

Abstract
Electrospun polyurethane fibers doped with nitric oxide (NO)-releasing silica particles are presented as novel macromolecular scaffolds with prolonged NO-release and high porosity. Fiber diameter (119–614 nm) and mechanical strength (1.7–34.5 MPa of modulus) were varied by altering polyurethane type and concentration, as well as the NO-releasing particle composition, size, and concentration. The resulting NO-releasing electrospun nanofibers exhibited ~83% porosity with flexible plastic or elastomeric behavior. The use of N-diazeniumdiolate- or S-nitrosothiol-modified particles yielded scaffolds exhibiting a wide range of NO release totals and durations (7.5 nmol mg−1–0.12 μmol mg−1 and 7 h to 2 weeks, respectively). The application of NO-releasing porous materials as coating for subcutaneous implants may improve tissue biocompatibility by mitigating the foreign body response and promoting cell integration.
Keywords: nitric oxide, controlled release, electrospun fiber, porous biomaterial, polyurethane
Introduction
Nitric oxide (NO) is a key physiological mediator of vasodilation, angiogenesis, wound healing, and phagocytosis, all of which are highly dependent on NO concentration.1 As many disease states and health ailments are mitigated by NO, exogenous NO donors are widely studied as potential therapeutic agents.2–5 In particular, macromolecular NO donor scaffolds have been the focus of much research due to their ability to store large amounts of NO and facilitate biological action. Indeed, the NO release achieved using xerogels,6–9 silica nanoparticles,10–12 dendrimers,13–16 biodegradable polyesters,17–20 and medical-grade polyurethanes21–23 has demonstrated utility to modulate wound healing,24, 25 kill bacteria and cancer cells,26–28 and improve the analytical performance of chemical sensors.29–31 Silica nanoparticles modified with NO donors represent an attractive NO-release vehicle due to straightforward synthesis, ability to achieve significant NO payloads and tunable NO-release kinetics, and their inherent low toxicity.9, 10 Previously, we employed polymers doped with NO donor-modified silica particles to prepare NO-releasing glucose sensor membranes.23 Nitric oxide release from the sensor membranes was tuned by altering the silica particle concentration, NO donor type, water uptake properties of the polyurethane, and the use of an overlaying polymer coating of variable thickness.23 Unfortunately, the utility of these membranes for sensor applications was limited due to an inverse relationship between NO-release duration and analyte (i.e., glucose) permeability.23 A more porous NO-releasing coating is thus desirable to maintain adequate analyte permeability.
Electrospinning of polymers is a straightforward method for preparing highly porous materials consisting of fibers.32, 33 The electrospinning process involves propelling an electrically charged viscoelastic jet of polymer solution to a grounded collector via a high voltage electrostatic field.33 As the jet of polymer solution travels through the air to the grounded collector, polymer nanofibers solidify upon solvent evaporation, resulting in a non-woven web or mat of fibers.33 Some advantages of polymeric fibers over bulk polymer films include large surface area to volume ratios, flexibility in surface functionality, and superior mechanical properties (e.g., stiffness and tensile strength).33–35 Additionally, the microporosity of the non-woven fiber mat is believed to be ideal for promoting tissue integration,36, 37 suggesting that these materials may be suitable as outer sensor membranes for subcutaneous glucose sensors.38 With physical properties that mimic the extracellular matrix, the use of electrospun fibers has been confirmed to promote cell proliferation and differentiation,36, 37, 39, 40 enhance tissue-scaffold integration, and decrease fibrous encapsulation compared to bulk polymer films.41, 42 Much research is now focused on developing electrospun fibers as tissue engineering scaffolds, wound dressings, and implant and medical prostheses coatings.33–35
The versatility of the electrospinning process has enabled the fabrication of fiber mats capable of releasing silver,43–45 dexamethasone,46 and NO.19, 47, 48 With respect to NO release, we previously reported on polyurethane and poly(vinyl chloride) fibers capable of NO release by doping a low molecular weight N-diazeniumdiolate NO donor (1-[(2-carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate or PROLI/NO) into the polymer solution prior to electrospinning.47 While the NO-release kinetics of the PROLI/NO-doped fibers proved to be variable depending on the polymer composition and fiber diameter, the NO payloads and release durations were limited.47 We hypothesize that the incorporation of macromolecular NO-release vehicles (i.e., silica particles) might enhance NO-release totals and durations compared to those obtained using PROLI/NO as a dopant. Herein, we report the fabrication of macromolecular NO release scaffold-doped fibers as a function of both the NO-releasing particle composition and polymer fiber characteristics (e.g., diameter and water uptake). Due to the size of the particle dopants (50–400 nm), careful attention is focused on the stability and mechanical properties of the ensuing fibers.
Experimental
Materials
Tecoflex (SG-85A) and Tecophilic (HP-93A-100) polyurethanes were gifts from Thermedics (Woburn, MA). Tecoplast (TP-470) polyurethane was provided by Lubrizol (Cleveland, OH). The following silanes for synthesizing silica particles were purchased from Gelest (Morrisville, PA): N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3), N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3), 3-mercaptopropyltrimethoxysilane (MPTMS), tetramethoxysilane (TMOS) and tetraethoxysilane (TEOS). All other salts and solvents were laboratory grade and purchased from Fisher Scientific (St. Louis, MO). Water (18.2 MΩ·cm; total organic content <6 ppb) was purified using a Millipore Milli-Q Gradient A-10 purification system (Bedford, MA). Nitrogen, argon, and nitric oxide gases were purchased from Airgas National Welders Supply (Durham, NC).
Synthesis of nitric oxide-releasing silica particles
Nitric oxide-releasing silica particles were synthesized as previously described via the co-condensation of an aminosilane (i.e., AEAP3 or AHAP3) or a mercaptosilane (i.e., MPTMS) at 65–75 mol% with a backbone silane (i.e., TEOS or TMOS).10, 11, 49 To form N-diazeniumdiolate NO donors, the amine-containing particles were exposed to 10 atm NO gas for 3 d in the presence of sodium methoxide at room temperature with constant stirring in a Parr pressure vessel.49 S-Nitrosothiol NO donor-modified particles were prepared by treating the thiol-containing nanoparticles with acidified nitrite for 2 h in the dark at 0 °C. All NO-releasing particle systems were stored in a vacuum-sealed, dark container at −20 °C until further use. Nitric oxide-release characteristics and sizes of the particles are provided in Supporting Information.
Nitric oxide-releasing silica particle-doped polyurethane fiber formation
Nitric oxide-releasing silica particle-doped electrospun fibers were fabricated using a custom electrospinning apparatus consisting of a Series 205B High Voltage Power Supply from Bertan Associates, Inc. (Hicksville, NY), a Kent Scientific Genie Plus syringe pump (Torrington, CT), and a circular steel disk (23 cm diameter) collector.47 Voltage was applied to a standard stainless steel blunt-tip needle (22 gauge and 0.508 mm ID; Jensen Global, Santa Barbara, CA) attached to a solution-filled syringe positioned atop the syringe pump. The grounded collector was covered in aluminum foil (for ease of sample collection) and mounted perpendicular to the direction of the syringe at a distance of 15 cm. Fiber mats were prepared by electrospinning the polymer solution at an applied voltage of 15 kV and a flow rate of 15 μL min−1. The resulting fiber mats were collected from the center of the disk collector for further evaluation. Polyurethane solutions containing NO-releasing silica particles were prepared by first dissolving the polymer in 1.6 mL of a 3:1 (v/v) tetrahydrofuran (THF): N,N-dimethylformamide (DMF) solution, then mixing in a suspension of NO donor-modified silica particles dispersed in methanol (400 μL). The final concentration of polymer in this cocktail ranged from 8–16% (w/v) with particles embedded at 1–10 wt% of the polymer mass. Solution viscosity was determined using a capillary-viscometer (Schott AVS 360; Hofheim, Germany) at room temperature. The conductivity of the polyurethane solutions was measured using a Malvern Nano Series Zetasizer (Malvern, England) operated in zeta potential mode, and consisted of an average of 5 measurements.
Characterization of NO-releasing silica particle-doped electrospun fibers
Electrospun fibers were imaged using an environmental scanning electron microscope (ESEM) (Quanta 200 field emission gun; FEI company; Hillsboro, OR) with a large-field detector (LFD) under low vacuum (i.e., 0.38 Torr). Samples were prepared without an additional metal coating in order to observe particles embedded in the fibers. Reported fiber diameters were measured with ImageJ software (NIH, Bethesda, MD) and reported as averages of at least 75 measurements per sample from three electrospun mats.
The surface area of the fiber mat was measured using a Micromeritics Tristar II 3020 Surface Area and Porosity Analyzer (Norcross, GA). The percent porosity of the fiber mat was calculated according to the following equations (1) and (2) below.50–52
| (1) |
| (2) |
The tensile strain-strength of the electrospun fiber mats was characterized using an Instron 5566 electromechanical tensile tester (Norwood, MA) at a cross-head speed of 10 mm min−1. Fiber mats were cut into strips (10 mm × 29 mm) for testing, with thicknesses determined by ESEM.53 Modulus was defined as the slope of the tensile stress-strain curve showing elastic deformation. The standard deviation was based on measurements from three different batches. Water uptake was evaluated by weighing a section of the fiber mat before and after soaking in PBS for 3 h.23 Leaching of silica particles from the fibers was evaluated by quantifying the concentration of silicon (Si) in solutions that the particle-doped electrospun fiber mats had been immersed (15 mL phosphate buffered saline (PBS) and incubated at 37 °C for 7 d). Silicon concentrations in the PBS soak solutions were determined using inductively coupled plasma-optical emission spectroscopy (ICP-OES; Teledyne Leeman labs; Hudson, NH) in an axial configuration at 251.611 nm. Prior to analysis, 0.05–10 ppm silica particle standard solutions (in PBS) were used to construct a calibration curve.
Nitric oxide release was measured using a Sievers chemiluminescence Nitric Oxide Analyzer (NOA, Model 280i; Boulder, CO). To determine NO flux, electrospun samples were placed in a solution of deoxygenated PBS (0.01 M, pH 7.4) at 37 °C. Liberated NO was carried to the NOA by continuously purging the solution and vessel head space with nitrogen gas at a controlled rate as previously described.11 The NOA was calibrated using a standard 26.80 ppm NO gas (balance nitrogen) and air passed through a Sievers NO zero filter. The sample vessel was shielded from light to prevent light-initiated NO release from S-nitrosothiol-based NO donors.11 Total NO payloads were determined spectrophotometrically by measuring the conversion of NO to nitrite using the Griess assay.54 After soaking NO-releasing fibers in PBS at 37 °C for a period exceeding their NO release, 50 μL of the sample solution was mixed with 50 μL of 1% (w/v) sulfanilamide in 5% (v/v) phosphoric acid and 0.1% (w/v) N-(1-naphthyl)ethylenediamine dihydrochloride, and incubated at room temperature for 10 min. The absorbance of this solution was then measured at 540 nm using a Labsystem Multiskan RC microplate spectrophotometer (Helsinki, Finland). Total nitrite concentration was determined using a calibration curve constructed with standard nitrite solutions. Of note, the total NO concentration measured by the Griess assay agreed with that obtained from chemiluminescence analysis, confirming that these materials released NO and not nitrite.54
Results and Discussion
Fabrication of nitric oxide-releasing silica particle-doped electrospun fiber mats
The therapeutic potential of active NO release from an implant surface has been widely discussed.3, 55 While we have previously published on polymeric biomaterials doped with NO-releasing silica particles, the utility of these materials as implant coatings has been somewhat limited due to insufficient porosity. The primary goal of the studies presented here was to fabricate stable NO-releasing silica particle-doped electrospun polyurethane fiber mats with porosities more apt for reducing the foreign body response when implanted subcutaneously,41 and thus allowing for improved analyte diffusion for sensor applications. Secondly, we aimed to achieve tunable NO-release properties from these fibers, as many of NO’s biological activities are concentration dependent.2 Three polyurethane compositions (i.e., Tecophilic, Tecoflex, and Tecoplast) of distinct hydrophobicity and water uptake properties were chosen since water uptake is known to influence NO release for N-diazeniumdiolate NO donors.23 The bulk densities of the Tecophilic, Tecoflex, and Tecoplast polyurethanes were 1.13, 1.05, and 1.18 g/cm3, respectively. Silica particles of varied size (50–400 nm), NO-donor class (N-diazeniumdiolate and S-nitrosothiol), NO payload (0.4–3.2 μmol mg−1), and NO-release duration (9.6 h to >2 d) were employed to tune the NO-release properties from the resulting fiber mats. Two sizes (50 and 100 nm) of N-diazeniumdiolated AHAP3/TEOS silica particles having similar NO-release properties were used to study the role of particle size on fiber mat incorporation and resulting NO release. A wide range of NO release was achieved by employing two N-diazeniumdiolate-based particles (AEAP3/TMOS and AHAP3/TEOS), and a S-nitrosothiol-modified silica scaffold (MPTMS/TEOS) (resulting in short, medium and long NO-release kinetics, respectively). Of note, the MPTMS/TEOS system was selected as it allowed for much longer NO-release durations despite having an altered composition and size relative to the AEAP3/TMOS and AHAP3/TEOS systems.3, 4 Indeed, S-nitrosothiol-modified silica particles have longer NO release duration relative to the N-diazeniumdiolate silica (>48 h vs. ~10 h, respectively).11 The polymer and silica particle concentration ranges (8–16 w/v% and 1–10 wt%, respectively) were selected to allow for the greatest amount of particle incorporation within the fibers without inhibiting the electrospinning process.
As shown in Figure 1, the particles were successfully embedded inside of the electrospun fibers at the concentration studied. Although the nanoparticles were not dispersed homogeneously within individual fibers, they were distributed throughout the entire electrospun fiber mat. The electrospun polyurethane (PU) fiber mats exhibited random open porous structures with interconnected nano/submicron fibers and surface areas of ~2 m2 g−1. The thickness of the mats as determined by ESEM was proportional to the feed volume (e.g., ~50 μm for 1 mL electrospinning solution). In the absence of silica, the percent porosities of the electrospun fiber mats were 80.3 ± 2.1, 85.8 ± 7.6, and 83.8 ± 3.1% for the 12% (w/v) Tecophilic, Tecoflex, and Tecoplast polyurethanes, respectively. Particle incorporation up to 10 wt% did not significantly influence fiber mat porosity. As expected based on the nature of the bulk polymer, Tecoplast fibers were characterized as having the lowest water uptake (0.8 ± 0.5 mg H2O/mg of PU fiber mat) followed by the Tecoflex (1.6 ± 0.2 mg H2O/mg of PU fiber mat), and Tecophilic (4.7 ± 1.0 mg H2O/mg of PU fiber mat) polymers.23 The fiber mats exhibited greater water uptake than bulk polymer films of similar thickness after equivalent soaking time, a feature attributed to the open/porous structure of the fiber mats.
Figure 1.

Environmental scanning electron microscope images of polyurethane electrospun fibers composed of 5 wt% N-diazeniumdiolated-AEAP3/TMOS nanoparticle-doped 8% (w/v) (A) Tecophilic, (B) Tecoflex, and (C) Tecoplast; and 12% (w/v) (D) Tecophilic, (E) Tecoflex, and (F) Tecoplast PU polymer. Scale bar indicates 1 μm.
As expected, the physical properties of the electrospun fiber mats including fiber diameter, mechanical properties, and stability (i.e., leaching of silica particles) were dependent on the polymer solution concentration, polymer type, and NO donor system (particle type and concentration). Since porosity and fiber diameter represent important factors in mitigating the inflammatory response,37, 42 the effects of a number of electrospinning parameters on fiber diameter, tensile stress-strain, and silica incorporation/stability of the ensuing fiber mat were determined. Varying the applied voltage, needle tip diameter, flow rate, and distance between the collector and needle did not significantly impact the fiber diameter or morphology (data not shown). In contrast, both the viscosity and conductivity of the polymer solution proved important for controlling the geometry of the fiber mat.47, 56 Bead formation due to insufficient solution cohesion and/or improper Taylor jet elongation33, 47, 56, 57 was suppressed with increasing the solution viscosity and conductivity. As shown in Figure 1C, bead formation was only observed for fibers electrospun using 8% (w/v) Tecoplast polymer solutions, which exhibited a lower kinematic viscosity (45.3 ± 3.3 mm2 s−1) compared to Tecoflex and Tecophilic PU (67.4 ± 1.0 and 146.6 ± 2.3 and mm2 s−1, respectively). Increasing the concentration of the Tecoplast polymer from 8 to 12% (w/v) increased the solution’s kinematic viscosity from 45.3 ± 3.3 to 94.0 ± 2.2 mm2 s−1, in turn eliminating bead formation.
As shown in Figure 2, the kinematic viscosity of the polymer solution directly affected the diameter of resulting fibers. For example, the average diameter of 5 wt% AEAP3/TMOS particle-doped 12% (w/v) PU electrospun fibers increased from 168 ± 34 to 462 ± 109 and 551 ± 71 nm as the kinematic viscosity increased from 94.0 ± 2.2 to 287.0 ± 1.7 and 405.8 ± 4.5 mm2 s−1 for Tecoplast, Tecoflex, and Tecophilic polyurethanes, respectively. Additionally, the fiber diameter of Tecophilic PU fibers was greater than Tecoflex and Tecoplast fibers regardless of type of dopant (Figure 3). Similarly, increasing the polyurethane concentration resulted in larger fiber diameters. For example, changing the concentration of Tecoflex PU in the electrospinning polymer cocktail from 8 to 12 and 16% (w/v) increased the size of the resulting fibers from 257 ± 66 to 462 ± 109 and 625 ± 156 nm, respectively. The largest polymer concentration investigated (i.e., 16% (w/v)) inhibited proper electrospinning of Tecophilic PU due to needle clogging. At this concentration, the viscosity of the polymer solution as 2206.6 ± 82.6 mm2 s−1. Such upper limit at 16% (w/v) was not observed for Tecoflex and Tecoplast as the polymer solution viscosities remained moderate (1576.7 ± 24.9 and 342.0 ± 3.0 mm2 s−1, respectively).
Figure 2.
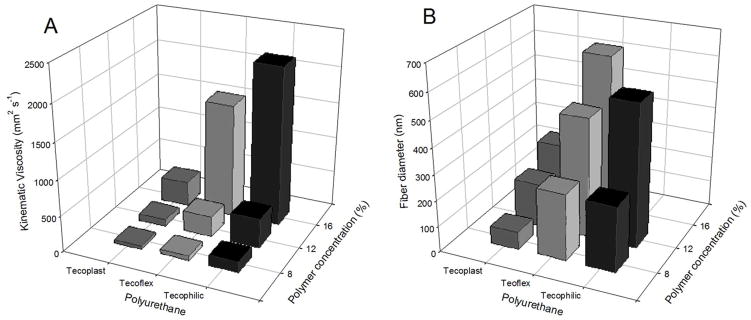
(A) Kinematic viscosity of polymer solution and (B) diameters of resulting fibers from polyurethanes doped with 5 wt% N-diazeniumdiolated AEAP3/TMOS silica nanoparticles as a function of polymer concentration and type.
Figure 3.

Diameters of 5 wt% particle-doped 12% (w/v) polyurethane electrospun fibers as a function of polyurethane type and NO-releasing silica particle dopant. Data is presented as mean ± standard deviation (n=3, >250 measurements).
Fiber diameter was also influenced by the conductivity of the polyurethane solution and the type of silica particle dopants employed. The zeta potential (i.e., surface charge) of S-nitrosothiol-modified silica particles is low/near zero, and thus the addition of such particles into the polymer solution did not significantly change the solution conductivity. Alternatively, N-diazeniumdiolated silica particles carry a large surface charges due to the negatively charged NO donor group. Thus, the addition of N-diazeniumdiolated AHAP3/TEOS and AEAP3/TMOS particles resulted in an increase in solution conductivity as shown in Table 1, which concomitantly also suppressed bead formation when using Tecoplast PU (SI Figure 2). Overall, the addition of N-diazeniumdiolated AEAP3/TMOS particles reduced fiber diameter relative to undoped and control (non-N-diazeniumdiolated AEAP3/TMOS particle-doped) fibers (Table 1) because greater solution conductivity elevated both the charge density on the Taylor cone and the elongation force along the elastic jet.56 Fiber diameter decreased further with a greater concentration of N-diazeniumdiolate particles (up to 10 wt%) for each of the PU systems, albeit slightly. Lastly, the fiber diameter was also influenced by the size of the particle dopants. Fibers prepared with 50 nm N-diazeniumdiolated AHAP3/TEOS particles were thinner compared to those doped with 100 nm particles (Figure 3). Such behavior is attributed to greater charge density per unit volume for polymer solution containing more N-diazeniumdiolated particles.
Table 1.
Conductivity of initial polymer solution and resulting fiber diameter as a function of dopant type.a
| Nitric oxide donor | Type of dopant | Particle size (nm) | Conductivity (μS cm−1) | Fiber diameter (nm) |
|---|---|---|---|---|
| None | 0.9 ± 0.3 | 558 ± 162 | ||
| Control particle | AEAP3/TMOS | 9.4 ± 2.2 | 491 ± 155 | |
| N-diazeniumdiolate | AEAP3/TMOS | 152 ± 2 | 44.3 ± 8.2 | 462 ±109 |
| AHAP3/TEOS | 56 ± 7 | 49.0 ± 7.5 | 387 ± 163 | |
| AHAP3/TEOS | 93 ± 14 | 48.4 ± 3.6 | 514 ± 190 | |
| S-nitrosothiol | MPTMS/TEOS | 416 ± 23 | 1.8 ± 0.9 | 573 ± 211 |
5 wt% particle-doped 12% (w/v) Tecoflex polyurethane electrospun fiber
To assess the suitability of the PU fiber mats as biomaterials, the mechanical properties of the particle-doped electrospun fibers were characterized in terms of modulus and elongation as a function of PU type and particle concentration. For tissue-based applications (e.g., subcutaneous implants), the mechanical properties of the scaffold should resemble native tissue to minimize shear stress and undesirable collagen deposition.58 As shown in Figure 4A, each type of polyurethane exhibited different mechanical strengths. For example, Tecoplast (12% (w/v)) fiber mats doped with 5 wt% AEAP3/TMOS were characterized by a modulus of 34.5 ± 18.7 MPa and elongation of 92.3 ± 60.7% tensile strain at break, exhibiting flexible plastic-like mechanical behavior. The 5 wt% AEAP3/TMOS Tecophilic and Tecoflex (12% w/v) fiber mats had lower moduli of 1.7 ± 0.5 and 4.9 ± 0.4 MPa, respectively, and greater elongations of 223.0 ± 32.1 and 211.52 ± 29.83 % tensile strain at break, respectively. To determine the effects of mechanical properties on water absorption and potential particle leaching, the tensile strain and stress were enumerated after incubating the fiber mats in PBS at 37°C for 24 h. Similar elongations of tensile strain at break were observed regardless of polyurethane composition. For wet (i.e., soaked) fibers, the tensile stress at break decreased by roughly 20 and 50% for Tecoplast and Tecoflex, respectively. In contrast, the tensile stress at break value increased by ~100% for wet Tecophilic fiber mats, highlighting the ability of the more hydrophilic polyurethane to absorb greater energy up to fracture. The Tecoplast-based fiber mats would thus likely be more useful for prosthetic and orthopedic applications. Pacemakers, wound dressings, and catheters might benefit more from the properties of the Tecophilic and Tecoflex fiber mats.59, 60 As might be expected, the fiber mat modulus and tensile strain were also influenced by the concentration of particles incorporated into the fibers. Elongation of the electrospun fiber mat decreased proportionally with increasing particle concentration from 1 to 10 wt% (Figure 4B). The modulus also increased with increasing particle dopant concentration due to decreased strength in the cross-sectional area of the load-bearing polymer matrix.61 For example, doping AEAP3/TMOS particles into 12% (w/v) Tecophilic polyurethane fiber mats at a concentration of 1 wt% resulted in a modulus of 0.9 ± 0.1 MPa, which was identical to electrospun fiber mats without additives (0.9 ± 0.2 MPa). However, the moduli of the electrospun fiber mats increased with increasing particle concentration, resulting in moduli of 1.7 ± 0.5 and 2.1 ± 0.3 MPa for 5 and 10 wt% particle concentrations, respectively. These data suggest that Tecoflex and Tecophilic fibers doped with low particle concentrations possess mechanical properties best suited for lessening the FBR at implant-tissue interfaces.
Figure 4.
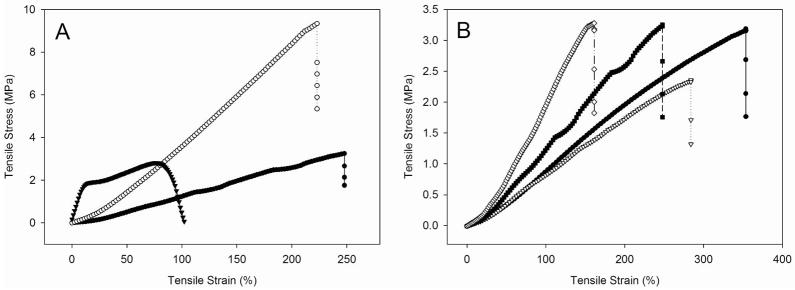
Tensile stress-strain curves of (A) 5 wt% AEAP3/TMOS particle-doped 12% (w/v) electrospun fiber mats as a function of polyurethane type: Tecophilic (●), Tecoflex (○), and Tecoplast (▼), and (B) 12% (w/v) Tecophilic electrospun fiber mats as a control (●) and a function of 1 (▽), 5 (■), and 10 wt% (◇) AEAP3/TMOS particle concentrations.
Although silica-based materials are generally regarded as non-toxic, leaching of particles from the fibers was evaluated to assess the stability of the particle-polymer composites. Particle-doped fiber mats were immersed in physiological media (PBS, pH 7.4, 37 °C), and silicon content in the soak solutions was measured after 7 d to assess the extent of particle leaching. As expected, stability was greatly dependent on the physical and chemical characteristics of the particle dopants as well as the water-uptake of the polymers. Smaller particle dopants showed lower stability as indicated by greater leaching from the fiber mats (SI Table 2). Nearly all of the 50 nm AHAP3/TEOS silica particles but only 70% of the 100 nm particles leached from the polyurethane fibers regardless of polymer composition, indicating smaller particles are more readily liberated upon swelling of the fibers. Fortunately, the particle concentrations doped within the fibers were low, such that even 100% leaching should not result local silica concentrations that are toxic.49, 62–64 Silica particle leaching further decreased for all polymer compositions as the size of the particle dopant increased, with the largest diameter particle (MPTMS/TEOS particles at 416 ± 23 nm) characterized by <2% leaching. Differences in polymer swelling due to water uptake also influenced the overall material stability. Tecoplast fibers, characterized by the lowest water uptake, exhibited the smallest level of particle leaching relative to the Tecophilic and Tecoflex polyurethanes for all dopant types. Overall, the lowest level of silica nanoparticle leaching (i.e., 0.7%) was achieved using the 5 wt% MPTMS/TEOS particles doped into 12% (w/v) Tecoplast electrospun fibers. Taken together, these data suggest that the greatest stability is achieved with lower water-uptake polymers and larger diameter particles.
Nitric oxide release from silica nanoparticle-doped electrospun fiber mat
While our previous report on electrospun fibers demonstrated controlled NO release using a low molecular weight NO donor (i.e., PROLI/NO), neither the NO-release kinetics nor duration of release proved tunable over a wide range.47 Since optimal mitigation of the FBR via NO release from subcutaneous implants requires at least 48 h of NO release and a large overall NO payload (>1 μmol/cm2), sustained and controlled NO release is an important aspect in developing NO-releasing biomaterials.65, 66 Four distinct NO-releasing silica particle systems were used to fabricate NO-releasing fibers with diverse NO-release totals (0.4–3.2 μmol mg−1) and durations (up to >48 h). Full characterization of the NO release from each particle system is provided in supporting information (SI Table 1). Since the NO-release mechanism of N-diazeniumdiolate NO donors is proton-initiated, the NO release is generally controlled by pH and the hydrophobicity of surrounding matrix.23 In contrast, the NO release for S-nitrosothiol systems is not dependent on pH or water uptake, but rather a function of heat, light, and/or the presence of copper ions.4 In the case of the N-diazeniumdiolated scaffolds, both size of the AHAP3/TEOS particles (50 and 100 nm) had similar NO-release properties and exhibited larger payloads over a slightly longer release durations than the AEAP3/TMOS particles. In contrast, the S-nitrosothiol-modified MPTMS/TEOS particles delivered the greatest NO payload (3.2 μmol mg−1) and had the longest release durations (>48 h) among all particle systems.
Analogous to the particle stability studies above, the NO release from the electrospun fiber mats was determined in PBS (pH 7.4) at 37 °C to mimic physiological conditions. Compared to previously reported PROLI/NO-doped electrospun fibers (NO-release duration of 8 min to 1.3 h), the silica particle-doped electrospun fibers exhibited substantially prolonged NO release with durations ranging from 7 h to 14 d (Table 2). Of note, the electrospinning process had no affect on the particles’ NO payload. For example, the total NO released from 5 wt% AHAP/TEOS particle-doped 12% (w/v) Tecoflex fiber mats was 98.3% of the theoretically calculated total NO (determined based on particle concentration in starting polymer cocktail). Although the NO release from fibers doped with N-diazeniumdiolated silica particles was limited to <1 d, the NO fluxes from these materials may still prove useful as thromboresistant coatings for blood-contacting biomedical devices (e.g., stents and catheters) since the N-diazeniumdiolated NO donor systems release NO at fluxes required to promote hemocompatibilty (i.e., 0.4–5.0 pmol cm−2 s−1).67–70 As expected, longer NO-release durations were achieved with the S-nitrosothiol-functionalized particles regardless of the type of polymer system employed (~2 weeks). Of note, the MPTMS/TEOS particle-doped electrospun fiber mats exhibited NO-release durations at or above that reported sufficient to mitigate the foreign body response for subcutaneous implants (i.e., NO release >72 h).66, 71
Table 2.
Nitric oxide release characteristics of 5 wt% NO-releasing silica particle-doped 12% (w/v) electrospun fiber mats.
| NO donor type | NO-releasing silica particle | Type of polyurethaneb | [NO]maxc (pmol mg−1 s−1) | tmaxd (min) | Total NO releasede (nmol mg−1) | tdf (h) |
|---|---|---|---|---|---|---|
| N-diazeniumdiolate | AHAP3/TEOSa (50 nm) | Tecophilic | 28.0 ± 9.2 | 8.4 ± 5.5 | 62.5 ± 36.1 | 7.2 ± 3.6 |
| Tecoflex | 13.1 ± 10.1 | 24.0 ± 16.2 | 69.4 ± 18.3 | 13.3 ± 4.1 | ||
| Tecoplast | 3.2 ± 2.6 | 28.5 ± 1.1 | 60.9 ± 15.2 | 29.9 ± 12.8 | ||
| AHAP3/TEOSa (100 nm) | Tecophilic | 17.6 ± 3.8 | 1.2 ± 0.1 | 57.0 ± 6.4 | 14.5 ± 0.2 | |
| Tecoflex | 22.3 ± 2.9 | 2.6± 1.9 | 37.3 ± 10.9 | 14.9 ± 0.8 | ||
| Tecoplast | 19.7 ± 1.7 | 1.9 ± 1.5 | 42.2 ± 19.8 | 14.1 ± 0.2 | ||
| AEAP3/TMOS | Tecophilic | 2.2 ± 1.0 | 1.7 ± 0.5 | 15.0 ± 5.0 | 15.3 ± 1.0 | |
| Tecoflex | 1.5 ± 0.9 | 0.8 ± 0.5 | 10.8 ± 1.0 | 19.6 ± 2.7 | ||
| Tecoplast | 1.3 ± 0.3 | 3.1 ± 2.7 | 7.5 ± 2.0 | 14.3 ± 1.6 | ||
| S-nitrosothiol | MPTMS/TEOS | Tecophilic | 15.2 ± 6.1 | 6.9 ± 0.4 | 124.7 ± 6.8 | 366.8 ± 78.2 |
| Tecoflex | 10.1 ± 0.8 | 0.9 ± 0.4 | 123.2 ± 16.1 | 296.6 ± 6.5 | ||
| Tecoplast | 11.7 ± 9.8 | 1.8 ± 1.0 | 86.8 ± 9.0 | 299.9 ± 2.8 |
AHAP3/TEOS particles have two distinct sizes but similar NO release properties;
Hydrophobicity increases from top to bottom of one particle system;
Maximum instantaneous concentration of NO released as measured with NOA;
Time required to reach [NO]max;
Total number of moles of NO released per mg of particle-doped electrospun fiber mat as measured by the Griess assay;
Duration of NO release (time to release 99% of total NO).
The effect of polymer composition on NO-release kinetics was most apparent with the 50 nm N-diazeniumdiolated AHAP3/TEOS-doped electrospun fibers (Table 2 and SI Figure 3). For example, 5 wt% N-diazeniumdiolate particle-doped hydrophobic Tecoplast fibers were characterized by the lowest maximum NO flux (3.2 ± 2.6 pmol mg−1 s−1) and longest NO-release duration (29.9 ± 12.8 h) due to lower water uptake. The more hydrophilic Tecophilic-based counterparts had an increased flux and shorter release duration (28.0 ± 9.2 pmol mg−1 s−1 and 7.2 ± 3.6 h, respectively). The total NO release was constant regardless of polyurethane type as expected. The NO-release kinetics proved less tunable for the larger particles systems (e.g., AHAP3/TEOS and AEAP/TMOS at 100 nm and 150 nm, respectively) due to the limited fiber diameter and decreased thickness of the water restricting layer around the particles, ultimately eliminating any water uptake-mediated effect on N-diazeniumdiolate NO donor decomposition (SI Figure 4).47 In this respect, increasing the distance water must diffuse through the polymer to reach the particle scaffolds may prove to be an importance method for fine-tuning NO-release kinetics. A future objective is to adopt a co-axial electrospinning strategy72 where fibers are composed of an inner layer containing the particle dopants and an outer shell comprised of undoped polymer of varied hydrophobicity and/or thickness.
The total NO payload and initial bolus of NO release from the fiber mats were further altered by changing the silica particle concentrations. As expected, increasing the concentration of silica particle dopant elevated both the maximum NO flux and total NO released from the electrospun fiber mats (Figure 5 and SI Figure 5). For example, electrospun fibers doped with 1, 5, and 10 wt% AEAP3/TMOS particle concentrations resulted in maximum NO fluxes of 0.6 ± 0.7, 2.2 ± 1.0, and 5.4 ± 3.5 pmol mg−1 s−1, respectively. Additionally, the total NO released from those electrospun fibers was 3.6 ± 3.3, 15.0 ± 5.0, and 22.3 ± 0.6 nmol mg−1 for 1, 5, and 10 wt% silica dopant concentrations, respectively. Similar trends were observed for all other particle compositions. Of importance, the greatest total NO release achieved from the particle-doped electrospun fiber mats was lower than previously reported proven to be cytotoxic or cause apoptosis.73, 74 Not surprisingly, neither the NO-release half-life or duration of N-diazeniumdiolated particle-doped electrospun fiber mats was greatly affected by the amount of particle dopant (1 to 10 wt%).
Figure 5.
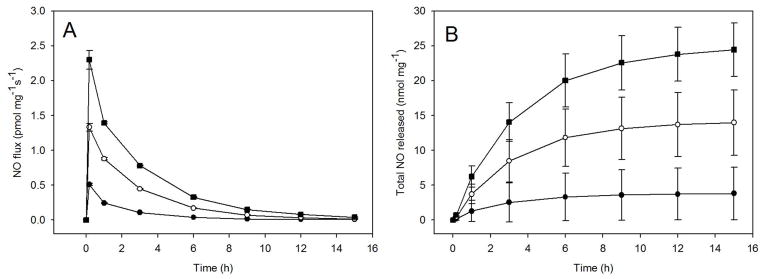
(A) Nitric oxide flux and (B) NO release totals from NO donor-modified AEAP3/TMOS particle-doped 12% (w/v) Tecophilic electrospun polyurethane fiber mats as a function of dopant concentration: 1 (●), 5 (○), and 10 (■) wt%.
Conclusion
The electrospun polyurethane fibers doped with NO donor-modified silica particles presented here have allowed us to overcome limitations of previously reported NO-release materials (e.g., short NO release duration and low porosity). The use of electrospun fibers provides a material with high porosity while maintaining mechanical strength compared to bulk polymers doped with NO-releasing silica particles. Moreover, the incorporation of NO-releasing silica particles into electrospun fibers enables greater NO release durations compared to electrospun fibers doped with a low molecular weight NO donor (1 h vs. 2 weeks). Changing the type of NO-releasing particle system, polyurethane water uptake, and dopant concentration resulted in a wide range of NO release characteristics (i.e., total NO payloads of 7.5–124.7 nmol mg−1 and durations from 7 h to 2 weeks). Of the systems studied herein, S-nitrosothiol-modified silica particles promoted the longest NO release and most stable particle-fiber composites. Other macromolecular scaffolds, such as NO-releasing dendrimers,13–16 may also prove advantageous as fiber dopants as a result of larger NO payloads that can be incorporated with improved polymer partitioning attributes. As a result of both flexible and open architectures, porous NO-releasing fibers represent ideal candidates for biomedical implant coatings.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (EB000708). The authors thank Wesley L. Storm for assistance in measuring the surface areas of the fiber mats. We also thank Dr. Bin Sun and Dr. Daniel Riccio for synthesizing NO-releasing particles.
Footnotes
Characterization of nitric oxide-releasing silica particles (size, composition, NO release data), additional ESEM images of particle-doped fibers, ICP-OES particle leaching data, and additional NO release characterization of particle-doped fibers were included in supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Williams DLH. Org Biomol Chem. 2003;1:441–449. doi: 10.1039/b209748f. [DOI] [PubMed] [Google Scholar]
- 2.Ignarro LJ. Nitric oxide : biology and pathobiology. Academic; San Diego, Calif.; London: 2000. [Google Scholar]
- 3.Carpenter AW, Schoenfisch MH. Chem Soc Rev. 2012;41:3742–3752. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riccio DA, Schoenfisch MH. Chem Soc Rev. 2012;41:3731–3741. doi: 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jen MC, Serrano MC, van Lith R, Ameer GA. Adv Funct Mater. 2012;22:239–260. doi: 10.1002/adfm.201101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mater. 2003;15:4193–4199. [Google Scholar]
- 7.Nablo BJ, Schoenfisch MH. Biomaterials. 2005;26:4405–4415. doi: 10.1016/j.biomaterials.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948–1956. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494–4502. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shin JH, Metzger SK, Schoenfisch MH. J Am Chem Soc. 2007;129:4612–4619. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 11.Riccio DA, Nugent JL, Schoenfisch MH. Chem Mater. 2011;23:1727–1735. doi: 10.1021/cm102510q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin JH, Schoenfisch MH. Chem Mater. 2008;20:239–249. doi: 10.1021/cm702526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y, Sun B, Li C, Schoenfisch MH. Chem Mater. 2011;23:4227–4233. doi: 10.1021/cm201628z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stasko NA, Fischer TH, Schoenfisch MH. Biomacromolecules. 2008;9:834–841. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stasko NA, Schoenfisch MH. J Am Chem Soc. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 16.Sun B, Slomberg DL, Chudasama SL, Lu Y, Schoenfisch MH. Biomacromolecules. 2012;13:3343–3354. doi: 10.1021/bm301109c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coneski PN, Rao KS, Schoenfisch MH. Biomacromolecules. 2010;11:3208–3215. doi: 10.1021/bm1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coneski PN, Schoenfisch MH. Polym Chem. 2011;2:906–913. doi: 10.1039/C0PY00269K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damodaran VB, Joslin JM, Wold KA, Lantvit SM, Reynolds MM. J Mater Chem. 2012;22:5990–6001. [Google Scholar]
- 20.Damodaran VB, Reynolds MM. J Mater Chem. 2011;21:5870–5872. [Google Scholar]
- 21.Frost MC, Meyerhoff ME. J Biomed Mater Res. 2005;72A:409–419. doi: 10.1002/jbm.a.30275. [DOI] [PubMed] [Google Scholar]
- 22.Frost MC, Reynolds MM, Meyerhoff ME. Biomaterials. 2005;26:1685–1693. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Koh A, Riccio DA, Sun B, Carpenter AW, Nichols SP, Schoenfisch MH. Biosens Bioelectron. 2011;28:17–24. doi: 10.1016/j.bios.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heck DE, Laskin DL, Gardner CR, Laskin JD. J Biol Chem. 1992;267:21277–21280. [PubMed] [Google Scholar]
- 25.Schäffer MR, Tantry U, Gross SS, Wasserkrug HL, Barbul A. J Surg Res. 1996;63:237–240. doi: 10.1006/jsre.1996.0254. [DOI] [PubMed] [Google Scholar]
- 26.Bogdan C. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 27.Mocellin S, Bronte V, Nitti D. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 28.Jones M, Ganopolsky J, Labbé A, Wahl C, Prakash S. Appl Microbiol Biotechnol. 2010;88:401–407. doi: 10.1007/s00253-010-2733-x. [DOI] [PubMed] [Google Scholar]
- 29.Shin JH, Schoenfisch MH. Analyst. 2006;131:609–615. doi: 10.1039/b600129g. [DOI] [PubMed] [Google Scholar]
- 30.Yan Q, Major TC, Bartlett RH, Meyerhoff ME. Biosens Bioelectron. 2011 doi: 10.1016/j.bios.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frost MC, Meyerhoff ME. Curr Opin Chem Biol. 2002;6:633–641. doi: 10.1016/s1367-5931(02)00371-x. [DOI] [PubMed] [Google Scholar]
- 32.Teo WE, Ramakrishna S. Nanotechnology. 2006;17:R89–R106. doi: 10.1088/0957-4484/17/14/R01. [DOI] [PubMed] [Google Scholar]
- 33.Huang ZM, Zhang YZ, Koraki M, Ramakrishna S. Compos Sci Technol. 2003;63:2223–2253. [Google Scholar]
- 34.Agarwal S, Wendorff JH, Greiner A. Polymer. 2008;49:5603–5621. [Google Scholar]
- 35.Lu X, Wang C, Wei Y. Small. 2009;5:2349–2370. doi: 10.1002/smll.200900445. [DOI] [PubMed] [Google Scholar]
- 36.Li WJ, Laurencin CT, Caterson EJ, Tuan RS, Ko FK. J Biomed Mater Res. 2002;60:613–621. doi: 10.1002/jbm.10167. [DOI] [PubMed] [Google Scholar]
- 37.Leung V, Ko F. Polym Adv Technol. 2011;22:350–365. [Google Scholar]
- 38.Wang N, Burugapalli K, Song W, Halls J, Moussy F, Ray A, Zheng Y. Biomaterials. 2013;34:888–901. doi: 10.1016/j.biomaterials.2012.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mo XM, Xu CY, Kotaki M, Ramakrishna S. Biomaterials. 2004;25:1883–1890. doi: 10.1016/j.biomaterials.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 40.Saino E, Focarete ML, Gualandi C, Emanuele E, Cornaglia AI, Imbriani M, Visai L. Biomacromolecules. 2011;12:1900–1911. doi: 10.1021/bm200248h. [DOI] [PubMed] [Google Scholar]
- 41.Cao H, McHugh K, Chew SY, Anderson JM. J Biomed Mater Res. 2010;93A:1151–1159. doi: 10.1002/jbm.a.32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saino E, Focarete ML, Gualandi C, Emanuele E, Cornaglia AI, Imbriani M, Visai L. Biomacromolecules. 2012;12:1900–1911. doi: 10.1021/bm200248h. [DOI] [PubMed] [Google Scholar]
- 43.Son WK, Youk JH, Lee TS, Park WH. Macromol Rapid Commun. 2004;25:1632–1637. [Google Scholar]
- 44.Hong KH. Polym Eng Sci. 2007;47:43–49. [Google Scholar]
- 45.Xu X, Yang Q, Wang Y, Yu H, Chen X, Jing X. Eur Polym J. 2006;42:2081–2087. [Google Scholar]
- 46.Vacanti NM, Cheng H, Hill PS, Guerreiro JDT, Dang TT, Ma M, Watson S, Hwang NS, Langer R, Anderson DG. Biomacromolecules. 2012;13:3031–3038. doi: 10.1021/bm300520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coneski PN, Nash JA, Schoenfisch MH. ACS Appl Mater Interfaces. 2011;3:426–432. doi: 10.1021/am101010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wold KA, Damodaran VB, Suazo LA, Bowen RA, Reynolds MM. ACS Appl Mater Interfaces. 2012;4:3022–3030. doi: 10.1021/am300383w. [DOI] [PubMed] [Google Scholar]
- 49.Carpenter AW, Slomberg DL, Rao KS, Schoenfisch MH. ACS Nano. 2011;5:7235–7244. doi: 10.1021/nn202054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koschwanez HE, Yap FY, Klitzman B, Reichert WM. J Biomed Mater Res. 2008;87A:792–807. doi: 10.1002/jbm.a.31824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu X, Cui W, Li X, Jin Y. Biomacromolecules. 2008;9:1795–1801. doi: 10.1021/bm800476u. [DOI] [PubMed] [Google Scholar]
- 52.He W, Ma Z, Yong T, Teo WE, Ramakrishna S. Biomaterials. 2005;26:7606–7615. doi: 10.1016/j.biomaterials.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 53.Huang ZM, Zhang YZ, Ramakrishna S, Lim CT. Polymer. 2004;45:5361–5368. [Google Scholar]
- 54.Coneski PN, Schoenfisch MH. Chem Soc Rev. 2012;41:3753–3758. doi: 10.1039/c2cs15271a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nichols SP, Koh A, Storm WL, Shin JH, Schoenfisch MH. Chem Rev. 2013;113:2528–2549. doi: 10.1021/cr300387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan SH, Inai R, Kotaki M, Ramakrishna S. Polymer. 2005;46:6128–6134. [Google Scholar]
- 57.Shawon J, Sung C. J Mater Sci. 2004;39:4605–4613. [Google Scholar]
- 58.Helton KL, Ratner BD, Wisniewski NA. J Diabetes Sci Tech. 2011;5:647–656. doi: 10.1177/193229681100500318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wise DL, Trantolo DJ, Altobelli DE, Yaszemski MJ, Gresser JD. Human biomaterials applications. Humana Press; Totowa: 1996. [Google Scholar]
- 60.Black J, Hastings GW. Handbook of biomaterial properties. Chapman & Hall; London; New York: 1998. [Google Scholar]
- 61.Landon G, Lewis G, Boden GF. J Mater Sci. 1977;12:1605–1613. [Google Scholar]
- 62.Kunzmann A, Andersson B, Thurnherr T, Krug H, Scheynius A, Fadeel B. Biochim Biophys Acta. 2011;1810:361–373. doi: 10.1016/j.bbagen.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 63.Hudson SP, Padera RF, Langer R, Kohane DS. Biomaterials. 2008;29:4045–4055. doi: 10.1016/j.biomaterials.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbé C, Bartlett J, Kong L, Finnie K, Lin HQ, Larkin M, Calleja S, Bush A, Calleja G. Adv Mater. 2004;16:1959–1966. [Google Scholar]
- 65.Nichols SP, Koh A, Brown NL, Rose MB, Sun B, Slomberg DL, Riccio DA, Klitzman B, Schoenfisch MH. Biomaterials. 2012;33:6305–6312. doi: 10.1016/j.biomaterials.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hetrick EM, Prichard HL, Klitzman B, Schoenfisch MH. Biomaterials. 2007;28:4571–4580. doi: 10.1016/j.biomaterials.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reynolds MM, Frost MC, Meyerhoff ME. Free Radical Biol Med. 2004;37:926–936. doi: 10.1016/j.freeradbiomed.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 68.Robbins ME, Hopper ED, Schoenfisch MH. Langmuir. 2004;20:10296–10302. doi: 10.1021/la048368n. [DOI] [PubMed] [Google Scholar]
- 69.Skrzypchak AM, Lafayette NG, Bartlett RH, Zhou Zhengrong, Frost MC, Meyerhoff ME, Reynolds MM, Annich GM. Perfusion. 2007;22:193–200. doi: 10.1177/0267659107080877. [DOI] [PubMed] [Google Scholar]
- 70.Zhang H, Annich GM, Miskulin J, Osterholzer K, Merz SI, Bartlett RH, Meyerhoff ME. Biomaterials. 2002;23:1485–1494. doi: 10.1016/s0142-9612(01)00274-5. [DOI] [PubMed] [Google Scholar]
- 71.Nichols SP, Le NN, Klitzman B, Schoenfisch MH. Anal Chem. 2011;83:1180–4. doi: 10.1021/ac103070t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moghe AK, Gupta BS. Polym Rev. 2008;48:353–377. [Google Scholar]
- 73.Brüne B. Cell Death Differ. 2003;10:864–869. doi: 10.1038/sj.cdd.4401261. [DOI] [PubMed] [Google Scholar]
- 74.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Proc Natl Acad Sci. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.