and c[KW]5-AuNPs (25 μM) after 1 h incubation.
Time-Dependent Antiproliferative Assay
To determine whether c[KW]5-AuNPs can be exploited for the delivery of biologically relevant doses of CPT to cells, the antiproliferative activity of CPT was evaluated in SK-OV-3 cells in the presence and absence of peptide-capped AuNPs in a time-dependent manner. The antiproliferative activity of CPT (5 μM) in the presence of the c[KW]5-AuNPs was improved by approximately 9%, 32%, and 33% compared to that of CPT alone after 24, 48, and 72 h incubation, respectively (Figure 12). Time-dependent inhibitory effect on the cell proliferation of SK-OV-3 cells suggests that the sustained intracellular release of CPT and improved efficacy of the compound. c[KW]5-AuNPs alone did not show any toxicity in SK-OV-3 cells under similar condition, suggesting that the higher antiproliferative is possibly related to the enhanced uptake of the drug in the presence of the peptide-capped AuNPs and intracellular release of CPT.
Figure 12.

Time-dependent antiproliferative assay of CPT in the absence and presence of c[KW]5-AuNPs.
Intracellular Release of Dox
To investigate the kinetics of drug release in cells, Dox was selected as a model drug. The intracellular release of Dox in the presence of the c[KW]5-AuNPs in CCRF-CEM cells was monitored by HPLC. The CCRF-CEM cells (1.2 × 107) were incubated with Dox (7.5 μM)-loaded c[KW]5-AuNPs (37.5 μM) for different times including 2, 12, 24, and 48 h. HPLC analysis at 490 nm and a specific time intervals after cellular lysis was used to measure the quantity of the released Dox. The data exhibited that the intracellular release of Dox occurred in a time-dependent manner. Dox was observed at 15.8-15.9 min in the HPLC profile (Figures S4-S9, Supporting Information). HPLC data showed that approximately 30, 35, 55, and 88% of Dox was released intracellularly within 2, 12, 24, and 48 h, respectively. These data suggest the sustained release of Dox contributes to overall activity of the Dox–loaded c[KW]5-AuNPs as a potential prodrug.
Conclusions
In conclusion, a new class of DDS containing AuNPs and peptides with tryptophan and lysine residues were generated under a mild reaction condition. Cyclic and linear peptide capped-AuNPs (l(KW)5-AuNPs and c[KW]5-AuNPs) exhibited entirely different morphology and sizes. Both l(KW)5-AuNPs and c[KW]5-AuNPs showed minimal cytotoxicity at 100 μM. P-AuNPs were able to entrap hydrophobic CPT through non-covalent interactions, and act as molecular transporters of fluorescence-labeled lamivudine, emtricitabine, and a phosphopeptide (GpYEEI) intracellularly. High cellular internalization of the labeled drugs by P-AuNPs suggests the potential application of P-AuNPs as molecular transporters. Confocal microscopy showed that l(KW)5-AuNPs and c[KW]5-AuNPs delivered cargos to different destinations in cells e.g. cytoplasm and nucleus, respectively. The present results provide insights for generation of a new class of peptide-capped metal nanoparticles as cellular delivery transporters.
Supplementary Material
Acknowledgments
We acknowledge the financial support from the American Cancer Society, Grant No. RSG-07-290-01-CDD, and from the US National Science Foundation, Grant No. CHE 0748555. We thank National Center for Research Resources, NIH, and Grant Number 1 P20 RR16457 for sponsoring the core facility.
Footnotes
Supporting Information Availble: Additional synthetic Scheme and Figures, UV-VIS spectroscopy, cytotoxicity, and encapsulation with CPT. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science. 2004;303:1818–1823. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 2.Van der Aa MAEM, Mastrobattista E, Oosting RS, Hennink WE, Koning GA, Crommelin DJA. The Nuclear Pore Complex: The Gateway to Successful Nonviral Gene Delivery. Pharm Res. 2006;23:447–459. doi: 10.1007/s11095-005-9445-4. [DOI] [PubMed] [Google Scholar]
- 3.(a) Liu Jn, Bu W, Pan Lm, Zhang S, Chen F, Zhou L, Zhao K, Peng W, Shi J. Simultaneous Nuclear Imaging and Intranuclear Drug Delivery by Nuclear-targeted Multifunctional Upconversion Nanoprobes. Biomaterials. 2012;33:7282–7290. doi: 10.1016/j.biomaterials.2012.06.035. (b) Shiraishi T, Hamzavi R, Nielsen PE. Targeted Delivery of Plasmid DNA Into the Nucleus of Cells via Nuclear Localization Signal Peptide Conjugated to DNA Intercalating Bis- and Trisacridines. Bioconjugate Chemistry. 2005;16:1112–1116. doi: 10.1021/bc050093f. (c) Hodoniczky J, Sims CG, Best WM, Bentel JM, Wilce JA. The Intracellular and Nuclear-targeted Delivery of an Antiandrogen Drug by Carrier Peptides. Biopolymers. 2008;90:595–603. doi: 10.1002/bip.20986.
- 4.Kubitscheck U, Grnwald D, Hoekstra A, Rohleder D, Kues T, Siebrasse JP, Peters RJ. Nuclear Transport of Single Molecules: Dwell Times at the Nuclear Pore Complex. Cell Biol. 2005;168:233–243. doi: 10.1083/jcb.200411005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meinema AC, Laba JK, Hapsari RA, Otten R, Mulder FA, Kralt A, Van den Bogaart G, Lusk CP, Poolman B, Veenhoff LM. Long Unfolded Linkers Facilitate Membrane Protein Import Through the Nuclear Pore Complex. Science. 2011;333:90–93. doi: 10.1126/science.1205741. [DOI] [PubMed] [Google Scholar]
- 6.Kang B, Megan AM, Mostafa AE. Nuclear Targeting of Gold Nanoparticles in Cancer cells Induces DNA Damage, causing Cytokinesis Arrest and Apoptosis. J Am Chem Soc. 2010;132:1517–1519. doi: 10.1021/ja9102698. [DOI] [PubMed] [Google Scholar]
- 7.Raurio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, S J. Prodrugs: Design and Clinical Applications. Nature Rev. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 8.(a) De M, Ghosh PS, Rotello VM. Applications of Nanoparticles in Biology. Adv Mater. 2008;20:4225–4241. (b) Subramani C, Yu X, Agasti SS, Duncan B, Eymur S, Tonga M, Rotello VM. Direct Photopatterning of Light-Activated Gold nanoparticles. J Mater Chem. 2011;21:14156–14158.
- 9.(a) Dekiwadia CD, Lawrie AC, Fecondo JV. Peptide-mediated Cell Penetration and Targeted Delivery of Gold Nanoparticles Into Lysosomes. J Peptide Sci. 2012;18:527–534. doi: 10.1002/psc.2430. (b) Kim CK, Ghosh P, Pagliuca C, Zhu ZJ, Menichetti S, Rotello VM. Entrapment of Hydrophobic Drugs in Nanoparticle Monolayers with Efficient Release Into Cancer Cells. J Am Chem Soc. 2009;131:1360–1361. doi: 10.1021/ja808137c.
- 10.(a) Langel U. Cell-penetrating peptides: Processes and Applications. CRC Press; Boca Rotan: 2002. (b) Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT Peptide on the Surface of Liposomes Affords Their Efficient Intracellular Delivery Even at Low Temperature and in the Presence of Metabolic Inhibitors. Proc Nat Acad Sci USA. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. (c) Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Different Mechanisms for Cellular Internalization of the HIV-1 Tat-derived Cell Penetrating Peptide and Recombinant Proteins Fused to Tat. Eur J Biochem. 2002;269:494–501. doi: 10.1046/j.0014-2956.2001.02671.x. (d) Thorén PE, Persson D, Isakson P, Goksör M, Onfelt A, Nordén B. Uptake of Analogs of Penetratin, Tat(48–60) and Oligoarginine in Live Cells. Biochem Biophys Res Commun. 2003;307:100–107. doi: 10.1016/s0006-291x(03)01135-5.
- 11.(a) Rana S, Bajaj A, Mout R, Rotello VM. Monolayer Coated Gold Nanoparticles for Delivery Applications. Advanced Drug Delivery Reviews. 2012;64:200–216. doi: 10.1016/j.addr.2011.08.006. (b) Saha K, Bajaj A, Duncan B, Rotello NM. Beauty is Skin Deep: a Surface Monolayer Perspective on Nanoparticle Interactions with Cells and Bio-macromolecules. Small. 2011;7:1903–1918. doi: 10.1002/smll.201100478. (c) Yuan H, Fales AM, Vo-Dinh T. TAT Peptide-Functionalized Gold Nanostars: Enhanced Intracellular Delivery and Efficient NIR Photothermal Therapy Using Ultralow Irradiance. J Am Chem Soc. 2012;134:11358–11361. doi: 10.1021/ja304180y.
- 12.(a) Torchilin VP. Structure and Design of Polymeric Surfactant-Based Drug Delivery Systems. J Controlled Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. (b) Lee CC, MacKay JA, Frechet JMJ, Szoka FC. Designing Dendrimers for Biological Applications. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171.
- 13.(a) Morgan MT, Nakanishi Y, Kroll DJ, Griset AP, Carnahan MA, Wathier M, Oberlies NH, Manikumar G, Wani MC, Grinstaff MW. Dendrimer-Encapsulated Camptothecins: Increased Solubility, Cellular Uptake, and Cellular Retention Affords Enhanced Anticancer Activity In Vitro. Cancer Res. 2006;66:11913–11921. doi: 10.1158/0008-5472.CAN-06-2066. (b) Duncan B, Kim C, Rotello VM. Gold Nanoparticles Platforms as Drug and Biomolecule Delivery Systems. J Controlled Release. 2010;148:122–127. doi: 10.1016/j.jconrel.2010.06.004.
- 14.(a) Mandal D, Nasrolahi Shirazi A, Parang K. Cell-penetrating Homochiral Cyclic Peptides as Nuclear-targeting Molecular Transporters. Angew Chem Int Ed. 2011;50:9633–9637. doi: 10.1002/anie.201102572. (b) Nasrolahi Shirazi A, Tiwari RK, Oh D, Banerjee A, Yadav A, Parang K. Efficient Delivery of Cell Impermeable Phosphopeptides by a Cyclic Peptide Amphiphile Containing Tryptophan and Arginine. Mol Pharm. 2013 doi: 10.1021/mp400046u.
- 15.Nasrolahi Shirazi A, Mandal D, Tiwari RK, Guo L, Lu W, Parang W. Cyclic Peptide-capped Gold Nanoparticles as Drug Delivery Systems. Molecular Pharmaceutics. 2013;10:500–511. doi: 10.1021/mp300448k. [DOI] [PubMed] [Google Scholar]
- 16.Nasrolahi Shirazi A, Tiwari RK, Brown A, Mandal D, Sun G, Parang K. Cyclic Peptides Containing Tryptophan and Arginine as Src Kinase Inhibitors. Bioorg Med Chem Lett. 2013;23:3230–3234. doi: 10.1016/j.bmcl.2013.03.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan YN, Lee JY, Wang DIC. Uncovering the Design Rules for Peptide Synthesis of Metal Nanoparticles. J Am Chem Soc. 2010;132:5677–5686. doi: 10.1021/ja907454f. [DOI] [PubMed] [Google Scholar]
- 18.Iosin M, Baldeck P, Astilean S. Study of Tryptophan Assisted Synthesis of Gold Nanoparticles by Combining UV-Vis, Fluorescence, and SERS Spectroscopy. J Nanoparticle Res. 2010;12:2843–2849. [Google Scholar]
- 19.Garcia-Carbonero R, Supko JG. Current Perspectives on the Clinical Experience, Pharmacology, and Continued Development of the Camptothecins. Cancer Res. 2002;8:641–661. [PubMed] [Google Scholar]
- 20.Kailasan A, Yuan Q, Yang H. Synthesis and Characterization of Thermoresponsive Polyamidoamine-polyethylene Glycol-Poly(D,L-lactide) Core-shell Nanoparticles. Acta Biomater. 2009;6:1131–1139. doi: 10.1016/j.actbio.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conner SD, Schmid SL. Regulated Portals of Entry Into the Cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 22.Alkilany AM, Murphy CJ. Toxicity and Cellular Uptake of Gold Nanoparticles: What We Have Learned So Far. J Nanopart Res. 2010;12:2313–2333. doi: 10.1007/s11051-010-9911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Massard J, Benhamou Y. Treatment of Chronic Hepatitis B in HIV Co-Infected Patients. Gastroenterol Clin Biol. 2008;32:S20–S24. doi: 10.1016/S0399-8320(08)73261-5. (b) Saag MS. Emtricitabine, a New Antiretroviral Agent with Activity Against HIV and Hepatitis B Virus. Clin Infect Dis. 2006;42:126–131. doi: 10.1086/498348. (c) Nelson M, Schiavone M. Emtricitabine (FTC) for the Treatment of HIV Infection. Int J Clin Pract. 2004;58:504–510. doi: 10.1111/j.1368-5031.2004.00100.x.
- 24.Agarwal HK, Chhikara BS, Hanley MJ, Ye G, Doncel GF, Parang K. Synthesis and Biological Evaluation of Fatty Acyl Ester Derivatives of (-)-2′,3′-Dideoxy-3′-thiacytidine. J Med Chem. 2012;55:4861–4871. doi: 10.1021/jm300492q. [DOI] [PubMed] [Google Scholar]
- 25.Agarwal HK, Chhikara BS, Bhavaraju S, Mandal D, Doncel GF, Parang K. Emtricitabine Prodrugs with Improved Anti-HIV Activity and Cellular Uptake. Molecular Pharmaceutics. 2013;10:467–476. doi: 10.1021/mp300361a. [DOI] [PubMed] [Google Scholar]
- 26.(a) Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 Domains Recognize Specific Phosphopeptide Sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. (b) Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, Cowburn D, Hanafusa H, Mayer BJ, Overduin M, Resh MD, Rios CB, Silverman L, Kuriyan J. Crystal Structure of the Phosphotyrosine Recognition Domain SH2 of v-src Complexed with Tyrosine-phosphorylated Peptides. Nature. 1992;358:646–653. doi: 10.1038/358646a0.
- 27.Cai H, Yao P. In-Situ Preparation of Gold Nanoparticle-Loaded Lysozyme-Dextran Nanogels and Applications for Cell Imaging and Drug Delivery. Nano Scale. 2013;5:2892–2900. doi: 10.1039/c3nr00178d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.