

Abstract
Oxa-Pictet-Spengler cyclization and microwave-assisted C-H arylation have been implemented as key steps in the synthesis of new isochroman heterocycles containing a 4,5,6a,7-tetrahydrodibenzo[de,g]chromene motif. These isochromans may be easily transformed to phenanthrene alkaloids via acidic cleavage of the isochroman ring and standard synthetic manipulations thereafter. The route described is attractive in that it provides access to two biologically interesting scaffolds in simple and high yielding synthetic steps.
Keywords: C-H activation, Direct arylation, Oxa-Pictet-Spengler, Phenanthrene, Isochroman
1. Introduction
Compounds containing the isochroman (3,4-dihydro-1H-benzo[c]pyran, 1) moiety have displayed a number of interesting biological properties including antiplatelet aggregation,1 androgenic receptor antagonist, 2 anti-diabetic,3 antioxidant,4 and cytotoxic activities. 5 As such, naturally occurring as well as synthetic isochromans have been studied over the years in the synthetic and biological realms. Examples of isochroman-containing natural products include pseudodeflectusin (2), an isolate of the seaweed parasite Aspergillus pseudodeflectus,5 penicisochromans D-E (3-4) from the sea-fan derived fungus Penicillum sp. PSU-F406 and panowamycins A and B (5-6) from Streptomyces sp. K07-0010.7 Synthetic isochromans include the isochroman-6-carboxamides PNU-109291 (7) and PNU-142633 (8), both selective 5-HT1D agonists and anti-migraine agents,8-10 and CJ-17,493 (9) a neurokinin-1 receptor antagonist with antiemetic properties.11,12
Phenanthrene natural products are common in plants of the Orchidaceae family, particularly the Dendrobium, Bulbophyllum, Eria, Maxillaria, Bletilla, Coelogyna, Cymbidium, Ephemerantha and Epidendrum genera.13 Compounds containing the phenanthrene nucleus have displayed a wide range of biological activities including anticancer,14-16 antimicrobial,17,18 anxiolytic and sedative activities.19
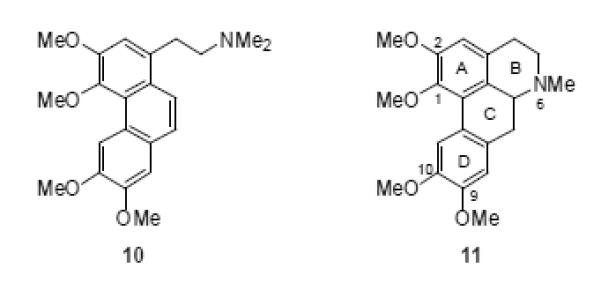
In nature, the aromatic rings of the phenanthrene nucleus are often decorated with oxygenated substituents - typically hydroxy methoxy or methylenedioxy groups. One sub-group of phenanthrene natural products are the phenanthrene alkaloids, (typified by N-methyl seco-glaucine, 10).20 These alkaloids contain an ethylamine unit attached to the phenanthrene core and are biogenetically related to aporphine alkaloids (eg. glaucine, 11).
Phenanthrene alkaloids such as 10 have been well studied in relation to their biological activity 21,22,23,24 and a number of methods for their synthesis have been documented. 25-28,20,29,30
A continuing interest in our laboratory is the evaluation of aporphine derivatives as central nervous system (CNS) receptor ligands and cytotoxic agents.31-33 Our work has led to the identification of a number of aporphine derivatives with promising 5-HT2A antagonist activity as well as cytotoxic activity. Our previous studies indicate that ring C of the tetracyclic aporphine nucleus is required for 5-HT2A receptor antagonism. However, the requirement for an intact ring B has not been determined in this regard.
Molecular docking studies indicate that the protonated amine nitrogen (N6) of aporphines is required for binding to the 5-HT2A receptor via interactions with an aspartate residue in the ligand cavity.32,34 However this requirement has not been extensively validated via synthesis and evaluation of analogs. Furthermore, with regards to cytotoxic activity of aporphines that we have studied, the requirement for the N6 nitrogen atom has not been thoroughly investigated. Further studies in these directions have necessitated the acquisition of other seco-aporphine derivatives as well as N6 isosteres. Such molecules would allow us to continue to probe the structure-activity relationships and utility of aporphines and aporphine derivatives as CNS receptor ligands and as cytotoxic agents.
Thus, we were motivated to develop a versatile synthetic route that would allow for diversification in providing libraries of compounds with a 4,5,6a,7-tetrahydrodibenzo[de,g]chromene isochroman motif as well as phenanthrene alkaloids for structure-activity relationship studies. Herein, we wish to report our successful efforts in the synthesis of the 4,5,6a,7-tetrahydrodibenzo[de,g]chromene framework and its transformation to the phenanthrene alkaloid structure.
2. Results and discussion
Our approach hinged on the preparation of the 4,5,6a,7-tetrahydrodibenzo[de,g]chromene motif (13) since this would facilitate synthesis of the phenanthrene alkaloid core (12). Compound 13 could be derived from 14 via cyclization under C-H activation conditions. For this key step, we envisaged that microwave-assisted C-H activation akin to that which we have employed in the synthesis of aporphines35 and C-homoaporphines,36 could be successfully deployed. The isochroman 14 in turn is derivable from alcohol 15 and aldehyde 16 via oxa-Pictet-Spengler cyclization.37-39
To initiate our study, we decided to target the synthesis of compound 24a (Scheme 1) since it was the oxygen-nitrogen isostere of a key aporphine molecule (nantenine) in our CNS ligand studies.33 We decided to prepare 24a via compound 22 since this would also allow for synthesis of a variety of other phenol ether analogs (24b - 24e). Thus, commercially available phenylacetic acid 17 was dibenzylated to yield 18. LAH reduction of 18 afforded alcohol 19 which was then condensed with aldehyde 20 40,41 in an oxa-Pictet-Spengler reaction42 to give isochroman 21. At this stage, we were set to attempt the key microwave-assisted C-H activation step to form the isochroman tetracycle. Our experience with this reaction in the synthesis of aporphines indicated that various ligands including 2′-(Diphenylphosphino)-N,N′-dimethyl-(1,1′- biphenyl)-2-amine (PhDavePhos), Bis(1,1-dimethylethyl)methylphosphine tetrafluoroborate and tricyclohexylphosphine were effective. We began by evaluating tricyclohexylphosphine tetrafluoroborate and were elated to find that the microwave-assisted direct arylation on 21 proceeded to give 22 in high yield, obviating the need for any optimization. Subsequent hydrogenolysis of the benzyl ether afforded phenol 23, which was alkylated to provide compounds 24a - 24e. In a manner similar to that depicted in Scheme 1, molecules with various substitution patterns in the lower aryl ring were synthesized (Scheme 2). The C-H activation step again gave excellent yields of cyclized products indicating excellent tolerance of a variety of substitution patterns in the lower aryl ring.
Scheme 1.
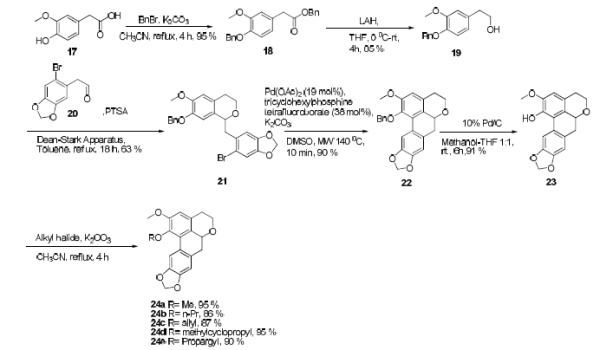
Synthesis of novel isochromans – C1 variants
Scheme 2.
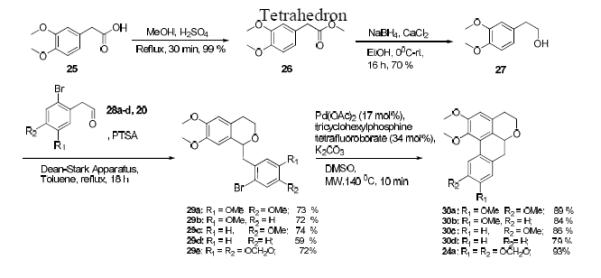
Synthesis of novel isochromans – ring D variants
We also examined the effect of catalyst loading on this reaction and found that in the case of cyclization of 29a to 30a there was generally a decrease in isolated yield when lower catalyst loadings were used. With a 5% Pd(OAc)2/10% tricyclohexylphosphine tetrafluoroborate catalytic system the isolated yield of 30a was 76%. However, this yield increased slightly (82%) with a 10% Pd(OAc)2/20% tricyclohexylphosphine tetrafluoroborate catalyst.
It would appear that tricyclohexylphosphine tetrafluoroborate (ligand C) is indeed optimal for cyclization of substrates of this type; cyclization of compound 29e with this ligand gave significantly higher yields than other ligands tried (Table 1).
Table 1.
Microwave-assisted C-H activation on compound 29e with various ligands 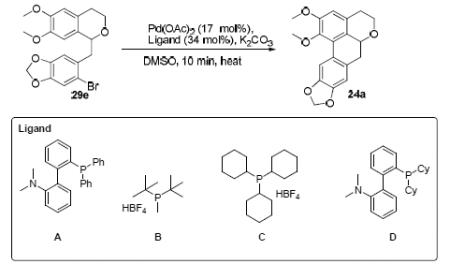
| Entry | Ligand | Temperature (°C) | Yield (%)a |
|---|---|---|---|
| 1 | A | 135 | 40 |
| 2 | B | 140 | 22 |
| 3 | C | 140 | 93 |
| 4 | D | 140 | No Reaction |
Isolated yield after purification
Having prepared the tetracyclic isochroman skeleton, our next task was to transform this structure to the phenanthrene alkaloid scaffold. The strategy here involved cleavage of the isochroman moiety to reveal a phenanthrene ethylalcohol moiety which could be subsequently modified to give the phenanthrene ethylamine structure. We decided to explore the utility of this method in the synthesis of the natural product 10.
An initial attempt at opening the isochroman ring of 30a with zinc dust and acetyl chloride gave a complex mixture of products which we could not separate chromatographically. After experimenting with a number of acidic cleavage conditions, we found that the desired reaction could be effected very rapidly by treatment with 33% HBr in acetic acid. This provided a mixture of the desired compound 32 along with its acetate 31. Treatment of this mixture with 20% aqueous NaOH at room temperature for 2 hours gave almost quantitative conversion to 32. Dess-Martin oxidation of the alcohol 32 gave an aldehyde which was immediately subjected to reductive amination (due to its apparent instability) to give the targeted alkaloid 10.
Dibenzo[de,g]chromanones (ie the lactone variants of compounds such as 30a) have been synthesized and utilized to prepare phenanthrenes.26 Access to the dibenzo[de,g]chromanone biaryl motif was accomplished via radical-mediated cyclization with toxic tin reagents. Our method avoids the use of toxic tin reagents for biaryl cyclization and is a good alternative in this regard. Aporphines have been synthesized via cyclization of phenanthrene alkaloids43,44 so that this route may also be extrapolated to aporphine synthesis.
To summarize, we have implemented oxa-Pictet-Splenger cyclization and microwave assisted C-H activation in the synthesis of a novel tetracyclic isochroman (4,5,6a,7-tetrahydrodibenzo[de,g]chromene) framework. The isochromans thus formed may be easily transformed to phenanthrene alkaloid natural products as demonstrated herein in the synthesis of compound 10.45,20 Thus the route affords flexibility in the preparation of diverse libraries of two biologically relevant scaffolds. The implementation of enantioselective oxa-Pictet Spengler reactions in the synthesis of enantiomerically pure 4,5,6a,7-tetrahydrodibenzo[de,g]chromenes, is an interesting direction which we plan to pursue in future. Biological evaluation of the molecules prepared herein will be presented elsewhere.
3. Experimental section
3.1. General experimental information
All glass apparatus were oven dried prior to use. A CEM Discover microwave reactor was used to carry out microwave-assisted C-H arylation reactions. HRESIMS spectra were obtained using an Agilent 6520 Q-TOF instrument. 1H NMR and 13C NMR spectra were recorded using a Bruker DPX-500 spectrometer (operating at 500 MHz for 1H; 125 MHz respectively for 13C) using CDCl3 as solvent. Tetramethylsilane (δ 0.00 ppm) served as an internal standard in 1H NMR and 13C NMR unless stated otherwise. Chemical shift (δ 0.00 ppm) values are reported in parts per million and coupling constants in Hertz (Hz). Splitting patterns are described as singlet (s), doublet (d), triplet (t) and multiplet (m). Melting points were obtained on a Mel-Temp capillary electrothermal melting point apparatus. Reactions were monitored by TLC with Whatman Flexible TLC silica gel G/UV 254 precoated plates (0.25 mm). TLC plates were visualized in UV light (254 nm) and by staining with phosphomolybdate spray reagent, vanillin or iodine. Flash column chromatography was performed with silica gel 60 (EMD Chemicals, 230-400 mesh, 0.04-0.063 μm particle size). Preparative thin layer chromatography was performed with silica gel GF plates (Analtech, catalog # 02003). All chemicals and reagents were obtained from Sigma-Aldrich and Fischer Scientific (USA) in reagent grade and were used without further purification.
3.2. Benzyl 2-(4-(benzyloxy)-3-methoxyphenyl)acetate (18)
A solution of commercially available acid 17 (5.0 g, 27.5 mmol), K2CO3 (7.58 g, 54.9 mmol) and benzyl bromide (6.5 mL, 54.9 mmol) in acetonitrile (150 mL) was refluxed for 4 h. The reaction was cooled to rt, filtered and the solvent evaporated under reduced pressure. The resulting crude product was purified on a silica gel column eluting in 10 % ethyl acetate-petroleum ether to afford 18 as a pale yellow solid. (9.4 g, 95 %). Mp: 50 - 53 °C; 1H NMR (500 MHz. CDCl3): δ 7.42 (m, 2H), 7.33 (m, 8H), 6.81 (d, 2H, J=7.9 Hz), 6.74 (dd, 1H, J=8.2, 1.9 Hz), 5.14 (s, 2H), 5.13 (s, 2H), 3.84 (s, 3H), 3.59 (s, 2H); 13C NMR (125 MHz. CDCl3): δ 171.5, 149.6, 147.3, 137.1, 135.8, 128.55 (×2), 128.54, 128.3, 128.2, 127.8, 127.2 (×2), 126.9, 121.4, 114.1 (×2), 112.9 (×2), 71.1, 66.6, 55.9, 40.9; HRESIMS: calcd. for C23H22O4Na [M+Na]+ 386.1444; found 386.1446.
3.3. 2-(4-(benzyloxy)-3-methoxyphenyl)ethanol (19)
Lithium Aluminum Hydride (1.09 g, 28.8 mmol) was added to a three neck round bottom flask under argon, and THF (70 mL) added. The resulting suspension was cooled to 0 °C and stirred for 30 min. A solution of 18 (2.0 g, 5.8 mmol) in THF (50 mL) was added dropwise to the mixture. The reaction was allowed to stir at 0 °C for another 30 min, and then at rt for 3 h. After 3 h, the reaction mixture was cooled to 0 °C, and water (2 mL) was added dropwise, followed by the addition of 10 ml of 2N NaOH solution. The reaction mixture was allowed to stir for 30 min, filtered through celite and the solvent evaporated under reduced pressure. Water (30 mL) was added and the solution extracted with three portions of dichloromethane (3 × 30 mL). The organic layers were collected, dried over Na2SO4, and evaporated under reduced pressure to give 19 as a white solid. (1.2 g, 85%). Data is in accordance with that previously reported.46
3.4. 7-(benzyloxy)-1-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-6-methoxyisochroman (21)
To a three neck round bottom flask attached to a Dean-Stark trap, a solution of 19 (1.20 g, 4.7 mmol), 2047 (0.56 g, 2.3 mmol) and PTSA (catalytic) in 100 mL toluene were added. The resulting mixture was refluxed for 18 h. After 18 h, the reaction mixture was cooled to rt and washed with water (30 mL). The aqueous layer was extracted with ethyl acetate (30 mL) and the organic extracts combined. The combined organic layer was dried over Na2SO4 and evaporated under reduced pressure. The resulting crude product was purified on a silica gel column eluting in 30% ethyl acetate-petroleum ether, to afford 21 as a white solid (a mixture of rotamers as evident from NMR data all signals observed are reported). (1.44 g, 63 %) Mp: 71 - 75 °C; 1H NMR (500 MHz, CDCl3): δ 7.42 (dd, 2H, J = 7.5, 7.5 Hz), 7.36 (m, 2H), 7.30 (m, 1H), 7.00 (s, 0.5 H), 6.81 (s, 0.5 H), 6.72 (m, 1H), 6.62 (m, 1H), 6.53 (m, 0.5 H), 5.94 (m, 2H), 5.13 (d, 1H, J=2.9 Hz), 5.07 (s, 1H), 4.81 (d, 1H, J = 8.3 Hz), 4.07 (m, 1H), 3.88 and 3.86 (s, 3H), 3.72 (m, 1H), 3.14 (dd, 1H, J = 14.4, 13 2.8 Hz), 2.7 (m, 2H), 2.64 (m, 1H); C NMR (125 MHz. CDCl3): δ 148.41, 148.40, 147.4, 147.10, 147.05, 146.5, 146.3, 145.9, 137.22, 137.21, 132.5, 131.4, 129.4, 128.6, 128.4, 127.87, 127.84, 127.32, 127.28 (C), 127.0, 126.7, 122.4, 114.8, 112.5, 112.0, 111.9, 111.8, 111.54, 111.50, 100.0, 108.0, 101.6, 100.8, 75.0, 71.4, 62.9, 62.5, 56.0, 42.6, 42.2, 28.7; HRESIMS: calcd. for C25H23BrO5 [M+H]+ 483.0807; found 483.0802
3.5. 1-(benzyloxy)-2-methoxy-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2 g]benzo[de]chromene (22)
Compound 21 (100 mg, 0.21 mmol), Pd(OAc)2, (9 mg, 0.04 mmol), K2CO3 (570 mg, 0.41 mmol) and tricyclohexylphosphine tetrafluoroborate (31 mg, 0.08 mmol) were dissolved in DMSO (1 mL) in a 10 mL microwave reaction vial. The reaction mixture was irradiated with microwaves at 140 ºC at 200 psi for 10 min. The resulting crude product was purified on a silica gel column using 20 % acetone- petroleum ether to afford 22 as a white solid (74 mg, 90 %). (The same reaction was for repeated on another 5 batches of compound to get a total of 375 mg of compound 22). Mp: 105 - 108 °C; 1H NMR (500 MHz. CDCl3): δ 7.99 (s, 1H), 7.36 (m, 2H), 7.30 (m, 3H), 6.72 (s, 1H), 6.63 (s, 1H), 5.96 (d, 1H, J=1.5 Hz), 5.93 (d, 1H, J=1.5 Hz), 4.84 (d, 1H, J= 10.5 Hz), 4.71 (d, 1H, J= 10.5 Hz), 4.48 (dd, 1H, J= 13.5, 5.0 Hz), 4.25 (dd, 1H, J=12.0, 6.5 Hz), 3.88 (s, 3H), 3.80 (td, 1H, J=10.0, 3.5 Hz), 3.13 (m, 1H), 2.83 (dd, 1H, J= 12.5, 7.5 Hz), 2.73 (t, 1H, J= 13.5 Hz), 2.61 (dd, 1H, J= 16.0, 3.0 Hz); 13C NMR (125 MHz. CDCl3): δ 152.6, 146.5, 143.1, 137.1, 129.2, 128.7 (×2), 128.1 (×2), 127.9, 127.48, 127.45, 126.8, 125.5, 110.9, 109.5, 108.5, 100.9, 74.7, 73.4, 64.8, 56.0, 36.6, 28.2; HRESIMS: calcd. for C25H22O5Na [M+Na]+ 425.1365; found 425.1360.
3.6. 2-methoxy-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2g]benzo[de]chromen-1-ol (23)
Compound 22 (300 mg, 0.75 mmol) was dissolved in 50 mL methanol-THF (1:1), and 10% Pd/C was added. The resulting suspension was stirred for 6 h under a hydrogen balloon. The mixture was filtered through celite and the filtrate evaporated in vacuo. The crude product thus produced was purified via silica gel column chromatography eluting in 20 % ethyl acetate – petroleum ether, to afford 23 as a white solid. (210 mg, 91 %) Mp: 108 - 112 °C; 1H NMR (500 MHz, CDCl3): δ 7.96 (s, 1H), 6.75 (s, 1H), 6.57 (s,1H), 6.12 (s, 1H), 5.97 (dd, 2H, J=7.5, 1.5 Hz), 4.54 (dd, 1H, J=13.5, 5.4 Hz), 4.25 (dd, 1H, J=11.4, 6.0 Hz), 3.92 (s, 3H), 3.81 (td, 1H, J=11.9, 3.6 Hz), 3.12 (m, 1H), 2.87 (dd, 1H, J=13.7, 5.3 Hz), 2.80, (t, 1H, J=13.5 Hz), 2.57 (dd, 1H, J= 16.2, 3.4 Hz); 13C NMR (125 MHz. CDCl3): δ 146.4, 146.2, 146.15, 146.12, 140.8, 128.9, 127.6, 125.6, 122.5, 118.6, 100.9, 73.5, 65.0, 56.3, 36.6, 28.0; HRESIMS: calcd. for C18H16O5 [M]+ 312.0998; found 312.0995.
3.7. General procedure for the synthesis of 24a – 24e
Compound 23 (0.03 g, 0.096 mmol) was dissolved in 10 mL acetonitrile and the appropriate alkyl halide (0.1152 mmol) and K2CO3 (0.198 g, 1.44 mmol) were added. The resulting reaction mixture was heated at reflux for 6 h. The reaction mixture was cooled to rt and was subjected to vacuum filtration. The filtrate was then evaporated and the resulting crude product was purified using on a silica gel preparative TLC plate using 30% ethyl acetate-petroleum ether as the eluting solvent to afford the respective compounds.
3.7.1. 1,2-dimethoxy-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2-g]benzo[de]chromene (24a)
White Solid; (29.4 mg, 95 %); Mp: 106 - 109 °C; 1H NMR (500 MHz. CDCl3): δ 7.99 (s, 1H), 6.78 (s, 1H), 6.64 (s,1H), 5.99 (d, 2H, J=4.4 Hz), 4.52 (dd, 1H, J=13.4, 5.25 Hz), 4.27 (dd, 1H, J=11.4, 6.5Hz), 3.91 (s, 3H), 3.84 (td, 1H, J=11.9, 3.5 Hz), 3.71 (s, 3H), 3.15 (m, 1H), 2.88 (dd, 1H, J=13.7, 5.4 Hz), 2.82 (t, 1H, J=13.5 Hz), 2.63 (dd, 1H, J=16.3, 2.9 Hz); 13C NMR (125 MHz. CDCl3): δ 152.5, 146.7, 146.5, 144.6, 129.4, 127.5, 127.3, 126.1, 125.4, 110.8, 108.9, 108.7, 100.9, 73.4, 64.8, 60.2, 55.9, 36.6, + 28.2; HRESIMS: calcd. for C19H18O5 [M]+ 326.1154; found 326.1151
3.7.2. 2-methoxy-1-propoxy-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2-g]benzo[de]chromene (24b)
White Solid; (29.2 mg, 86 %); Mp: 110 - 113 °C; 1H NMR (500 MHz. CDCl3): δ 8.03 (s, 1H), 6.77 (s, 1H), 6.63 (s,1H), 6.01 (s, 1H), 5.98 (s, 1H), 4.51 (dd, 1H, J=13.3, 5.2 Hz), 4.27 (dd, 1H, J=11.6, 6.5 Hz), 3.89 (s, 3H), 3.85 (m, 2H), 3.64 (m, 1H), 3.15 (m, 1H), 2.87 (t, 2H, J=13.7 Hz), 2.63 (dd, 1H, J=16.3, 3.0 Hz), 1.7 (m, 2H), 0.99 (t, 3H, J=7.4 Hz); 13C NMR (125 MHz. CDCl3): δ 152.6, 146.5, 146.4, 143.8, 129.2, 127.5, 127.1, 126.4, 125.7, 110.9, 109.3, 108.6, 100.9, 74.8, 73.4, 64.8, 56.0, 36.6, 28.2, 23.5, 10.5; HRESIMS: calcd. for C21H22O5 [M]+ 354.1467; found 354.1463.
3.7.3. 1-(allyloxy)-2-methoxy-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2-g]benzo[de]chromene (24c)
White Solid; (28.7 mg, 87 %); Mp: 120 - 124 °C; 1H NMR (500 MHz. CDCl3): δ 8.02 (s, 1H), 6.77 (s, 1H), 6.63 (s,1H), 6.07 (m, 1H), 6.00 (s, 1H), 5.98 (s, 1H), 5.31 (dd, 1H, J=17.2, 1.4 Hz), 5.20 (d, 1H, J=10.3 Hz), 4.52 (dd, 1H, J =5.35, 5.15 Hz), 4.41 (dd, 1H, J= 12.0, 5.9 Hz), 4.27 (m, 2H), 3.89 (s, 3H), 3.83 (td, 1H, J=11.9, 3.6 Hz), 3.14 (m, 1H), 2.88 (dd, 1H, J=13.6, 5.2 Hz), 2.80 (t, 1H, J= 13.5 Hz), 2.63 (dd, 1H, J=16.3, 3.0 Hz); 13C NMR (125 MHz. CDCl3): δ 152.6, 146.53, 146.50, 143.1, 134.1, 129.3, 127.5, 127.4, 126.5, 125.5, 117.6, 110.8, 109.2, 108.6, 100.9 73.7, 73.4, 64.8, 56.0, 36.6, 28.2, 23.5; HRESIMS: calcd. for C21H20O5Na [M+Na]+ 375.1209; found 375.1202
3.7.4 1-(cyclopropylmethoxy)-2-methoxy-4,5,6a,7-tetrahydro [1,3]dioxolo[4',5':4,5]benzo[1,2-g]benzo[de]chromene (24d)
White Solid; (33.2 mg, 95 %); Mp: 95 - 98 °C; 1H NMR (500 MHz. CDCl3): δ 8.13 (s, 1H), 6.76 (s, 1H), 6.62 (s,1H), 6.00 (s, 1H), 5.98 (s, 1H), 4.52 (dd, 1H, J=12.8, 4.2 Hz), 4.27 (dd, 1H, J=11.5, 6.75 Hz), 3.89 (s, 3H), 3.85 (m, 1H), 3.76 (m, 1H), 3.45 (t, 1H, J= 8.5 Hz), 3.15 (m, 1H), 2.87 (dd, 1H, J =13.7, 4.9 Hz), 2.80 (t, 1H, J= 13.5 Hz), 2.62 (d, 1H, J=16.3 Hz), 1.21 (m, 1H), 0.53 (d, 2H, J=8.1 Hz), 0.20 (m, 2H); 13C NMR (125 MHz. CDCl3): δ 152.6, 146.5, 146.4, 143.4, 129.2, 127.4, 127.2, 126.6, 126.7, 110.7, 109.4, 108.5, 100.9, 77.9, 73.4, 64.8, 55.9, 36.6, 28.2, 11.0, 3.4, 3.1; HRESIMS: calcd. for C22H22O5Na [M+Na]+ 389.1365; found 389.1363
3.7.5. 2-methoxy-1-(prop-2-yn-1-yloxy)-4,5,6a,7-tetrahydro-[1,3]dioxolo[4',5':4,5]benzo[1,2-g]benzo[de]chromene (24e)
White Solid; (29.7 mg, 90 %); Mp: 160 - 164 °C; 1H NMR (500 MHz. CDCl3): δ 8.01 (s, 1H), 6.76 (s, 1H), 6.64 (s, 1H), 6.01 (s, 1H), 5.99 (s, 1H), 4.60 (dd, 1H, J=15.2, 2.4 Hz), 4.50 (m, 1H), 4.27 (dd, 1H, J=11.3, 6.5 Hz), 3.90 (s, 3H), 3.82 (td, 1H, J=11.9, 3.6 Hz), 3.15 (m, 1H), 2.87 (dd, 2H, J=13.6, 5.2 Hz), 2.80 (t, 1H, J=13.5 Hz), 2.63 (dd, 1H, J=16.4, 2.9 Hz), 2.39 (t, 1H, J= 2.3 Hz); 13C NMR (125 MHz. CDCl3): δ 152.4, 146.6, 142.0, 129.4, 128.0, 127.5, 126.9, 125.4, 110.8, 109.3, 108.3, 101.0, 79.1, 75.1, 75.4, 64.7, 59.8, 55.9, 36.6, 28.2; HRESIMS: calcd. for C21H18O5Na [M+Na]+ 373.1052; found 373.1046
3.8. General procedure for the synthesis of 29a – 29e (Using 29a as a representative)
To a three-neck round bottom flask attached with a Dean-Stark apparatus, a solution of 27 (0.75 g, 4.12 mmol), aldehyde 28a (0.54 g, 2.06 mmol) and PTSA (catalytic) in toluene (100 mL) was added. The resulting reaction mixture was refluxed for 18 h. After 18 h, the reaction mixture was cooled to rt and transferred to a separatory funnel containing 30 mL water. The organic layer was extracted, and the aqueous layer extracted with a further 30 mL of ethyl acetate. The combined organic layer was dried over Na2SO4 and evaporated under reduced pressure. The resulting crude product was purified on a silica gel column using 10-30 % ethyl acetate-petroleum ether to afford 29a.
3.8.1. 1-(2-bromo-4,5-dimethoxybenzyl)-6,7-dimethoxyisochroman (29a)
White Solid; (0.64 g, 73 %); Mp: 107-110 °C; 1H NMR (500 MHz. CDCl3): δ 7.06 (s, 1H), 6.89 (s, 1H), 6.72 (s, 1H), 6.63 (s, 1H) 5.00 (d, 1H, J=6.1 Hz), 4.16 (m, 1H), 3.89 (s, 3H), 3.88 (s, 3H), 3.87 (s, 3H), 3.86 (s, 3H), 3.78 (m, 1H), 3.35 (dd, 1H, J = 14.4, 3.3 Hz), 3.06 (dd, 1H, J=14.4, 9.0 Hz ); 13C NMR (125 MHz. CDCl3): δ 148.2, 148.1, 147.7, 147.4, 130.3, 129.4, 126.1, 115.3, 114.7, 114.6, 111.4, 108.4, 75.3, 62.8, 56.13, 56.10, 56.0, 55.9, 42.4, 28.7; HRESIMS: calcd. for C20H23BrO5Na [M+Na]+ 445.0621; found 445.0618.
3.8.2. 1-(2-bromo-4,5-dimethoxybenzyl)-6,7dimethoxyisochroman (29b)
White Solid; (0.46 g, 72 %); Mp: 92-95 °C; 1H NMR (500 MHz, CDCl3): δ 7.47 (d, 1H, J = 8.8 Hz), 6.94 (d, 1H, J = 3.1 Hz), 6.71 6.71 (m, 2H), 6.64 (s, 1H), 5.03 (dd, 1H, J = 9.3, 3.0 Hz), 4.16 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H), 3.79 (m, 1H), 3.36 (dd, 1H, J = 14.3, 3.3 Hz), δ 3.08 (dd,1H, J=14.3, 9.3 Hz), 2.90 (m, 1H), δ 2.71 (td, 1H, J=16.1, 4.6 Hz); 13C NMR (125 MHz. CDCl3): δ 158.7, 147.7, 147.4, 139.3, 133.1, 129.5, 126.1, 117.9, 115.3, 113.8, 111.5, 108.4, 74.8, 62.8, 56.0, 55.9, 55.4, 43.0, 28.6; HRESIMS: calcd. for C19H21BrO4Na [M+Na]+ 415.0515; found 415.0516.
3.8.3. 1-(2-bromo-4-methoxybenzyl)-6,7-dimethoxyisochroman (29c)
White Solid; (0.52 g, 74 %); Mp: 98-103 °C; 1H NMR (500 MHz. CDCl3): δ 7.27 (d, 1H, J = 8.4 Hz), 7.16 (d, 1H, J=2.5 Hz), 6.85 (dd, 1H, J = 8.4, 2.5 Hz), 6.71 (s, 1H), 6.63 (s, 1H), 4.99 (dd, 1H, J = 9.1, 2.2 Hz), 4.14 (m, 1H), 3.89 (s, 3H), 3.85 (s, 3H), 3.81 (s, 3H), 3.79 (m, 1H), 3.34 (dd, 1H, J = 14.4, 3.3 Hz), 3.06 (m, 1H), 2.88 (m, 1H), δ 2.70 (dt, 1H, J=16.0, 4.5 Hz); 13C NMR (125 MHz. CDCl3): δ 158.7, 147.7, 147.4, 132.5, 130.3, 129.6, 126.1, 124.8, 117.8, 113.8, 113.5, 111.4, 108.4, 62.5, 56.0, 55.9, 55.5, 42.0, 28.6; HRESIMS: calcd. for C19H21BrO4Na [M+Na]+ 415.0515; found 415.0514.
3.8.4. 1-(2-bromobenzyl)-6,7-dimethoxyisochroman (29d)
Colourless Oil; (0.65 g 59 %); 1H NMR (500 MHz. CDCl3): δ 7.60 (d, 1H, J = 8.0 Hz), 7.37 (dd, 1H, J = 1.5, 7.6 Hz), 7.32 (m, 1H), 7.13 (ddd, 1H, J = 7.6, 7.6, 1.5 Hz), 6.70 (s, 1H), 6.64 (s, 1H), 5.05 (dd, 1H, J=9.3, 3.0 Hz), 4.15 (m, 1H), 3.89 (s, 3H), 3.80 (s, 3H), 3.78 (m,1H), 3.40 (dd,1H, J = 14.3, 3.3 Hz), 3.12 (m, 1H), 2.89 (m,1H), 2.73 (td, 1H, J=13.3, 7.6 Hz); 13C NMR (125 MHz. CDCl3): δ 147.7, 147.4, 138.3, 132.7, 132.3, 129.6, 128.1, 127.3, 126.3, 126.1, 114.4, 108.4, 74.7, 62.5, 55.9, 55.9, 42.9, 28.6; HRESIMS: calcd. for C18H19BrO3Na [M+Na]+ 385.0410; found 385.0409.
3.8.5. 1-((6-bromobenzo[d][1,3]dioxol-5-yl)methyl)-6,7-dimethoxyisochroman (29e) (a mixture of rotamers as evident from NMR data - all signals observed are reported)
White Solid; (0.18 g, 72 %; Mp: 91-94 °C; 1H NMR (500 MHz, CDCl3): δ 7.02 (s, 1H), 6.86 (s, 1H), 6.71 (s, 1H), 6.61 (s, 1H), 5.96 (d, 1H, J = 1.3 Hz), 5.95 (d, 1H, J = 1.3 Hz), 4.93 (dd, 1H, J = 9.3, 1.7 Hz), 4.12 (m, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.75 (m, 1H), 3.30 (dd, 1H, J= 3.0, 14.5 Hz), 2.97 (dd, 1H, J = 14.5, 9.6 Hz), 2.87 (m, 1H), 2.68 (td, 1H, J=16.0, 4.4 Hz); 13C NMR (125 MHz, CDCl3): δ 147.7, 147.4, 147.2, 147.1, 131.4, 129.4, 126.1, 112.5, 111.7, 111.4, 108.3, 101.6, 75.0, 62.6, 56.0, 55.9, 42.7, 28.6; HRESIMS: calcd. for C19H19BrO5Na [M+Na]+ 429.0314; found 429.0312.
3.9. General procedure for synthesis of 30a – 30d and 24a (Using 30a as a representative)
Compound 29a (0.05 g, 0.12 mmol), Pd(OAc)2 (0.006 g, 0.02 mmol), K2CO3 (0.032 g, 0.24 mmol) and tricyclohexyl phosphine tetrafluoroborate (0.014 g, 0.04 mmol) were dissolved in DMSO (1 mL) in a microwave reaction vial. The reaction mixture was then irradiated with microwaves at 140 ºC, 200 psi for 10 min. The resulting crude product was purified directly on a silica gel column using 20 % acetone- petroleum ether to afford 30a.
3.9.1. 1,2,9,10-tetramethoxy-4,5,6a,7-tetrahydrodibenzo[de,g]chromene (30a)
White Solid, (35.9 mg, 89 %); Mp: 110-112°C; 1H NMR (500 MHz. CDCl3): δ 8.12 (s, 1H), 6.77 (s, 1H), 6.62 (s, 1H), 4.54 (dd, 1H, J=12.8, 6.0 Hz), 4.26 (dd, 1H, J= 11.4, 6.4 Hz), 3.92 (s, 3H), δ 3.91 (s, 3H) 3.90 (s, 3H), 3.82 (ddd, 1H, J=11.8, 11.8, 3.6 Hz), 3.68 (s, 3H), 3.14 (m, 1H), δ 2.89 (m, 2H), 2.62 (dd, 1H, J=11.4, 3.2 Hz); 13C NMR (125 MHz. CDCl3): δ 152.5, 148.2, 147.6, 144.4, 127.9, 127.6, 127.4, 126.1, 124.4, 111.5, 111.2, 110.6, 73.5, 64.9, 60.2, 55.9, 55.88, 55.85, 36.1, 28.3; HRESIMS: calcd. for C20H22O5Na [M+Na]+ 365.1365; found 365.1360.
3.9.2. 1,2,9-trimethoxy-4,5,6a,7-tetrahydrodibenzo[de,g]chromene (30b)
White Solid, (33 mg, 84 %); Mp: 107-110°C; 1H NMR (500 MHz. CDCl3): δ 8.37 (d, 1H, J= 8.8 Hz), 6.88 (dd, 1H, J = 8.8, 2.5 Hz), 6.83 (d, 1H, J=2.5 Hz), 6.63 (s, 1H), 4.57 (dd, 1H, J=13.0, 5.4 Hz), 4.29 (dd, 1H, J=11.4, 6.5 Hz), 3.91 (s, 3H), 3.87 (s, 3H), 3.83 (m, 1H), 3.70 (s, 3H), 3.15 (m, 1H), 2.94 (m, 2H), 2.64 (dd, 1H, J = 16.3, 2.8 Hz); 13C NMR (125 MHz. CDCl3): δ 158.8, 152.5, 144.6, 136.9, 129.7, 127.5, 127.4, 126.0, 124.7, 113.8, 112.4, 110.6, 73.3, 64.9, 60.1, 55.9, 55.2, 37.0, 28.2; HRESIMS: calcd. for C19H20O5 [M+H]+ 313.1440; found 313.1434.
3.9.3. 1,2,10-trimethoxy-4,5,6a,7-tetrahydrodibenzo[de,g]chromene (30c)
Brown Oil, (34.1 mg, 86 %); 1H NMR (500 MHz. CDCl3): δ 8.09 (d, 1H, J=2.1 Hz), 7.20 (d, 1H, J = 8.3 Hz), 6.84 (dd, 1H, J=8.3, 2.1 Hz), 6.69 (s, 1H), 4.54 (dd, 1H, J=13.5, 4.95 Hz), 4.29 (dd, 1H, J =11.4, 6.6 Hz), 3.92 (s, 3H), 3.86 (s, 3H), 3.82 (dd, 1H, J = 11.5, 3.45 Hz), 3.73 (s, 3H), 3.17 (m, 1H), 2.95 (dd, 1H, J = 13.5, 5.1 Hz), 2.83 (t, 1H, J=13.5 Hz), 2.65 (dd, 1H, J = 16.2, 2.5 Hz); 13C NMR (125 MHz. CDCl3): δ 158.7, 152.4, 145.2, 132.8, 129.0, 128.3, 127.6, 127.2, 126.1, 113.8, 112.2, 110.6, 73.6, 64.8, 60.3, 55.9, 55.4, 35.7, 28.3; HRESIMS: calcd. for C19H20O5 [M+H]+ 313.1440; found 313.1434.
3.9.4. 1,2-dimethoxy-4,5,6a,7-tetrahydrodibenzo[de,g]chromene (30d)
Colorless Oil, (30.6 mg, 79 %); 1H NMR (500 MHz. CDCl3): δ 8.42 (d, 1H, J = 7.9 Hz), 7.28 (m, 3H), 6.68 (s, 1H), 4.57 (dd, 1H, J=13.5, 5.2 Hz), 4.29 (dd, 1H, J =11.4, 6.4 Hz), 3.92 (s, 3H), 3.84 (ddd, 1H, J=11.8, 11.8, 3.6 Hz), 3.71 (s, 3H) δ 3.18 (m, 1H), 3.00 (dd, 1H, J = 13.7, 5.15 Hz), 2.91 (t, 1H, J=13.6 Hz), 2.65 (dd, 1H, J = 16.4, 3.2 Hz); 13C NMR (125 MHz. CDCl3): δ 151.4, 150.4, 134.0, 128.4, 128.3, 127.7, 127.3, 127.0, 126.9, 124.2, 111.6, 106.8, 73.4, 65.5, 59.8, 56.5, 36.6, 30.0; HRESIMS: calcd for C18H16O3 [M]+ 280.1096; found 280.1094.
3.10. 2-(3,4,6,7-tetramethoxyphenanthren-1-yl)ethanol (32)
33 % HBr-AcOH (20 mL) was added to a two neck round bottom flask containing compound 30a (0.5 g, 1.46 mmol) at 10 °C. The resulting reaction mixture was allowed to stir at 10 °C for 30 min after which water (10 mL) was added. The mixture was extracted with dichloromethane (2 ×15 mL), and the combined organic layer filtered, dried over Na2SO4 and evaporated to afford a crude residue. The residue was dissolved in methanol (20 mL) and 20 % aqueous NaOH solution (20 mL) added. The reaction mixture was stirred at rt for 2h, after which the methanol was evaporated. The reaction mixture was extracted with dichloromethane (3 ×10 mL). The combined organic layer was dried over Na2SO4, and evaporated under reduced pressure to get compound 32 (0.18 g) as a pale yellow solid representing an overall yield of 98 % (0.49 g) from 30a. (In a separate experiment the mixture of 31 and 32 was separated by column chromatography eluting in 30-70% ethyl acetate-hexanes. Hydrolysis of 31 as previously described for the mixture gave quantitative conversion to 32).
3.11. 2-(3,4,6,7-tetramethoxyphenanthren-1-yl)ethyl acetate (31)
Brown Oil; 1H NMR (500 MHz. CDCl3): δ 9.28 (s, 1H), 7.80 (d, 1H, J = 9.1 Hz), 7.56 (d, 1H, J = 9.1 Hz), 7.21 (s, 1H), 7.20 (s, 1H), 4.41 (t, 2H, J = 7.5 Hz), 4.08 (s, 3H), 4.05 (s, 3H), 4.03 (s, 3H), 3.92 (s, 3H), 3.41 (t, 2H, J = 7.5 Hz), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 173.4, 152.6, 151.1, 150.8, 147.5, 132.8, 130.5, 128.2, 127.3, 127.0, 126.6, 122.8, 116.6, 111.2, 110.1, 66.9, 62.3, 58.9, 58.0, 35.4, 32.0, 23.3; HRESIMS: calcd. for C20H24O6Na [M+Na]+ 407.1471; found 407.1462.
3.12. 2-(3,4,6,7-tetramethoxyphenanthren-1-yl)ethanol (32)
Pale Yellow Solid; Mp: 134 - 136 °C; H NMR (500 MHz. CDCl3): δ 9.21 (s, 1H), 7.75 (d, 1H, J = 9.0 Hz), 7.50 (d, 1H, J = 9.0 Hz), 7.19 (s, 1H), 7.17 (s, 1H), 4.06 (s, 3H), 4.02 (s, 3H), 4.01 (s, 3H), 3.99 (t, 2H, J = 6.6 Hz), 3.89 (s, 3H), 3.33 (t, 2H, J = 6.6 Hz); 13C NMR (125 MHz. CDCl3): δ 150.2, 148.8, 148.4, 131.4, 128.2, 125.8, 124.8, 124.8, 124.3, 120.6, 114.5, 108.8, 107.7, 63.3, 60.0, 56.5, 55.8, 55.7, 31.2; HRESIMS: calcd. for C20H22O5Na [M+Na]+ 365.1365; found 365.1358.
3.13. N,N-dimethyl-2-(3,4,6,7-tetramethoxyphenanthren-1-yl)ethanamine (10)
To a solution of compound 32 (0.11g, 0.33 mmol) in dichloromethane (30 mL), Dess-Martin periodinane (0.15g, 0.36 mmol) was added. The resulting reaction mixture was allowed to stir at rt for 15 min, after which it was filtered through a bed of silica gel. The filtrate was evaporated to afford the aldehyde as a brown residue. The crude aldehyde was then dissolved in anhydrous dichloromethane (40 mL) and cooled to 0 °C. Dimethylamine (1N THF solution, 0.82 mL, 0.81 mmol) was added and the reaction mixture was allowed to stir for 15 min at 0 °C, after which Na(AcO)3BH (0.172 g, 0.81 mmol) was added. The solution was stirred at 0 °C for a further 1h and then at rt for 1h. Water (15 mL) was then added and the mixture was extracted with dichloromethane (2 × 20 mL). The combined organic layer was dried over Na2SO4, and evaporated to get a crude residue which was purified on a silica gel column using 5 % - 20 % methanol-dichloromethane, to afford 10 as a brown oil (70 mg, 59%). Spectral data are in accordance with literature values.20
Supplementary Material
Fig. 1.
Examples of natural products and synthetic compounds containing an isochroman motif
Fig. 2.
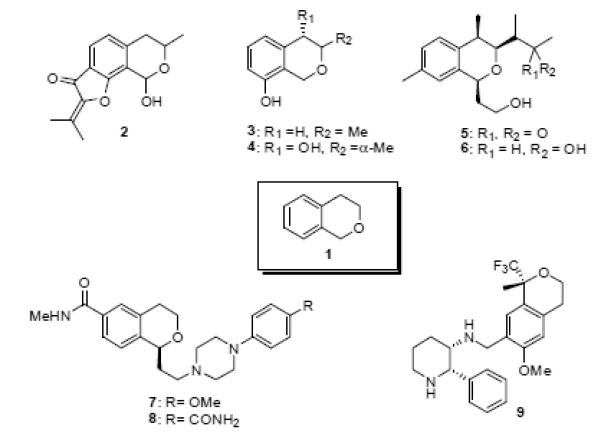
Structures of the phenanthrene alkaloid N-methyl seco-glaucine (10) and the typical aporphine alkaloid glaucine (11)
Fig. 3.
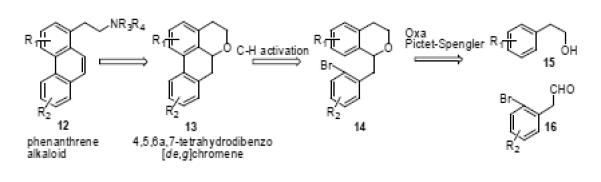
Retrosynthetic strategy for preparation of 4,5,6a,7-tetrahydrodibenzo[de,g]chromene en route to phenanthrene alkaloids
Scheme 3.
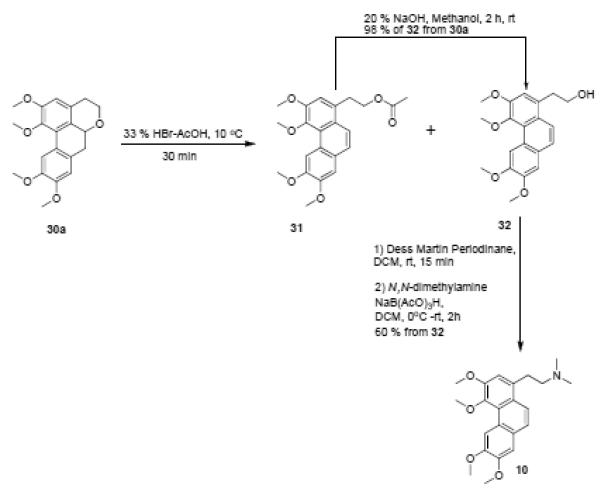
Isochroman cleavage and synthesis of compound 10
Acknowledgments
This publication was made possible by Grant Numbers 1SC1GM092282 and G12RR003037 from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Experimental procedures for synthesis of key intermediates and 1H and 13C NMR spectra of new compounds.
References and notes
- 1.Togna GI, Togna AR, Franconi M, Marra C, Guiso M. J. Nutr. 2003;133:2532–2536. doi: 10.1093/jn/133.8.2532. [DOI] [PubMed] [Google Scholar]
- 2.Schreurs RHMM, Sonneveld E, van d. S. P. T., van d. B. B., Seinen W. Toxicol. Lett. 2005;156:261–275. doi: 10.1016/j.toxlet.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Lakshminarayana N, Rajendra PY, Gharat L, Thomas A, Ravikumar P, Narayanan S, Srinivasan CV, Gopalan B. Eur. J. Med. Chem. 2009;44:3147–3157. doi: 10.1016/j.ejmech.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Bernini R, Crisante F, Fabrizi G, Gentili P. Curr. Org. Chem. 2012;16:1051–1057. [Google Scholar]
- 5.Ogawa A, Murakami C, Kamisuki S, Kuriyama I, Yoshida H, Sugawara F, Mizushina Y. Bioorg. Med. Chem. Lett. 2004;14:3539–43. doi: 10.1016/j.bmcl.2004.04.050. [DOI] [PubMed] [Google Scholar]
- 6.Trisuwan K, Rukachaisirikul V, Sukpondma Y, Phongpaichit S, Preedanon S, Sakayaroj J. Tetrahedron. 2010;66:4484–4489. [Google Scholar]
- 7.Hashida J, Niitsuma M, Iwatsuki M, Mori M, Ishiyama A, Namatame M, Nishihara-Tsukashima A, Matsumoto A, Ara I, Takahashi Y, Yamada H, Otoguro K, Shiomi K, Omura S. J. Antibiot. 2012;65:197–202. doi: 10.1038/ja.2011.139. [DOI] [PubMed] [Google Scholar]
- 8.Fleishaker JC, Pearson LK, Knuth DW, Gomez-Mancilla B, Francom SF, McIntosh MJ, Freestone S, Azie NE. Int. J. Clin. Pharmacol. Ther. 1999;37:487–92. [PubMed] [Google Scholar]
- 9.Gomez-Mancilla B, Cutler NR, Leibowitz MT, Spierings EL, Klapper JA, Diamond S, Goldstein J, Smith T, Couch JR, Fleishaker J, Azie N, Blunt DE. Cephalalgia. 2001;21:727–32. doi: 10.1046/j.1468-2982.2001.00208.x. [DOI] [PubMed] [Google Scholar]
- 10.Ennis MD, Ghazal NB, Hoffman RL, Smith MW, Schlachter SK, Lawson CF, Im WB, Pregenzer JF, Svensson KA, Lewis RA, Hall ED, Sutter DM, Harris LT, McCall RB. J. Med. Chem. 1998;41:2180–3. doi: 10.1021/jm980137o. [DOI] [PubMed] [Google Scholar]
- 11.Shishido Y, Wakabayashi H, Koike H, Ueno N, Nukui S, Yamagishi T, Murata Y, Naganeo F, Mizutani M, Shimada K, Fujiwara Y, Sakakibara A, Suga O, Kusano R, Ueda S, Kanai Y, Tsuchiya M, Satake K. Bioorg. Med. Chem. 2008;16:7193–205. doi: 10.1016/j.bmc.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 12.Caron S, Do NM, Sieser JE, Arpin P, Vazquez E. Org. Process Res. Dev. 2007;11:1015–1024. [Google Scholar]
- 13.Kovacs A, Vasas A, Hohmann J. Phytochemistry (Elsevier) 2008;69:1084–1110. doi: 10.1016/j.phytochem.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 14.Song JI, Kang YJ, Yong H-Y, Kim YC, Moon A. Oncol. Rep. 2012;27:813–818. doi: 10.3892/or.2011.1551. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y-C, Guh J-H, Teng C-MJ. Biomed. Sci. (Dordrecht, Neth.) 2005;12:113–121. doi: 10.1007/s11373-004-8171-y. [DOI] [PubMed] [Google Scholar]
- 16.Lee YH, Park JD, Baek NI, Kim SI, Ahn BZ. Planta Med. 1995;61:178–80. doi: 10.1055/s-2006-958043. [DOI] [PubMed] [Google Scholar]
- 17.Boger DL, Mitscher LA, Mullican MD, Drake SD, Kitos PJ. Med. Chem. 1985;28:1543–7. doi: 10.1021/jm00148a031. [DOI] [PubMed] [Google Scholar]
- 18.Okwu DE, Nnamdi FU. J. Chem. Pharm. Res. 2011;3:27–33. [Google Scholar]
- 19.Wang Y-G, Wang Y-L, Zhai H-F, Liao Y-J, Zhang B, Huang J-M. Nat. Prod. Res. 2012;26:1234, 1239. doi: 10.1080/14786419.2011.561491. [DOI] [PubMed] [Google Scholar]
- 20.Blanco O, Castedo L, Cid M, Seijas JA, Villaverde C. Heterocycles. 1990;31:1077–80. [Google Scholar]
- 21.Teng C-M, Hsueh C-M, Chang Y-L, Ko F-N, Lee S-S, Liu KC-S. J. Pharm. Pharmacol. 1997;49:706, 711. doi: 10.1111/j.2042-7158.1997.tb06096.x. [DOI] [PubMed] [Google Scholar]
- 22.Li S, Han L, Sun L, Zheng D, Liu J, Fu Y, Huang X, Wang Z. Molecules. 2009;14:5042–5053. doi: 10.3390/molecules14125042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Estelles R, Milian L, Abu NYN, Mateo T, Cerda-Nicolas M, Losada M, Ivorra MD, Issekutz AC, Cortijo J, Morcillo EJ, Blazquez MA, Sanz M-JJ. Leukocyte Biol. 2005;78:696–704. doi: 10.1189/jlb.0105048. [DOI] [PubMed] [Google Scholar]
- 24.Mollataghi A, Coudiere E, Hadi AHA, Mukhtar MR, Awang K, Litaudon M, Ata A. Fitoterapia. 2012;83:298–302. doi: 10.1016/j.fitote.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Martinez E, Estevez JC, Estevez RJ, Castedo L. Tetrahedron. 2001;57:1981–1986. [Google Scholar]
- 26.Martinez E, Estevez JC, Estevez RJ, Castedo L. Tetrahedron. 2001;57:1973–1979. [Google Scholar]
- 27.Kini SV, Ramana MMV. etrahedron Lett. 2004;45:4171–4173. [Google Scholar]
- 28.Seijas JA, De LAR, Villaverde MC, Castedo L. J. Chem. Soc., Chem. Commun. 1985:839–40. [Google Scholar]
- 29.Estevez JC, Villaverde MC, Estevez RJ, Seijas JA, Castedo L. Can. J. Chem. 1990;68:964–8. [Google Scholar]
- 30.Nimgirawath S, Chaturonrugsamee S. J. Chin. Chem. Soc. (Taipei, Taiwan) 2006;53:443–447. [Google Scholar]
- 31.Ponnala S, Chaudhary S, Gonzalez-Sarrias A, Seeram NP, Harding WW. Bioorg. Med. Chem. Lett. 2011;21:4462–4464. doi: 10.1016/j.bmcl.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pecic S, Makkar P, Chaudhary S, Reddy BV, Navarro HA, Harding WW. Bioorg. Med. Chem. 2010;18:5562–5575. doi: 10.1016/j.bmc.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaudhary S, Pecic S, Le GO, Navarro HA, Harding WW. Bioorg. Med. Chem. Lett. 2009;19:2530–2532. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Indra B, Matsunaga K, Hoshino O, Suzuki M, Ogasawara H, Ishiguro M, Ohizumi Y. Can. J. Physiol. Pharmacol. 2002;80:198–204. doi: 10.1139/y02-019. [DOI] [PubMed] [Google Scholar]
- 35.Chaudhary S, Pecic S, Le GO, Harding WW. Tetrahedron Lett. 2009;50:2437–2439. doi: 10.1016/j.tetlet.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhary S, Harding WW. Homoaporphine synthesis via microwave-assisted direct arylation. 2010. [DOI] [PMC free article] [PubMed]
- 37.Larghi EL, Kaufman TS. Eur. J. Org. Chem. 2011;2011:5195–5231. [Google Scholar]
- 38.Guiso M, Betrow A, Marra C. Eur. J. Org. Chem. 2008:1967–1976. [Google Scholar]
- 39.Larghi EL, Kaufman TS. Synthesis. 2006:187–220. [Google Scholar]
- 40.Stanislawski PC, Willis AC, Banwell MG. Org. Lett. 2006;8:2143–2146. doi: 10.1021/ol060642c. [DOI] [PubMed] [Google Scholar]
- 41.Liu Q, Ferreira EM, Stoltz BM. J. Org. Chem. 2007;72:7352–7358. doi: 10.1021/jo0710883. [DOI] [PubMed] [Google Scholar]
- 42.Guiso M, Bianco A, Marra C, Cavarischia C. Eur. J. Org. Chem. 2003:3407–3411. [Google Scholar]
- 43.Lewis FD, Reddy GD, Cohen BE. Tetrahedron Lett. 1994;35:535–8. [Google Scholar]
- 44.Seijas JA, Rodriguez d. L. A., Villaverde C, Castedo L. Heterocycles. 1985;23:3079–84. [Google Scholar]
- 45.Wijeratne EMK, Lankananda BD, Tezuka Y, Nagaoka T, Gunatilaka AA. L. J. Nat. Prod. 2001;64:1465–1467. doi: 10.1021/np0102399. [DOI] [PubMed] [Google Scholar]
- 46.Battersby AR, LeCount DJ, Garratt S, Thrift RI. Tetrahedron. 1961;14:46–53. [Google Scholar]
- 47.Zhang Q-W, Xiang K, Tu Y-Q, Zhang S-Y, Zhang X-M, Zhao Y-M, Zhang T-C. Chem.--Asian J. 2012;7:894–898. doi: 10.1002/asia.201101029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.