

Abstract
Evidence is emerging that the closely related ROCK1 and ROCK2 serine/threonine kinases support the invasive and metastatic growth of a spectrum of human cancer types. Therefore, inhibitors of ROCK are under preclinical development. However, a key step in their development involves the identification of genetic biomarkers that will predict ROCK inhibitor anti-tumor activity. One identified mechanism for ROCK activation in cancer involves the loss of function of the DLC1 tumor suppressor gene, which encodes a GTPase activating protein (RhoGAP) for the RhoA and RhoC small GTPases. DLC-1 loss may lead to hyperactivation of RhoA/C and its downstream effectors, the ROCK kinases. We therefore determined whether loss of DLC-1 protein expression identifies non-small cell lung carcinoma (NSCLC) cell lines whose growth and invasion phenotypes are sensitive to ROCK inhibition. We identified and characterized a novel small molecule pharmacologic inhibitor of ROCK and additionally applied genetic approaches to impair ROCK1 and/or ROCK2 activity, and we determined that although NSCLC anchorage-dependent growth was ROCK-independent, both anchorage-independent growth and Matrigel invasion were ROCK-dependent. However, loss of DLC-1 expression did not correlate with ROCK activation or with OXA-06 sensitivity. Unexpectedly, suppression of ROCK1 or ROCK2 expression alone was sufficient to impair anchorage-independent growth, supporting their non-overlapping roles in oncogenesis. Mechanistically, the block in anchorage-independent growth was associated with accumulation of cells in the G0/G1 phase of the cell cycle, but not increased anoikis. We conclude that ROCK may be a useful therapeutic target for NSCLC.
Keywords: DLC-1, Rho kinase, non-small cell lung cancer (NSCLC), kinase inhibitors, Rho small GTPases, cancer therapeutics
Introduction
There is considerable and growing evidence for the importance of the ROCK serine/threonine kinases (ROCK1/ROKβ and ROCK2/ROKα) kinases in oncogenesis (1,2). The highly related ROCK1 and ROCK2 kinases function as key downstream effectors of the RhoA small GTPase (3). ROCK regulates diverse cellular processes such as actomyosin contractility, focal adhesion assembly, cytokinesis, and cell proliferation. ROCK has also been implicated in colorectal, breast, gastric, and glioblastoma proliferation or anchorage-independent growth (4–7) and in prostate, bladder, fibrosarcoma, melanoma, and hepatocellular cancer metastatic growth (8–13). An evaluation of ROCK as a therapeutic target for lung cancer has not been done.
Although preclinical development of ROCK inhibitors is ongoing, a number of issues need to be resolved to facilitate their clinical development. First, genetic or biochemical determinants that identify cancers responsive to ROCK inhibitors need to be identified. Second, biomarkers that correlate with inhibitor anti-tumor response are needed for effective clinical evaluation. Although the phosphorylation status of key substrates of ROCK is widely utilized, their value as biomarkers for ROCK inhibition remains unresolved. Third, the majority of studies implicating ROCK in cancer growth utilized the Y-27632 ROCK inhibitor (1,2). Since Y-27632 can inhibit other protein kinases in vitro, whether the anti-tumor activities ascribed to this inhibitor are target-based is unresolved.
One candidate molecular determinant for ROCK inhibitor sensitivity is the loss of expression of the DLC1 tumor suppressor (14). DLC1 mRNA expression was lost in 95% of NSCLC patient tumors and 58% of NSCLC cell lines (15,16). Due at least in part through its function as a Rho GTPase activating protein and thus negative regulator of RhoA and the related RhoB and RhoC, restoration of DLC-1 expression in DLC1 deficient NSCLC lines resulted in reduction of cell migration, proliferation, anchorage-independent growth, in vitro invasion, and tumorigenicity in nude mice (16–18), supporting its role as a tumor suppressor (19). It is well-established that aberrant RhoA and RhoC activation can promote tumorigenic, invasive and metastatic growth (20–22). Thus, by analogy to the loss of the neurofibromin RasGAP or the tuberous sclerosis RhebGAP in cancer (23,24), loss of DLC-1 results in hyperactivation and persistent RhoA/C effector signaling (15,25). However, like Ras, Rho GTPases are not tractable molecules for drug discovery (14). Instead, in further analogy to Ras, where inhibitors of the Raf-MEK-ERK effector protein kinase pathway are being considered for anti-Ras drug development (26), inhibitors of RhoA/C downstream effector protein kinases, in particular ROCK, may also be attractive therapeutic targets for DLC1-deficient NSCLC. In support of this possibility, ectopic expression of DLC-1 suppressed ROCK activation and ROCK-dependent motility in DLC-1 deficient hepatocellular carcinoma cell lines (27)., and suppression of DLC-1 expression sensitized liver cancer cells to reduced colony formation by pharmacologic inhibition of ROCK (19).
In light of the frequent loss of DLC1 expression in NSCLC, we speculated that DLC1 loss-induced activation of RhoA/C will in turn cause ROCK activation-driven NSCLC tumorigenic and malignant growth. However, previous studies implicating ROCK in cancer growth have relied primarily on the use of Y-27632 ATP competitive ROCK1/2 kinase inhibitor, which has additional off-target inhibitory activity for other kinases such as PKN and MSK1 (28). To offset this concern, we utilized a structurally distinct and more potent and selective small molecule ROCK1/2 inhibitor together with RNA interference depletion of ROCK1/2 expression to validate a role for ROCK in DLC1 deficient NSCLC growth. We determined that NSCLC anchorage-dependent growth was ROCK-independent, but anchorage-independent growth and Matrigel invasion were ROCK-dependent. However, loss of DLC-1 expression did not correlate with ROCK dependence. ROCK inhibition of growth was associated with inhibition of cell cycle progression rather than enhancement of cell death. Our studies provide validation of ROCK for NSCLC therapy.
Materials and Methods
Identification and characterization of the OXA-06 ROCK inhibitor
ROCK1/2 cDNA sequences were subcloned into a baculovirus expression vector for protein expression as a C-terminal fusion protein with His6 in insect cells. The expressed protein (comprising residues 2–238 of ROCK1 fused to residues 255–548 of ROCK2) was purified and used in a fluorescence polarization assay-based high-throughput screening campaign to identify ROCK kinase inhibitors within OSI Pharmaceuticals compound library. The screening assay buffer consisted of 10 mM Tris HCl (pH 7.2), 10 mM MgCl2, 0.1% BSA, 1 mM DTT and contained 100 nM substrate peptide (5′-FAM-AKRRRLSSLRA-COOH), 1 μM ATP and 12.5 nM ROCK kinase domain. A series of azaindole-based compounds was selected for medicinal chemistry optimization, and synthetic routes for this series are described within patent application WO2007084667. To determine the IC50 values of OXA-06 and Y-27632 against ROCK, we utilized the above fluorescence polarization assay with 12.5 nM ROCK fusion protein and 1.4 μM ATP (roughly the measured Km value) as well as at 100 μM ATP. The IC50 for OXA-06 was measured 18 times, with the average and standard deviation reported, and Y-27632 was measured three times, with the average and standard deviation reported.
Protein kinase selectivity assays
Kinase selectivity assays were performed using ProfilerPro Kinase Selectivity Kits (Caliper Life Sciences, Inc.) according to the manufacturer’s instructions. The extent of inhibition by each compound is measured directly by quantifying the level of both unphosphorylated and phosphorylated peptide substrate after electrophoretic separation. Assays were performed at room temperature following a one h pre-incubation of compounds with the enzymes. Each assay was performed on two separate occasions.
Quantitative assays of MYPT1 phosphorylation and cell migration in PANC-1 cells
Analysis of MYPT1 phosphorylation in PANC-1 cells by quantitative ELISA was performed as described previously (29). PANC-1 cell migration was quantitated using 96-well modified Boyden chamber migration assay plates (Chemicon, Temecula, CA). Cells were applied to the upper chamber in growth medium supplemented with 0.5% fetal calf serum (FCS), either in the presence or absence of inhibitor, and the cells were allowed to migrate towards growth medium supplemented with 10% FCS for 16 h. The number of migrated cells was then quantitated by labeling with Cyquant GRR (Invitrogen).
Cell Culture
NSCLC lines were obtained directly from the ATCC (Rockville, MD) and grown in RPMI-1640 supplemented with 10% FBS.
Soft agar colony formation, Matrigel invasion, and anoikis assays
Anchorage-independent growth soft agar assays were done as previously described (30). For inhibitor treatments, vehicle (dimethyl sulfoxide; DMSO) or OXA-06 was also added to the medium at the final indicated concentrations. For siRNA treatments, cells were suspended in soft agar 48 h post-transfection and maintained at 37 °C for 14–30 days, when viable colonies were stained with the MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) viability stain. The total number per plate of viable colonies >10 cells were quantified by counting the number of colonies in five representative fields of view within each plate. Results are expressed as mean ± SD.
Invasion assays were performed with Growth Factor Reduced Matrigel Invasion Chambers (BD Biosciences, San Jose, CA) according to the manufacturer’s protocol. For inhibitor treatments, vehicle or OXA-06 were also added to the upper chamber. RPMI-1640 containing 3% FBS as a chemoattractant was added to the bottom well. Cells were allowed to invade for 22 h at 37 °C and then non-invaders were removed. Invading cells were fixed and stained with the Diff-Quik Stain Set (Dade Behring Inc., Newark, DE). Five fields were counted for each chamber, and the total number of cells counted per chamber was used for calculating the average number of invading cells. Results are expressed as mean ± SD.
For the MTT anchorage-dependent growth viability assay, 2 × 103 cells per well were seeded in 96-well plates in octuplet. For ROCK inhibitor studies, growth medium supplemented with vehicle or OXA-06 was also added to the wells. Cells were incubated for two days for the siRNA and four days for inhibitor analyses. Optical density at 560 nm was recorded and normalized to either vehicle control non-specific siRNA control. Values shown are the mean ± SD.
For the anoikis assays, cells were plated on Ultra-Low Attachment plates (Corning, Lowell, MA) with RPMI-1640 plus 10% FCS, and either vehicle, 10 μM OXA-06, or 10 μM staurosporine (positive control for induction of apoptosis). Cells with staurosporine were harvested and lysed after 6 h. All other cells were harvested and lysed after 48 h, and then immunoblotted for PARP and caspase-3 as described below in western blot analyses section.
Flow cytometry cell cycle analysis
NSCLC cells were plated on Ultra-Low Attachment Plates (Corning) with RPMI-1640 supplemented with 10% FCS, and either vehicle or 10 μM OXA-06 for 48 h. After treatment, cells were collected, washed twice with phosphate-buffered saline (PBS) and fixed in 70% ethanol overnight. After centrifugation, cells were stained with a buffer containing 20 μg/ml propidium iodide (Roche, Indianapolis, IN) and 200 μg/ml RNAse A (Qiagen) in PBS. Analyses were performed on a CyAn flow cytometer (Beckman-Coulter, Brea, CA). Cell cycle distribution was determined using FlowJo (TreeStar, Inc., Ashland, OR). Experiments were performed two independent times and values are shown as means ± S.D.
Statistical analysis
Data were analyzed using an unpaired t test. Values are shown as means ± S.D. A P value <0.01 was considered significant.
Western blot analyses
For OXA-06 treatment immunoblots, cells were treated with 1 h with vehicle or the indicated OXA-06 concentration as this treatment time was previously shown to be sufficient for maximal ROCK inhibition effect (29). The following antibodies were then used for immunoblot analysis: pMYPT1(T853) (US Biologicals, Marblehead, MA), MYPT1 (Covance, Princeton, NJ), pCofilin (Ser3) and Cofilin (Cell Signaling Technology, Danvers, MA), β-actin (Sigma-Aldrich, St. Louis, MO). Pull down analyses to determine steady-state levels of RhoA-GTP were done as we have described previously using a glutathione S-transferase fusion protein containing the RhoA-GTP binding domain of Rhotekin and anti-RhoA antibody (Cell Signaling Technology) (31). For anoikis immunoblot analyses, suspension cell cultures were treated with staurosporine (Calbiochem, San Diego, CA) for 6 h, or with vehicle or OXA-06 for 48 h. The following antibodies were then used to detect the indicated proteins: PARP (Cell Signaling Technology), caspase-3 (Cell Signaling Technology), and vinculin (Sigma-Aldrich). For cell cycle immunoblot analyses, suspension cells were treated with vehicle or 2 or 10 μM OXA-06 for 48 h. The following antibodies were used to detect: phosphorylated (Ser608) Rb (Cell Signaling Technology), total Rb (Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin. For the siRNA immunoblot analyses, immunoblot analysis of ROCK1, ROCK2, phospho-MYPT1(T853), MYPT1, phospho-cofilin (Ser3), cofilin, and β-actin was done.
siRNA suppression of ROCK1 and ROCK2 expression
siRNA oligonucleotides (Stealth siRNA, as we described previously (29) were diluted to 20 μM in nuclease-free water and the transfection was performed with Lipofectamine2000 (Invitrogen) according to manufacturer’s protocols and as described previously (29).
Forty-eight h after transfection, transfected cells were harvested for either analyses of anchorage-independent growth or for western blot analyses. Data were analyzed using a paired t test. Values are shown as means ± S.D. A P value <0.01 was considered highly significant.
Results
OXA-06 is a novel and potent ROCK inhibitor
In light of the off-target activities of the Y-27632 ROCK inhibitor, we initiated a high-throughput screening of the OSI Pharmaceuticals compound library using recombinant chimeric ROCK1/2. A series of compounds was identified for medicinal chemistry optimization, resulting in the identification of a potent ATP-competitive ROCK inhibitor, designated OXA-06, that is structurally distinct from the widely used Y-27632 ROCK1/2 inhibitor (Fig. 1). We first compared the in vitro potency of OXA-06 with Y-27632 for ROCK in vitro. OXA-06 exhibited an IC50 value of 0.01 ± 0.005 μM, while Y-27632 measured 0.24 ± 0.09 μM when measured with an ATP concentration of 1.4 μM, making OXA-06 roughly 25-fold times more potent in vitro. When performed at 100 μM ATP concentrations, IC50 values of OXA-06 were similarly about 25-fold more potent than Y-27632. To evaluate selectivity, we chose 200 nM for OXA-06 and 10 μM for Y-27632, concentrations at which roughly 95% of ROCK1 and ROCK2 are inhibited by each respective compound. We performed kinase selectivity assays using the ProfilerPro Kinase Selectivity Kits comprised of 216 pharmacologically relevant wild type or mutant protein kinases. Consistent data were generated for 183 kinases within this analysis (assays that failed in one or more assay, or which did not generate data with CV values below 25% are not included) (Supplementary Table 1). Clear selectivity differences were noted between the two compounds. OXA-06 showed >50% inhibition of 9 out of 167 (5.4%) whereas Y-27632 inhibited 17 out of 167 (10.2%) (Fig. 1B). Both shared activity for five additional protein kinases, whereas OXA-06 was active on two other kinases and Y-27632 was active on 10 additional kinases. These results demonstrate the greater selectivity of OXA-06 over Y-27632. Additionally, this degree of selectivity is comparable or better than highly selective protein kinase inhibitors (e.g., imatinib, gefitinib) currently in clinical use (32,33). We conclude that the different and improved kinase inhibition selectivity profile, together with being structurally distinct from Y-27632, support the usefulness of OXA-06 to investigate the role of ROCK kinase activity in cellular processes.
Figure 1. OXA-06 is structurally distinct from Y-27632 and is a more potent and selective inhibitor of ROCK1 and ROCK2 in vitro.
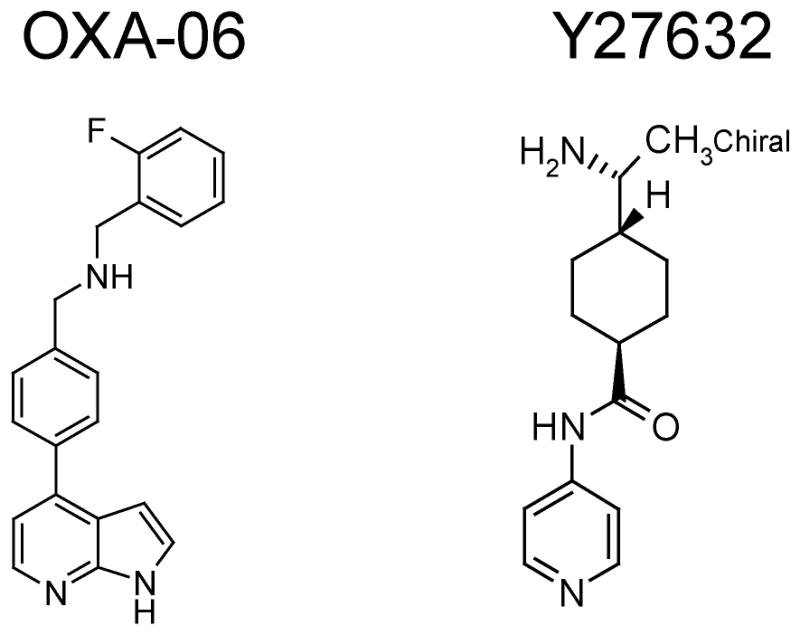
Structures of OXA-06 and Y-27632.
We next determined if OXA-06 could block ROCK activity in intact cells. We determined previously that siRNA suppression of ROCK1 and ROCK2 expression, and additionally treatment with three different ROCK1/2 inhibitors, reduced the phosphorylation of MYPT1 at T853 was ROCK-dependent in PANC-1 pancreatic tumor cells (29). PANC-1 migration was also shown previously to be ROCK-dependent (34). We determined that OXA-06 treatment of PANC-1 cells showed dose-dependent inhibition of MYPT1 phosphorylation (IC50 300 nM) and migration in vitro (Supplementary Fig. 1). By comparison Y-23732 showed a reduced potency to inhibit MYPT1 phosphorylation (IC50 1.4 μM) and migration.
OXA-06 blocks anchorage-independent growth and invasion of NSCLC lines
We next utilized OXA-06 to validate the importance of ROCK1/2 as therapeutic targets. Western blot analyses of 16 NSCLC cell lines determined that all lines express ROCK1 and ROCK2 protein (Fig. 2A). Based on previous studies with other cancer types (19,27), we hypothesized that the absence of DLC-1 expression might be a predictor of sensitivity to inhibition of ROCK1 and/or ROCK2. For these analyses, we evaluated the A549, H23 and H358 cell lines which lack DLC-1 protein expression, and the H1299 and H1703 cell lines that express high levels of DLC-1 (18).
Figure 2. OXA-06 treatment blocks anchorage-independent growth and invasion in NSCLC cell lines, independent of DLC1 status.

A, ROCK1 and ROCK2 proteins are expressed all NSCLC cell lines. The indicated NSCLC cell lines were lysed and resolved by SDS-PAGE, and then blotted with ROCK isoform-specific antibodies. B, Cell viability was measured using the MTT assay after treatment of the indicated cell lines with vehicle (DMSO) or the indicated concentrations of OXA-06 for 4 d. Data shown are the O.D. values relative to vehicle and represent the mean ± SD of octuplet wells and are representative of two independent experiments. C, Colony formation of NSCLC cell lines in soft agar in growth medium supplemented with vehicle (DMSO) or the indicated concentrations of OXA-06. The number of viable proliferating colonies were stained by MTT and counted after 30 d, except for A549 and H1299 cells, which were counted after 14 d. Data shown are the percent colonies relative to vehicle, are the average ± SD of triplicate wells, and are representative of at least two independent experiments. D, Invasion of the indicated NSCLC cell lines was assayed by Matrigel transwell assays with vehicle (DMSO) or the indicated concentrations of OXA-06 after 22 h. Data shown are the percent invaded cells relative to vehicle, are the average ± SD of triplicate chambers and are representative of at least two independent experiments. For panels B–D, DLC-1 protein expression as determined by western blot analyses are indicated as detectable (DLC-1 positive) or undetectable (DLC-1 negative, as we determined previously. E, Elevated steady-state RhoA activity levels do not correlate with loss of DLC-1 expression. Pull down analyses, followed by blot analysis for RhoA, was done to measure the level of activated RhoA-GTP and total RhoA expression. β-actin blot analysis was done to verify equivalent total protein loading.
Surprisingly, none of the cell lines was inhibited in anchorage-dependent proliferation by treatment with 2 or 10 μM OXA-06 (Fig. 2B). However, at the highest concentration of OXA-06 used, 10 μM, greater than 90% anchorage-independent growth inhibition was achieved in all the lines tested (Fig. 2C). While 400 nM was sufficient to cause ~90% reduction in the colony formation activity of the DLC-1 positive H1299 cell line, this concentration did not cause statistically significant inhibition of the DLC-1 negative A549 and H358 or the DLC-1 positive H1703 cell lines.
Since OXA-06 also inhibited seven other kinases in vitro (e.g., PKA), we also assessed the ability of Y-27632 to inhibit anchorage-independent growth of H1299 and A549 cells. In both lines, Y-27632 also caused a decrease in anchorage-independent growth, suggesting a ROCK-dependent for anchorage-independent growth, although Y-27632 had much weaker potency than OXA-06 (Fig. 3). This increased potency of OXA-06 compared to Y-27632 in blocking anchorage-independent growth is consistent with its increased potency in vitro and in ROCK-dependent cell-based assays (Fig. 1B, Supplementary Table 1 and Supplementary Fig. 1). Similar to their sensitivity to inhibition of anchorage-independent growth by OXA-06, both DLC-1 negative and positive NSCLC cell lines showed comparable dose-dependent sensitivities to OXA-06 suppression of Matrigel invasion, with ~70% inhibition seen at 2 μM (Fig. 2D). The observation that Matrigel invasion and anchorage-independent growth were inhibited at similar concentrations of OXA-06 is consistent with the inhibitor blocking both tumor phenotypes through inhibition of the same target(s). We conclude that loss of DLC-1 did not correlate with increased sensitivity to OXA-06.
Figure 3. OXA-06 exhibits greater potency than Y-27632 for inhibition of NSCLC anchorage-independent growth.

Colony formation of A549 (Panel A) or H1299 (Panel B) NSCLC cell lines in soft agar in growth medium supplemented with vehicle (DMSO) or the indicated concentrations of OXA-06 or Y-27632. The number of viable proliferating colonies were stained by MTT and counted after 14 d. Data shown are the percent colonies relative to vehicle, are the average ± SD of triplicate wells, and are representative of at least two independent experiments.
One possible basis for the lack of correlation between DLC-1 loss of expression and increased sensitivity to OXA-06 is that DLC-1 expression alone does not determine the steady-state level of RhoA activation. The activities of RhoGEFs as well as RhoGAPs will also influence RhoA steady-state levels. Consistent with this possibility, we performed pull down analyses to quantitate the steady-state levels of active RhoA-GTP in a panel of NSCLC cell lines. We found no direct correlation between elevated RhoA-GTP levels the absence of DLC-1 protein expression (Fig. 2E).
OXA-06 anti-tumor activity correlates with suppression of Cofilin phosphorylation
To investigate the mechanism of OXA-06-mediated inhibition of growth and invasion, we determined if the anti-tumor activity of OXA-06 correlated with inhibition of ROCK activity. Two well-characterized ROCK-dependent phosphorylation activities are direct ROCK phosphorylation of the MYPT1 and the indirect phosphorylation of Cofilin through ROCK phosphorylation and activation of LIMK1/2. However, since both MPYT1 and Cofilin can be phosphorylated by ROCK-independent mechanisms, their phosphorylation state may or may not accurately monitor ROCK activity.
We found that OXA-06 treatment reduced pMYPT1 and pCofilin levels at concentrations that coincided with that required to block anchorage-independent growth. These data are consistent with ROCK inactivation as a key mechanism for OXA-06 anti-tumor activity. However, the extent of phosphorylation reduction and the inhibitor concentration required for this effect varied between different cell lines (Fig. 4). Generally, we found that pCofilin provided the most accurate marker for OXA-06 growth inhibition activity for all five NSCLC cell lines evaluated. For example, H1299 cells were the most sensitive to OXA-06 colony suppression (Fig. 2C) and pCofilin reduction was seen at the lowest concentrated used, 80 nM. In contrast, H358 cells were the most resistant to OXA-06 colony suppression and even at the highest concentration studied (10 μM), incomplete reduction in pCofilin was seen. For the remaining three NSCLC cell lines, 10 μM was required for near-complete suppression of pCofilin as well as soft agar growth. In contrast, pMYPT1 levels were less informative, largely because two cell lines lacked detectable phosphorylation in untreated cells. For A549, but not H358 cells, this was most likely due to the barely detectable levels of MYPT1 expression. Where it could be detected, pMYPT1 did show dose-dependent reduction that coincided with OXA-06 concentrations needed for growth inhibition. Thus, Cofilin phosphorylation may provide a useful biomarker for ROCK inhibition in NSCLC.
Figure 4. OXA-06 treatment reduces steady-state levels of phospho-Cofilin and phospho-MYPT1 in NSCLC lines.

DLC-1 negative or positive cell lines were treated with vehicle (DMSO) or the indicated concentrations of OXA-06 for 1 h. Western blot analysis using phospho-specific antibodies for pMYPT1 (pT853) and pCofilin (pS3) and for total MYPT1 and Cofilin.
Genetic suppression of ROCK1 and/or ROCK2 expression phenocopies the biochemical and biological activities of OXA-06
Although ROCK1/2 were the protein kinases most potently inhibited by OXA-06 in vitro (Table 1), it is possible that the effects of OXA-06 on NSCLC anchorage-independent growth and invasion resulted from inhibition of other kinases. To confirm that ROCK kinase inhibition contributes to the anti-tumor activity of OXA-06, we specifically reduced ROCK1 and ROCK2 expression with siRNA validated previously to be selective inhibitors of ROCK1 and ROCK2 (29). We utilized two independent siRNA sequences each for ROCK1 and ROCK2 in H1299 cells that caused near complete knockdown of ROCK1 and/or ROCK2 protein expression (Fig. 5A). Interestingly, neither ROCK1 nor ROCK2 depletion individually was able to strongly reduce pMYPT1 or pCofilin, and instead, concurrent depletion of both was required to reduce pMYPT1 and to a lesser extent pCofilin (Fig. 5A). This result demonstrates that both ROCK isoforms are functionally redundant for these signaling activities.
Table 1.
Protein kinase inhibition profiles of OXA-06 and Y-27632a
| Protein kinase | Compound
|
|
|---|---|---|
| OXA-06 | Y27632 | |
| ROCK2 | 95 | 95 |
| ROCK1 | 94 | 94 |
| PKA | 98 | 29 |
| PKG1b | 73 | 68 |
| PKG | 72 | 64 |
| STK10 | 58 | 87 |
| PRKX | 56 | 78 |
| ARK5 | 56 | 1 |
| IKKbeta | 56 | 82 |
| PKCdelta | 36 | 55 |
| PKCeta | 27 | 81 |
| PKCepsilon | 26 | 84 |
| MST1 | 19 | 59 |
| RSK2 | 17 | 70 |
| MNK1 | 16 | 78 |
| PKCtheta | 12 | 82 |
| RSK3 | 10 | 82 |
| RSK1 | 6 | 51 |
| PKCbeta1 | −3 | 52 |
Percent inhibition of a panel of protein kinases performed using ProfilerPro Kinase Selectivity Kits with 10 μM Y-27632 or 200 nM OXA-06. This table shows all kinases inhibited by >50% (grey boxes) by either OXA-06 or Y-27632 compiles from Supplementary Table 1. Data shown are representative of two independent experiments.
Figure 5. Concurrent suppression of ROCK1 and ROCK2 expression does not impair cell viability, but is required to block downstream phosphorylation but not anchorage-independent growth.

H1299 NSCLC cells were transfected with non-specific (NS) siRNA or two different siRNA sequences targeting ROCK1 or ROCK2. A, Concurrent suppression of ROCK1 and ROCK2 expression blocks phospho-MYPT1 and to a lesser degree phospho-Cofilin. After 48 h, lysates were harvested and western blot analysis was performed using the indicated antibodies. B, Suppression of ROCK1 or ROCK2 expression alone is sufficient to impair anchorage-independent growth. Forty-eight h post-transfection, cells were suspended in soft agar. The number of viable proliferating colonies were stained by MTT and counted after 14 d. Data shown are the percent colonies relative to vehicle, are the average ± SD of triplicate wells and are representative of at least two independent experiments. C, Concurrent suppression of ROCK1 and ROCK2 expression does not affect cell viability. Forty-eight h post-transfection, cells were split at 5 × 103 cells/well and cell viability was measured MTT assay analysis. Data shown are the O.D. values relative to non-specific siRNA and represent the mean ± SD of octuplet wells and are representative of two independent experiments.
In contrast, we found that depletion of either ROCK1 or ROCK2 alone, using two independent siRNA sequences each, was sufficient to potently (~90%) inhibit the anchorage-independent growth of H1299 cells. Concurrent suppression of both ROCK1 and ROCK2 more effectively suppressed growth with a near complete suppression of colony formation (Fig. 5B). Thus ROCK1 and ROCK2 each contribute non-redundant activities to support anchorage-independent growth. Finally, we determined that, similar to OXA-06 treatment, concurrent depletion of ROCK1 and ROCK2 did not significantly impair anchorage-dependent proliferation (Fig. 5C). That siRNA for ROCK1 and ROCK2 phenocopied the biological activities of OXA-06 suggests that the anti-tumor activities seen with OXA-06, despite having other protein kinase targets, are likely due primarily to inhibition of ROCK1 and ROCK2.
OXA-06 stimulates accumulation in G0/G1 phase but not apoptosis in nonadherent NSCLC cells
Although we found that prolonged OXA-06 treatment did not reduce NSCLC cell line viability on plastic (Fig. 3B), this result did not exclude the possibility that OXA-06 rendered NSCLC cells more sensitive to suspension-induced apoptosis (anoikis). We therefore investigated possible apoptotic effects of OXA-06 during blockade of anchorage-independent growth of NSCLC cells. We saw no evidence for caspase-3 cleavage in A549 or H1299 cells, and only partial PARP cleavage in A549 but not H1299 cells in suspension cells, even when treated with the high concentration of 10 μM OXA-06, while the positive control of 10 μM staurosporine demonstrated clear PARP and caspase-3 cleavage (Supplementary Fig. 2A). Thus increased anoikis sensitivity is not a mechanism for the decreased anchorage-independent growth seen with OXA-06.
We next determined if OXA-06 impaired soft agar colony formation by disruption of cell cycle progression under anchorage-independent conditions. We used propidium iodide staining and flow cytometry to investigate possible cell cycle distribution effects of OXA-06 on two NSCLC cell lines treated with inhibitor in suspension. OXA-06 treatment at 1 μM in suspension (a concentration that is around the cellular IC50 value for reduced anchorage-independent growth, invasion, and downstream ROCK target phosphorylation) caused a statistically significant reduction in S phase and an increase in G0/G1 (Fig. 6A).
Figure 6. OXA-06 and suppression of ROCK1/2 expression causes a block in G0/G1 in NSCLC lines when in suspension.

The indicated NSCLC cell lines were plated on Ultra-Low Attachment plates and incubated with A, DMSO (Vehicle) or 1 μM OXA-06 for 48 h, or B, transfected with non-specific (NS) siRNA or ROCK1 siRNA #1 and ROCK2 siRNA #1 sequences targeting ROCK1 or ROCK2. Forty-eight h post-transfection, the cells were plated on Ultra-Low Attachment plates (Corning). The cell-cycle dependent DNA content for the indicated treatments is shown on the left (each representative of the three experiments), and the means and standard deviations of cell-cycle percentages for the three independent experiments are shown on the right.
To confirm that ROCK inhibition is required for these effects of OXA-06, we used siRNA to reduce ROCK1 and ROCK2 protein expression and we found that concurrent suppression of ROCK1 and ROCK2 expression also caused a very similar cell cycle phenotype (Fig. 6B). This result suggests that these activities of OXA-06 on the cell cycle are due to inhibition of ROCK. Furthermore, we did not detect any sub-G0 peaks in the analysis of OXA-06 at 1 μM (Fig. 6) or at 10 μM (data not shown), or ROCK1/ROCK2 siRNA (Fig. 6), which when taken together with lack of full caspase-3 or PARP cleavage, excludes anoikis or necrosis as a mechanism for the ROCK inhibition-dependent block in anchorage-independent growth.
Finally, we evaluated the effects of OXA-06 treatment on two key regulators of G1 progression, the Rb tumor suppressor and cyclin D1. Using western blot analysis on OXA-06 treated A549 or H1299 cells in suspension at 2 or 10 μM, we did not observe increased Rb phosphorylation at S608 (a site phosphorylated by cyclin/CDK complexes) nor decreased levels of cyclin D1 (Supplementary Fig. 2B). We also did analysis for two regulators of Rb phosphorylation, p21/CIP1 and p27/KIP1 and we found no consistent and significant changes in their levels of expression upon OXA-06 treatment (data not shown). These results indicate that OXA-06 blocks anchorage-independent growth by causing cell cycle arrest in G0/G1 through a mechanism involving other regulators of G1 progression.
Discussion
In light of their role as key effectors of the RhoA and RhoC small GTPases which have validated roles in oncogenesis (20,21), ROCK1/2 are being considered as therapeutic targets for cancer (1,2). The first goal of our study was to rigorously validate ROCK as a useful therapeutic target for NSCLC. The majority of previous studies with this goal utilized Y-27632 or its close structural relatives Wf-536 or fasudil, which are known to also potently block the activities of other protein kinases (28). To overcome this limitation with Y-27632, we utilized a second novel ROCK inhibitor. OXA-06 exhibited increased potency to block ROCK-dependent signaling in cell-based assays, and additionally exhibited less off-target protein kinase inhibitory activities in vitro. Therefore, when used in conjunction with a structurally distinct ROCK inhibitor with a distinct off-target activity profile, concurrently with two independent siRNAs each targeting ROCK1 and/or ROCK2, we feel that our analyses provide an accurate assessment of ROCK function in lung cancer growth. Generally, RNAi depletion of a protein kinase, where there is a loss of entire protein expression, may not accurately model pharmacologic inhibition of the catalytic activity alone, where a kinase-inactive protein persists and may retain non-kinase functions. However, our finding that RNAi depletion and pharmacologic inhibition of ROCK exhibited essentially identical consequences on NSCLC growth suggests that the anchorage-independent growth and Matrigel invasion inhibition seen is due to loss of ROCK protein kinase function. Furthermore, as discussed below, our findings that OXA-06 anti-tumor activity correlated well with OXA-06 inhibition of phosphorylation of known direct and indirect ROCK substrates and that siRNA for ROCK phenocopied the effects of OXA-06 on anchorage-dependent and -independent growth strongly support our conclusion that ROCK kinases represent functionally relevant targets of OXA-06 in NSCLC growth. Thus, while OXA-06 can inhibit other protein kinases, our siRNA results argue that the anti-tumor activity observed in NSCLC cell lines may be due primarily to ROCK inhibition. The FDA approval of the dasatinib and sunitinib protein inhibitors for cancer treatment, both of which show activity (Kd <100 nM) for greater than 15% of the 290 protein kinases tested (38), clearly demonstrate that multi-kinase inhibitors can be developed successfully for therapeutic use. OXA-06 does not have sufficient pharmacokinetic/pharmacodynamic properties for use in animal studies (unpublished observations), but it provides a more potent and selective inhibitor than the widely used Y-27632 compound to study ROCK function in cell culture. More importantly, our results support the discovery and development of potent and selective ROCK inhibitors in animal models of NSCLC.
The second goal of our study was to determine if DLC-1 loss of expression is a reliable biomarker for NSCLC tumor cell ROCK dependency. In contrast to two recent studies that found ROCK important for DLC-1 tumor suppressive effects in hepatocellular carcinoma (19,27), we found that DLC-1 expressing and deficient cells were comparably sensitive to OXA-06 and additionally showed comparable levels of phosphorylation of ROCK substrates. This disconnect is not entirely surprising for several reasons. First, there exist a multitude of GEFs and GAPs for RhoA/C (14). Thus, the steady-state activation of RhoA-C and ROCK may not be dictated solely by loss of DLC-1 alone. Consistent with this possibility, we found no direct correlation between the level of DLC-1 protein reduction and increased cellular RhoA-GTP levels. Furthermore, in studies of a DLC-1 mutant that failed to associate with focal adhesions, this disrupted subcellular localization was not associated with a loss in the ability to lower total cellular RhoA-GTP levels (39). Thus, it is not surprising that total RhoA-GTP levels are not tightly linked to the level of DLC-1 expression. Second, RhoA/C may utilize non-ROCK effectors to promote tumorigenesis (37). Finally, DLC-1 can also serve as a GAP for other Rho family proteins and DLC-1 tumor suppressor function involves both Rho-dependent and Rho-independent mechanisms (18). These additional complexities of DLC-1 function may prevent a simple and direct relationship between DLC-1 expression loss and ROCK-dependent growth. Perhaps the combined use of DLC-1 loss together with hyperphosphorylation of ROCK substrates will provide a more reliable marker to predict ROCK dependency.
The third goal of our study was to determine if the widely studied substrates for ROCK activity provided accurate biochemical markers that correlated with OXA-06-mediated growth inhibition. While our studies found that Cofilin, rather than MYPT1, provided a more accurate biomarker for pan-ROCK inhibitor treatment, our use of isoform-specific siRNA to selectively silence ROCK1 or ROCK2 found that inhibition of either isoform alone inhibited anchorage-independent growth, yet inhibition of both ROCK1 and ROCK2 were required to suppress Cofilin phosphorylation. Thus, ROCK1 and ROCK2 may be functionally overlapping for Cofilin phosphorylation but serve non-overlapping roles in NSCLC growth, although whether they are truly functionally distinct remains to be rigorously evaluated. These results support the value of investigation of isoform-specific ROCK inhibitors, which may provide reduced off-target activities, and the need for additional biomarkers for ROCK isoform-selective inhibitors. Nevertheless, our observation that combined inhibition of ROCK1 and ROCK2 has strong anti-cancer activity in NSCLC lines supports the further development of ROCK dual-selective inhibitors for lung cancer.
One surprising observation was the failure of OXA-06 to impair anchorage-dependent NSCLC tumor cell growth at a concentration (10 μM) that potently blocked soft agar colony formation and Matrigel invasion. This activity begins to shed some light on the role of ROCK in NSCLC tumor growth. When evaluated on cells in suspension, we found that OXA-06 treatment did not increase apoptosis or necrosis and instead caused a G0/G1 arrest, although it is possible that other less-well understood types of cell death in combination with the cell-cycle arrest seen are responsible for the striking inhibition of anchorage-independent growth seen with ROCK inhibition and knockdown. While our analyses did not establish a mechanism for why ROCK dependency was not evident for cells grown on plastic, previous studies suggest that loss of integrin-dependent signaling events (38,39) are a likely basis for ROCK dependency for cells in suspension. Finally, how tumor cells will respond to ROCK inhibition within the context of host stromal tissue will be an important future question to address using both pharmacologic inhibitors and conditional ROCK knockouts in mouse models of lung cancer.
In summary, our studies provide strong validation of ROCK1 and/or ROCK2 as useful targets for NSCLC therapy. Furthermore, despite the potentially significant off-target activities of our newly identified OXA-06 ROCK inhibitor, our demonstration that OXA-06 NSCLC anti-tumor activity correlates well with ROCK inhibition argues that, as with other multi-kinase inhibitors FDA-approved for cancer treatment, a highly selective ROCK inhibitor may not be essential for clinical success of ROCK inhibitors. Finally, our failure to validate DLC-1 loss as a genetic marker for ROCK dependency or pCofillin as a biomarker for ROCK inhibition defines these two areas where more progress will be needed if ROCK inhibitors will be developed successfully.
Supplementary Material
Acknowledgments
Financial Support: This work was supported by grants from the National Institutes of Health (CA67771 and CA129610 to C. Der) and by postdoctoral fellowships from the American Cancer Society to D. Vigil and from Susan Komen to T.Y. Kim.
We thank the UNC Flow Cytometry Facility for technical expertise and data interpretation. We thank Adrienne Cox for helpful discussions and Lanika DeGraffenreid and Jenni Sells for assistance in manuscript preparation.
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare no conflict of interest.
References
- 1.Olson MF. Applications for ROCK kinase inhibition. Curr Opin Cell Biol. 2008;20:242–8. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cell Mol Life Sci. 2010;67:171–7. doi: 10.1007/s00018-009-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4:446–56. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 4.Sahai E, Ishizaki T, Narumiya S, Treisman R. Transformation mediated by RhoA requires activity of ROCK kinases. Curr Biol. 1999;9:136–45. doi: 10.1016/s0960-9822(99)80067-0. [DOI] [PubMed] [Google Scholar]
- 5.Ying H, Biroc SL, Li WW, Alicke B, Xuan JA, Pagila R, et al. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol Cancer Ther. 2006;5:2158–64. doi: 10.1158/1535-7163.MCT-05-0440. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Tang Q, Xu F, Xue Y, Zhen Z, Deng Y, et al. RhoA regulates G1-S progression of gastric cancer cells by modulation of multiple INK4 family tumor suppressors. Mol Cancer Res. 2009;7:570–80. doi: 10.1158/1541-7786.MCR-08-0248. [DOI] [PubMed] [Google Scholar]
- 7.Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar-Uniyal M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009;29:119–23. [PubMed] [Google Scholar]
- 8.Somlyo AV, Bradshaw D, Ramos S, Murphy C, Myers CE, Somlyo AP. Rho-kinase inhibitor retards migration and in vivo dissemination of human prostate cancer cells. Biochem Biophys Res Commun. 2000;269:652–9. doi: 10.1006/bbrc.2000.2343. [DOI] [PubMed] [Google Scholar]
- 9.Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin Cancer Res. 2003;9:2632–41. [PubMed] [Google Scholar]
- 10.Nakajima M, Katayama K, Tamechika I, Hayashi K, Amano Y, Uehata M, et al. WF-536 inhibits metastatic invasion by enhancing the host cell barrier and inhibiting tumour cell motility. Clin Exp Pharmacol Physiol. 2003;30:457–63. doi: 10.1046/j.1440-1681.2003.03855.x. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima M, Hayashi K, Egi Y, Katayama K, Amano Y, Uehata M, et al. Effect of Wf-536, a novel ROCK inhibitor, against metastasis of B16 melanoma. Cancer Chemother Pharmacol. 2003;52:319–24. doi: 10.1007/s00280-003-0641-9. [DOI] [PubMed] [Google Scholar]
- 12.Xue F, Takahara T, Yata Y, Xia Q, Nonome K, Shinno E, et al. Blockade of Rho/Rho-associated coiled coil-forming kinase signaling can prevent progression of hepatocellular carcinoma in matrix metalloproteinase-dependent manner. Hepatol Res. 2006;38:810–817. doi: 10.1111/j.1872-034X.2008.00333.x. [DOI] [PubMed] [Google Scholar]
- 13.Wong CC, Wong CM, Tung EK, Man K, Ng IO. Rho-kinase 2 is frequently overexpressed in hepatocellular carcinoma and involved in tumor invasion. Hepatology. 2009;49:1583–94. doi: 10.1002/hep.22836. [DOI] [PubMed] [Google Scholar]
- 14.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:824–57. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durkin ME, Yuan BZ, Zhou X, Zimonjic DB, Lowy DR, Thorgeirsson SS, et al. DLC-1:a Rho GTPase-activating protein and tumour suppressor. J Cell Mol Med. 2007;11:1185–207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, Reynolds SH. DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene. 2004;23:1405–11. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- 17.Qian X, Li G, Asmussen HK, Asnaghi L, Vass WC, Braverman R, et al. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci USA. 2007;104:9012–7. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Healy KD, Hodgson L, Kim TY, Shutes A, Maddileti S, Juliano RL, et al. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2008;47:326–37. doi: 10.1002/mc.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xue W, Krasnitz A, Lucito R, Sordella R, Vanaelst L, Cordon-Cardo C, et al. DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 2008;22:1439–44. doi: 10.1101/gad.1672608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–9. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–42. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- 22.Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–8. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 23.McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191–216. doi: 10.1146/annurev.pathol.2.010506.091940. [DOI] [PubMed] [Google Scholar]
- 24.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim TY, Vigil D, Der CJ, Juliano RL. Role of DLC-1, a tumor suppressor protein with RhoGAP activity, in regulation of the cytoskeleton and cell motility. Cancer Metastasis Rev. 2009;28:77–83. doi: 10.1007/s10555-008-9167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 27.Wong CC, Wong CM, Ko FC, Chan LK, Ching YP, Yam JW, et al. Deleted in liver cancer 1 (DLC1) negatively regulates Rho/ROCK/MLC pathway in hepatocellular carcinoma. PLoS One. 2008;3:e2779. doi: 10.1371/journal.pone.0002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garton AJ, Castaldo L, Pachter JA. Quantitative high-throughput cell-based assays for inhibitors of ROCK kinases. Methods Enzymol. 2008;439:491–500. doi: 10.1016/S0076-6879(07)00433-8. [DOI] [PubMed] [Google Scholar]
- 30.Cox AD, Der CJ. Biological assays for cellular transformation. Methods Enzymol. 1994;238:277–94. doi: 10.1016/0076-6879(94)38026-0. [DOI] [PubMed] [Google Scholar]
- 31.Kim TY, Healy KD, Der CJ, Sciaky N, Bang YJ, Juliano RL. Effects of structure of Rho GTPase-activating protein DLC-1 on cell morphology and migration. J Biol Chem. 2008;283:32762–70. doi: 10.1074/jbc.M800617200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–45. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 34.Kaneko K, Satoh K, Masamune A, Satoh A, Shimosegawa T. Expression of ROCK-1 in human pancreatic cancer: its down-regulation by morpholino oligo atisense can reduce the migration of pancreatic cancer cells in vitro. Pancreas. 2002;24:251–7. doi: 10.1097/00006676-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CA, Campbell BT, et al. A quantitative analysis of kinase inhibitor specificity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 36.Qian X, Asmussen HK, Asnaghi L, Vass WC, Braverman R, Yamada KM, et al. Oncogenic inhibition by a deleted in liver cancer gene requires cooperation between tensin binding and Rho-specific GTPase-activating protein activities. Proc Natl Acad Sci. 2007;104:9012–7. doi: 10.1073/pnas.0703033104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holeiter G, Heering J, Erlmann P, Schmid S, Jahne R, Olayioye MA. Deleted in liver cancer 1 controls cell migration through a Dia1-dependent signaling pathway. Cancer Res. 2008;68:8743–51. doi: 10.1158/0008-5472.CAN-08-0984. [DOI] [PubMed] [Google Scholar]
- 38.Howe AK, Juliano RL. Regulation of anchorage-independent signal transduction by protein kinase A and p21-activated kinase. Nat Cell Biol. 2000;2:593–600. doi: 10.1038/35023536. [DOI] [PubMed] [Google Scholar]
- 39.Del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internatlization of membranedomains. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.