

Abstract
The brain is a highly metabolically active tissue that critically relies on oxidative phosphorylation as means for maintaining energy. One by-product of this process is the production of potentially damaging radicals such as the superoxide anion (O2.-). Superoxide has the capacity to damage components of the electron transport chain and other cellular constituents. Eukaryotic systems have evolved defenses against such damaging moieties, the chief member of which is superoxide dismutase (SOD2), an enzyme that efficiently converts superoxide to the less reactive hydrogen peroxide (H2O2), which can freely diffuse cross the mitochondrial membrane. Loss of SOD2 activity can result in numerous pathological phenotypes in metabolically active tissues; particularly within the central nervous system. We will review SOD2’s potential involvement in the progression of neurodegenerative disease such as stroke, Alzheimer’s, Parkinson’s, as well as its potential role in “normal” age-related cognitive decline. We will also examine in vivo models of endogenous oxidative damage based upon loss of SOD2 and associated neurological phenotypes in relation to human neurodegenerative disorders.
Keywords: Mitochondria, Superoxide dismutase, Neurodegeneration, Oxidative stress, Aging
Introduction
Oxidative stress and reactive oxygen species (ROS) are often implicated in a wide range of pathological processes throughout the body. Historically, much of the research which has focused on endogenous oxidative stress can be traced back to the mitochondrial free radical theory of aging proposed by Harman in 1972 (1). This theory implicated the production of endogenous damaging free radicals as a by-product of mitochondrial metabolism leading to progressive damage and ultimately age-related pathology. While the theory provided a potential framework for discussing late onset degenerative disease; it has been contested as the primary cause of aging (2), which may be due to an overall lack of specificity with regards to the specifics of the theoretical framework. The theory is generally vague in a number of key aspects, such as:
What levels of free radicals elicit deleterious phenotypes?
Are all radicals equally deleterious?
Are there specific proteins/lipids/nucleic acids that are especially vulnerable to damaging radicals?
To what extent do different cell types/tissues contribute radicals under different metabolic states?
We feel that questions such as these, and many other more nuanced iterations to a large extent make the theory untestable. Current theories about key mechanisms which attempt to explain the pathobiology of aging such as age-related neurodegenerative disease frequently include oxidative stress as part of larger constellation of pathological processes, rather than being focused on a single cause (3-6). In general, contemporary views of common neurodegenerative disorders including Alzheimer’s disease, Parkinson’s disease, stroke, and Age-related cognitive decline (7), include a more nuanced view not overwhelmingly reliant on a single biochemical process. Here, we will examine the role of oxidative stress in neurodegeneration in relation to the mitochondria’s principal antioxidant enzyme Superoxide Dismutase 2 (SOD2).
The SOD2 enzyme is the particular focus of this article because its cellular localization and function arguably makes its presence more critical to neurodegenerative diseases than the other superoxide dismutase isoforms found in mammals. Localization of the enzyme determines the particular source of superoxide each isoform acts against in neuronal tissues (Figure 1). For instance, SOD3 is secreted to the extracellular matrix in most tissues including the central nervous system (8,9). While this enzyme is not directly located within the cell, it can participate in metabolic regulation of cells in the central nervous system by altering vascular tone and blood flow to the brain (10-13). Similarly, SOD1 can also play an important role in the prevention of damage to the central nervous system with its cytoplasmic localization (14), and to a lesser extent the mitochondrial intermembrane space where it can also be found (15-18). Mutations in the SOD1 gene are responsible in part for damage to the mitochondria leading to the onset of the progressive neurodegenerative disorder Amyotrophic Lateral Sclerosis (ALS) (19-21). It is clear that the maintenance of superoxide levels in the extracellular and cytosolic environment are important to maintaining the CNS with SOD1 and SOD3; however in this review will limit our discussion to the most critical defense against mitochondrial superoxide, SOD2.
Figure 1. Function of Superoxide dismutatses.

Schematic of a cell showing the localization and function of the 3 superoxide dismutase isoforms. SOD1 is primarily localized to the cytosol of the cell (yellow) as well in the intramembrane space of the mitochondria, while SOD3 is secreted to the extracellular space (blue). The SOD2 isoform is specifically localized to the inner mitochondrial matrix at the site of superoxide production from the mitochondrial election transport chain (purple). Shown in this diagram is a representation of molecular oxygen diffusing into the mitochondria where it is converted into superoxide as a byproduct of oxidative phosphorylation. SOD2 converts this damaging agent into hydrogen peroxide where it can diffuse from the mitochondria and be further detoxified to water by antioxidant enzymes such as Catalase (CAT) or Glutathione Peroxidase (GPx) (both in the mitochondria in some cases, as well as in the cell).
SOD2 is located within the mitochondrial matrix, the main site of free radical production from the electron transport chain (22). This enzyme catalyzes the reaction of superoxide (O2.-) to the less reactive hydrogen peroxide (H2O2) (which is not considered a free radical) at diffusion limiting rates (23,24), before it can oxidize macromolecules such as DNA, proteins, or lipid (25-29). This conversion to hydrogen peroxide also facilitates a passive diffusion of hydrogen peroxide away from the mitochondrial matrix preventing a high accumulation of superoxide close to the site of ATP production (24). We also discuss the current in vivo model systems for examining endogenous mitochondrial oxidative stress in central nervous system by modulating SOD2 function. Finally, we examine the benefits and limitations of model systems in relation to clinical features of neurodegeneration.
SOD2 in Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of advanced dementia, and currently has no effective clinical intervention to alleviate AD symptoms. The classic clinical hallmark in the diagnosis of this disease has been the accumulation of neurofibrillary tangles and plaques in the brain (30,31). These aggregates of insoluble protein consist primarily of hyper-phosphorylated tau and misprocessed amyloid-β isoforms in conjunction with metals, likely arising from a dysregulation of metal homeostasis with age (6). Further, specific genes have been implicated in familial forms of the disease, primarily relating to amyloid processing (32). The progression of Alzheimer’s can also be exacerbated by oxidative stress (33,34). Clinical evidence in post-mortem samples from patients with Alzheimer’s disease reveals that there are elevated markers of oxidative stress within the tissue including protein and lipid peroxidation (35,36). This has lead to the suggestion that markers of oxidative stress such as oxidized forms of nucleic acids or specific proteins could be used as markers for AD risk factors (37). Butterfield and colleagues proposed that HNE produced by Aβ-induced lipid peroxidation could be a useful prognostic marker in cerebrospinal fluid (38). However, teasing out cause and effect with regards to oxidative stress is always problematic, did the stress come first, or is it a result of some other event as a compensatory process?
The interaction of ROS and AD is also supported by clinical findings that revealed an up-regulation of antioxidant enzymes such as SOD2 early in the disease progression (39). However, there is no evidence to support the idea that dysregulation of SOD2 activity itself is responsible for increased oxidative stress with age. Rather, up regulation of SOD2 is likely a compensatory mechanism against elevated oxidative stress due to an age-related oxidative challenge of unknown etiology, and altered mitochondrial homeostasis. There is some evidence supporting the idea that altered metal homeostasis in the brain with age may be the cause of this endogenous oxidative stress (5). Other contributing factors to altered mitochondrial function with age in the brain may be interactions with amyloid itself (which also accumulates with age) (40,41). Amyloid has been shown to directly interfere with mitochondrial respiration in vitro (42). This progression could participate in a complex degenerative cycle in which mitochondrial function is reduced leading to enhanced plaque formation, which in turn promotes further mitochondrial dysfunction. There are many variants on this model of a “vicious cycle” of mitochondrial dysfunction, which has long been speculated on in the literature (43,44). However, there now exist many mouse models which could be used to test the idea that mitochondrial dysfunction is synergistic to pathological outcomes (“mitochondrial dysfunction begets more mitochondrial dysfunction”, and so on…), so this question could be answered definitively with an appropriate experimental design.
While there are numerous reports indicating a loss of mitochondrial function in Alzheimer’s disease as well as in mouse models of this important age related disorder, a recent study has reported that a number of prominent mouse models of AD contain no synaptic mitochondrial defects despite having ongoing neurodegeneration in association with the prototypical pathology associated with overexpression of mutant Aβ (45). This finding suggests that mitochondrial dysfunction at the synapse is not associated with aberrant Aβ pathology due to overexpression of multiple transgenes involved in plaque formation. More importantly, this emphasizes that these commonly employed animal models of AD incompletely recapitulate the mitochondrial aspects of the disease phenotype (45), which have long been reported in tissues from patients suffering from AD. Over-reliance on animal models may lead to spurious therapies that correct plaque formation, but do not address potentially important respiratory defects or oxidative damage aspects of AD that may be critical to the pathophysiology.
A number of mouse models have been created that affect the level of endogenous mitochondrial oxidative stress by modulating SOD2 function in the presence of plaque driven by transgenes (46,47). The creation of a transgenic mouse with one deleted copy of the Sod2 gene crossed to an established model of Alzheimer’s (human APP/J20) accelerated the AD pathology in the mouse (48). Interestingly, this reduction in SOD2 decreased the deposition of amyloid-β in the parenchyma and increased amyloidosis in the vasculature of the brain. Similar results were also reported in other mouse models of AD, such as the Tg2576 and Tg19959 mice, which over-express mutant forms of human amyloid-β (46,47). This genetic manipulation also enhanced deposition of amyloid plaque in response to reduced levels of SOD2. In a recent report, the overexpression of mutant amyloid prevented long-term potentiation in cultured neurons which was rescued with administration of MitoQ and Euk-134 (an antioxidant and SOD mimetic, respectively) (49). This evidence strongly suggests a synergistic action between the mitochondrial oxidative stress and the progression of plaque deposition. It is therefore possible that mitochondrial dysfunction and/or endogenous oxidative stress is a prerequisite for neuronal loss in Alzheimer’s disease. There is no clear genetic link between a normal age-related decline of neuronal function and the neuronal loss from abnormalities in amyloid plaque and neurofibrillary tangles. However, a decline in bioenergetic capacity and mitochondrial metabolism may be a common precursor to both of these conditions. This emphasizes the importance of minimizing oxidative damage within the brain to inhibit the loss of neurons over time across multiple disease paradigms.
SOD2 in Parkinson’s Disease
Parkinson’s disease (PD) can be caused by either genetic or environmental insults that result in the selective loss of dopaminergic neurons (50). The sporadic form of Parkinson’s disease is a complex disorder caused by an as yet unknown mechanisms. Roughly 10% of PD cases are partially explained by a specific gene mutation that results in the Parkinsonian phenotype (51). Both the sporadic and familial forms progress with the selective loss of dopaminergic neurons within the pars compacta of the substantia nigra of affected individuals. The result of this neuronal death are clinical presentations of tremor, and a profound loss of motor control. The progression of Parkinson’s is accompanied by perturbations in specific biochemical pathways that include a loss of normal mitochondrial function. To date a number of specific gene mutations in mitochondrial related genes including PINK1, DJ1, PARKIN, and HTRA2 that have all been linked to hereditary forms of PD (52-56). All of these mutations have been studied in relation to a common molecular hallmark, the loss of complex I activity within the electron transport chain in association with PD. As with Alzheimer’s disease, reduction in metabolic efficiency within the mitochondria may cause a selective vulnerability of specific subpopulations of neurons that may contribute to the disease.
While there is a substantial amount of evidence for biochemical defects in mitochondria of Parkinson’s patients, there is little evidence to suggest that SOD2 function is directly involved in the clinical progression of this disease. There have been reports that indicate that the levels of SOD2 may be increased in the frontal cortex of Parkinson’s patients although the numbers of patients recruited in the studies were too small to be conclusive (57,58). Similarly, the activity of SOD2 in the plasma of Parkinson’s patients was found to be elevated over age-matched controls even when they are in the asymptomatic phase of the disease (59). Other studies have contradicted these findings, supporting the notion that there is no change in SOD2 activity in PD (60). While the change in SOD2 in relation to PD remains debatable, there are characteristic hallmarks of oxidative stress within the substantia nigra of patients with PD (61-63). There is however a relative lack of detailed data in this respect, likely relating to the inherent difficulty in examining ongoing changes in the disease process, and a necessity to rely on material collected at the end stage of the disease after cell damage and death has occurred. Like AD, the issue of cause and effect is also problematic; do the oxidative changes occur in response to something else? Is oxidative damage directly involved in the pathogenesis? This presents a real challenge in mechanistically ordering the key events in age-related neurodegenerative disease (64). To some extent, this can be better modeled using animal models of PD, where cause and effect, and temporal sequencing of pathologies critically involved in the pathogenesis of PD may be better understood.
Because of complex variations in mutations that can cause different forms of heritable PD, there are several challenges in making appropriate animal models which capture the phenotypes observed in PD. There have been several model systems developed which target selective destruction of dopaminergic neurons within the brain. One of the most common animal models used to study PD, was inspired by a serendipitous finding in drug addicts who had consumed the opiate MPPP (1-Methyl-4-phenyl-4-propionoxypiperidine) contaminated with the toxin MPTP (1-methyl 4-phenyl 1,2,3,6 tetrahydropyridine) (65). Use of this narcotic resulted in a rapidly developing Parkinsonian like phenotype, after repeated use of the drug. The symptoms were caused by selective death of the dopaminergic neurons by the MPTP contaminating the recreational drug. The mechanisms behind the drug’s neurotoxic effects have been well characterized since the initial report. The conversion of MPTP to MPP+ (1-methyl 4-phenylpyridinium) within the CNS causes the compound to be taken up by the dopamine reuptake machinery at the synaptic terminal. Once within the cell, MPP+ acts as a Complex I inhibitor of the mitochondrial respiratory chain, causing bioenergetic collapse and neuronal cell death (66). The exposure to MPTP therefore closely mimics the loss of neurons in PD from a variety of genetic lesions.
MPTP treatment of rodents and lower primates has uncovered a number of important aspects in the progression of PD with respect to the reduction of mitochondrial activity. However, simply selectively challenging the dopaminergic neurons with neurotoxins does not fully explain how PD progresses in humans. There are a number of biochemical abnormalities that must occur prior to, or concurrently with the loss of mitochondrial function. A recent study demonstrated that contrary to a widely held belief, the dopaminergic neuronal population is not selectively vulnerable to bioenergetic challenge over that of the non-dopaminergic neurons (67). Furthermore, this study reported that in mice, the ability of mitochondria to maintain their membrane potential is not affected by age in either neuronal population. This suggests that the initiation of Parkinson’s is not simply a loss of energy production, but rather the selective loss of neurons from a unique environment (the substantia nigra) which are biochemically distinct (68). Glutamate induced toxicity in a culture model demonstrated that the expression level of SOD2 was sufficient to prevent cell death, where SOD1 does not affect neuron survival (69). Additionally, these dopaminergic neurons contain a number of substances such as high iron load and cytoplasmic dopamine with the potential to generate endogenous oxidative stress beyond that generated during normal cellular respiration (70).
Other genetic models of PD have been developed beyond the neurotoxic approach, uncovering potentially important mechanisms (62). One such example is the DJ-1 knockout mouse, which has revealed that the loss of this protein increased H2O2 levels within the brain (71,72), further implicating oxidative stress in PD. However, these mice do not demonstrate either a loss of dopaminergic neurons or mitochondrial damage. Therefore, increased levels of H2O2 may be due to a compensatory up-regulation of antioxidant defenses that include SOD2, thereby giving rise to increased levels of H2O2. Exogenous DJ-1 treatment was protective of dopaminergic neurons when injected into the fore-brains of mice which were administered neurotoxins (73). The authors of this study also found that the levels of SOD2 were increased as a result of DJ-1 inactivation. Modulation of this disease phenotype has also been noted in unrelated models where indirect up-regulation of SOD2 via the transcription factor FOXO prevented the loss of dopaminergic neurons in PINK1 null drosophila (74). Therefore although SOD2 level does not appear to be directly tied to PD, the antioxidant defense system is critical is preventing neuronal death in the face of disparate cellular stresses.
SOD2 in Ischemic Stroke
The sequence of events during a stroke can lead to a complex cascade ultimately setting up neuronal death within the affected region (75). Initially during an ischemic stroke, also called a “cerebrovascular accident”, a vessel supplying the brain becomes occluded, thereby preventing the normal supply of blood delivering oxygen to meet the metabolic demand of the tissue. This immediately sets up a situation where the neuronal tissue distal to vessel(s) becomes ischemic, causing a cascade of bioenergetic collapse (76,77). Typically, a symptomatic patient presents in the clinic where they are administered agents to dissolve the impeding clot and restore blood flow to the ischemic region (78). This reperfusion of the area with oxygenated blood prevents neuronal death due to lack of oxygen and metabolic substrates. However, the rapid resumption of metabolism in the ischemic area causes a secondary wave of reactive oxygen species generated from enzymes such as xanthine oxidase and the mitochondria further damaging the affected area (79). Therefore, the antioxidant defense systems in neuronal tissue including SOD2 are critical to preventing cell death from ischemia and reperfusion injury subsequent to a stroke.
The role of oxidative stress and mitochondria in the neurodegenerative processes of stoke are somewhat clearer than that in Alzheimer’s and Parkinson’s disease because of the acute nature of the traumatic injury to the brain (80). Consistent with this notion, antioxidants have been proposed as potential acute therapeutics in recovery from stroke and more generally as preventative agents (79). Potential biomarkers for the efficacy of such antioxidant treatments such as the matrix metalloproteinase MMP9 activity are also under investigation (81). This strategy may prove to be effective in lessening the damage after stroke as it has been demonstrated in humans that there is a diminished capacity for antioxidant activity in patients following an ischemic stroke (82,83). Because of our ability to follow both the initial and subsequent oxidative damage from a specific traumatic event; a number of valuable translation models have been developed to model what occurs in human stroke.
From these translational models of stoke we can also appreciate SOD2 as a significant player in counteracting neurodegeneration. If antioxidants are given in the critical period during the beginning of ischemia, SOD2 is markedly unchanged in cerebral tissue (84). Jung et al. also demonstrated during reperfusion the regulation of SOD2 expression under the control of STAT3 is crucial to neuroprotection (85). Similar genetic control in response to ischemia was seen in genomic studies of brain tissue from ipsilateral and contralateral strokes identified Sod2 as one of the most dramatically altered genes after a stroke (86). Overall the clinical and basic studies of oxidative damage during and following a stroke indicate that treating the metabolic symptoms may be important and part of a larger regimen to restore blood flow, reduce inflammation, and prevent scar tissue to promote the best outcome for the patient (87,88).
SOD2 in Aging
While much research has been devoted to studying specific diseases or neurological disorders associated with advancing age, a decline in cognitive function is nearly a universal attribute of aging. Age-related cognitive impairment (ARCI) typically begins within the third decade of life, although it is usually not apparent until a much later age. This decline is not thought to be due to a large decline in the numbers of neurons in the brain, but rather a loss of connectedness in the neural networks resulting in the loss of our ability to learn, store and process information (89). Because ARCI is nearly universally present, it often makes it difficult to differentiate a typical age-related cognitive decline from early pre-dementia conditions. This often has implications for the physician, requiring them to make a judgment call as to what a normal or acceptable rate of cognitive loss is for a patient against their chronological age (90). Neuronal loss and disease development can be viewed as a function of the aging process where the loss of cellular homeostasis and function eventually leads to a bioenergetic crisis point or threshold for each neuron (Figure 2). The exact order of events leading to this stochastic neuronal loss is not precisely known, and is further complicated by the interrelated nature of cellular functions in various neuron types. What is clear is that given enough time of other pathologies; we all would experience some form of neuronal loss leading to reduced cognitive function.
Figure 2. Aging and neurodegenerative disorders.
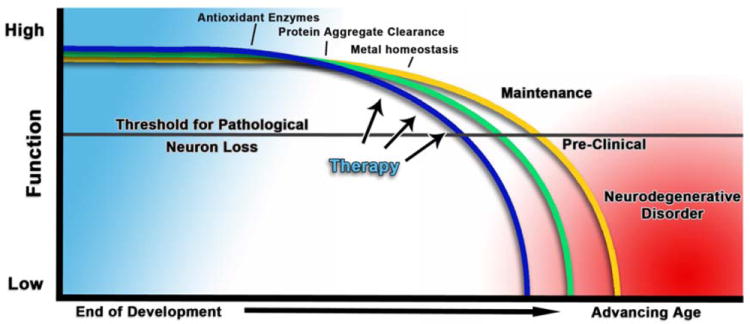
A simplistic model of how aging impacts the development of neurodegenerative disorders. As we age, there is a decline in a multitude of interrelated cellular processes, including antioxidant defenses of mitochondria, causing a cascade of functional losses ultimately reaching a threshold for bioenergetic collapse and cell death. On a long enough time scale free from morbidity in other tissues; everyone would eventually see the effects of this functional loss prior to death. Therefore examining the progression and timing of the causes and developing therapies to counteract or maintain functional loss are critical in maintaining healthy brain aging.
While the loss of cognitive function in ARCI is not as severe as in Alzheimer’s disease, ARCI can be considered a mild but progressive form of a neurodegenerative disorder. As we continue to extend the median and maximal lifespan of individuals, the maintenance of cognitive function will be of increasing concern to maintain the quality of life of the aging community (91). The maintenance of neuroplasticity comes with a high energy cost to maintain synaptic connections. This constant metabolic demand can result in endogenous oxidative stress as our defenses decline with age. This is complicated by the fact that we know very little about the rate of oxidative stress within specific neuronal subpopulations of the brain in vivo. Although it is clear that mitochondria have the potential to produce more oxidative stress when metabolically quiescent, rather than when actively respiring, it is unclear how age-related changes in the brain impact the potential for a pro-oxidative environment. Mitochondria within neurons have been shown to be more susceptible to stress with age, particularly in synaptic mitochondria (92). Similarly, endogenous oxidative stress can severely restrict the bioenergetic capacity of the mitochondria in the synapse (93). This increased oxidative stress has been shown to reduce cognitive function in a number of animal studies (94-96). Studies in human beings have also found some correlation between markers of oxidative stress and cognitive function (97,98). The ability to modulate a decline in cognitive function with dietary supplements has met with mixed success, and this may be attributable in part to the complexity of cognitive assessment, differences in study design, efficacy of the antioxidants in question, and the difficulty of effectively delivering antioxidants to the brain (99). While the results in human trials are not conclusive, aged C57Bl6 mice treated with catalytic antioxidants maintain their cognitive function better than that of untreated mice (100). While the promise of antioxidant supplements for the maintenance of cognitive function is an attractive prospect; there may be a more practical approach to decreasing oxidative stress and maintaining function as we age. Both exercise and diet can modulate the level of oxidative stress in an organism. Through the use of lifestyle, changes in activity or diet can slow the onset of cognitive decline with age (101-105). This not only has benefits to the individual, but also provides a low cost solution to the expanding costs within the healthcare system. Although there is a great deal of data on the benefits of exercise in reducing age-related pathologies in humans, corresponding studies in animal models are much less explored, and this remains a potentially vibrant area for exploration from a mechanistic perspective. Overall, future studies will determine if lifestyle alone can maintain normal cognitive function throughout our entire lifespan or pharmaceutical intervention will be necessary to achieve “successful” aging.
Neuronal effects in animal models of SOD2 dysfunction
We have examined a variety of animal models relevant to age-related neurodegenerative disease, in the context of our knowledge about the role of SOD2 in maintaining low levels of superoxide in the mitochondria. However, one can directly manipulate SOD2 by genetic approaches, and a number of animal models in this context have been created (Table 1). These models provide interesting phenotypes demonstrating how modulating the antioxidant capacity within the CNS can affect the progression of neurodegenerative disorders. The brain consumes approximately 20% of the total available blood flow for oxidative respiration despite accounting for only 3% of the total body mass (106). These data convey just how metabolically active the brain is, and how tightly regulated the neutralization of potentially damaging radicals from mitochondrial respiration must be in order to avoid cell toxicity.
Table 1.
In vivo models of Sod2’s role in Neuronal disease
| Genetic modification | Details | Phenotype | References |
|---|---|---|---|
| Constitutive Sod2 -/- | Strain dependent embryonic or neonatal lethality. | Neurodegeneration and spongiform encephalopathy prevalent, and modulated by antioxidant intervention. Selective regional loss of neurons. | (107-111,115) |
| Constitutive Sod2 +/- | Mouse is developmentally normal and lives into adulthood. | No overt phenotype phenotype, but there is a susceptibility to oxidative stress and increased risk of cancer. | (116) |
| Constitutive Sod2 over expression | Over expression of SOD2 enzyme in whole animal. | Mixed results in the extension of lifespan. Did not improve neuronal function | (117,118) |
| Constitutive Sod2 over expression in Tg2576 mice | SOD2 increased in a model common model of Alzheimer’s Disease | Over expression prevented some pathological changes and improved brain function. | (133) |
| Constitutive Sod2 +/- with Neurotoxins. | MPTP, 3-NP, and Malonate treatment of Sod2 null heterozygous mice. | Neurotoxic stress is enhanced by reduction in Sod2. | (113) |
| Targeted Sod2 knockout in all Nestin positive cells. | CRE-Lox system driven by Nestin promoter | Severe CNS degeneration with no major PNS effects. | (134) |
| Targeted Sod2 knockout in motor neurons | CRE-Lox system driven by vesicular acetylcholine transporter for adult cholinergic neurons | No altered function in motor neurons. | (135) |
| Mutant APP with Sod2 +/- | Sod2 +/- mice with Alzheimer’s models of plaque. | Reduction in SOD2 activity accelerates the severity of the AD phenotype. | (46-48) |
One of the first mammalian models to examine the effects of endogenous mitochondrial oxidative stress in mice was the constitutive Sod2 -/- mouse (107). Constitutive inactivation of the gene for superoxide dismutase 2 results in severe pathologies in multiple organ systems (107-115), including neurodegeneration ((111), an earlier report of neurodegeneration proved to be non-reproducible). The extent of neurodegeneration due to loss of Sod2 can be modulated by treating the mice with different doses of catalytic antioxidants to prevent the oxidative damage (108,110,111); despite the fact that the respiratory capacity of mitochondria remains severely impaired (93). Endogenous oxidative stress in this mouse model also promotes the hyperphosphorylation of tau at multiple phosphorylation sites, similar to that observed in Alzheimer’s disease (46). This study directly implicated mitochondrial dysfunction from endogenous oxidative damage in the etiology of one of the hallmarks of AD, the neurofibrillary tangle.
While the Sod2 -/- mouse is a useful model to frame questions about the consequences of endogenous mitochondrial oxidative stress, these animals experience acute damage that is not seen in the slow progressive decline of age-related neurodegenerative disorders. In an attempt to further refine this model system; there were a number of studies reported which crossed mice hemizygous for Sod2 with “traditional” models of Alzheimer’s disease. Interestingly, mice heterozygous for Sod2 have only a mild phenotype and live a normal lifespan (116), in contrast to the homozygous null which is neonatal lethal in multiple genetic backgrounds. However, the heterozygotes still undergo a low level of oxidative stress, which when combined with other transgenic neurodegeneration models can accelerate the progression of the disease. This was seen in the Tg2576 and human APP transgenic models of Alzheimer’s disease discussed earlier.
If reduced SOD2 activity was deleterious to the cell and to the development of age-related disease, then it seems reasonable to posit that constitutive over expression of this gene must be protective against such disorders. The results from such studies concluded that increased SOD2 could not consistently extend the lifespan of these mice (117,118). Similarly, there was no improvement or protection against age-related neuronal decline as compared to normal mice. Despite these findings, It was found that in the context of the Tg2576 Alzheimer’s mouse model, the over expression of Superoxide dismutase 2 is protective, and can prevent some of the pathological features characteristic of that mouse (119). There are several possibilities that may help explain these apparently disparate results. First, it is possible that there may be a distinction in the role of SOD2 in neuronal disease verses other morbidities. Another possibility is the effect of genetic background, as it has been known for some time that genetic background can have profound effects on phenotypic outcomes (120). Therefore, caution must be observed in over interpretation of studies on single genetic backgrounds, as radically different results from inactivation of a gene may arise giving apparent clear-cut results, which may in fact be due to compensatory processes and novel gene/environment interactions.
The level of ROS within the cell must be maintained and balanced within any metabolically active tissue. Over expression of SOD2 in a normal aging mouse will compensate for that increased antioxidant capacity, just as a heterozygous Sod2 mouse will compensate for the loss of that missing gene copy. The key to the function of SOD2 is to help maintain balance and produce the ideal metabolic environment for the cell. It is overly simplistic to assume that all ROS is “bad”, and reducing the levels of ROS within the cell to zero would result in a better cellular environment, and a corresponding increase in cellular efficiency. Adding to this overall complexity is our lack of knowledge of ROS metabolism between differing tissues and cell types. There is a general paucity of high quality information as to the levels of ROS resulting from differing physiological states such as aging. It is conceivable that imbalances in bioenergetic status, or oxidative stress could lead to a cellular crisis which may explain the slow stochastic loss of neurons in age related neurodegenerative disease (121). It is only when the system is perturbed, such as in the context of a mutant amyloid protein, loss of function in a biochemical enzyme, or exposure to neurotoxins that the activity of SOD2 appears to modulate the severity of the disease model.
Clinical relevance of oxidative stress in neurodegeneration
To date, attempts to stem the progression of neurodegenerative disease symptoms with antioxidants have not been successful. For Alzheimer’s disease a series of clinical trials for Idebenone (a synthetic analog of coenzyme Q10) indicated promising results but was ultimately unsuccessful in slowing the cognitive decline in patients (122-124). These results have been echoed in studies using Vitamin E and C in the prevention of mild cognitive impairment (MCI) and Alzheimer’s disease (125). Double-blinded studies have shown some significant benefits to delay the onset of advanced Alzheimer’s, but could not prevent the progression or rate of decline from MCI to fully developed Alzheimer’s disease (126). Similarly, clinical antioxidant treatments in Parkinson’s disease have not yet yielded beneficial treatment.
Two different antioxidant compounds have been tried in double-blinded clinical trials. Coenzyme Q10 was well tolerated in the patient group, but there was no benefit observed after 3 months of treatment (127). The efficacy of antioxidant therapy in the progression was again tested three years later in another clinical trail of “MitoQ”, a mitochondrially targeted form of CoQ10 in PD patients. This therapy too was not effective at slowing the progression of Parkinson’s (128). These data indicate that oxidative stress may not be a causative factor in PD, but may be symptomatic of the ongoing dysfunction in the cellular machinery regulating mitochondria. Further examination of antioxidants in combination with other treatments may yet provide additional benefits in neuroprotection.
While the regulation of oxidative stress alone does not appear to prevent specific neurodegenerative disorders, it may provide some benefit in slowing the progression of these diseases and help to maintain the bioenergetic function of neurons. This oxidative stress paradigm may be useful in maintaining function in the context of aging. A number of prospective clinical studies have seen modest benefits in delaying mild cognitive impairment with prophylactic treatment of antioxidants (129,130). However these results remain controversial within the literature (131,132). In general, these studies raise more questions than answers, and further stimulate enquiries into what else contributes to neurodegenerative disease, and how can age-related changes within the brain be dealt with on multiple fronts beyond oxidative damage and mitochondrial dysfunction. One step towards the development of multifaceted treatments is a deeper understanding of how to maintain the balance between bioenergetic demand, and the capacity to recycle the metabolic byproducts of the neuron. By understanding SOD2’s role in this equilibrium, or disequilibrium in the case of disease, we will come closer to developing treatments for age-related disease, and potentially a better understanding of mechanisms which drive the pathophysiology of aging itself.
Highlights.
Sod2 is critical to cellular function in metabolically active tissues, particularly neurons.
Neurodegenerative disorders show the importance of the SOD2’s role against pathological stresses.
Model systems portray mechanisms behind oxidative stress and in vivo neurodegenerative disease.
Acknowledgments
This work was supported in part by the Larry L. Hillblom Foundation (SM), and National Institutes of Health through the Nathan Shock Center [P30AG025708], AG18675(SM), and PO1AG025901(SM). JMF was supported by [T32AG000266] awarded to the Buck Institute for Research on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- 1.Harman D. The biologic clock: The mitochondria? J Am Geriatr Soc. 1972 Apr;20(4):145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Pérez VI, Bokov A, Remmen HV, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochimica Et Biophysica Acta (BBA) - General Subjects. 2009 Oct;1790(10):1005–14. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA. The role of metallobiology and amyloid-β peptides in Alzheimer’s disease. Journal of Neurochemistry. 2011 Nov 28;120:149–66. doi: 10.1111/j.1471-4159.2011.07500.x. [DOI] [PubMed] [Google Scholar]
- 4.James SA, Volitakis I, Adlard PA, Duce JA, Masters CL, Cherny RA, Bush AI. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radical Biol Med. 2012 Feb 15;52(2):298–302. doi: 10.1016/j.freeradbiomed.2011.10.446. [DOI] [PubMed] [Google Scholar]
- 5.Greenough MA, Camakaris J, Bush AI. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochemistry International. 2013 May;62(5):540–55. doi: 10.1016/j.neuint.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Bush AI. The metal theory of Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S277–81. doi: 10.3233/JAD-2012-129011. [DOI] [PubMed] [Google Scholar]
- 7.Andersen JK. Oxidative stress in neurodegeneration: Cause or consequence? Nature Medicine. 2004 Jul;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 8.Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest. 1984 Oct;74(4):1398–403. doi: 10.1172/JCI111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strålin P, Karlsson K, Johansson BO, Marklund SL. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995 Nov;15(11):2032–6. doi: 10.1161/01.atv.15.11.2032. [DOI] [PubMed] [Google Scholar]
- 10.Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest. 2000 Jun;105(11):1631–9. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radical Biology Med. 2003 Aug 1;35(3):236–56. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 12.Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxidants and Redox Signaling. 2011 Sep 15;15(6):1583–606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. Journal of Cerebral Blood Flow and Metabolism. 2001 Feb;21(1):2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Crapo JD, Oury T, Rabouille C, Slot JW, Chang LY. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proceedings of the National Academy of Sciences of the United States of America. 1992 Nov 1;89(21):10405–9. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. The Journal of Biological Chemistry. 2001 Oct 12;276(41):38084–9. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 16.Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005 Apr 9;25(10):2463–70. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Human Molecular Genetics. 2008 Nov 1;17(21):3303–17. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-sod in mitochondria. The Journal of Biological Chemistry. 2001 Oct 19;276(42):38388–93. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 19.Barrett EF, Barrett JN, David G. Mitochondria in motor nerve terminals: Function in health and in mutant superoxide dismutase 1 mouse models of familial ALS. Journal of Bioenergetics and Biomembranes. 2011 Dec;43(6):581–6. doi: 10.1007/s10863-011-9392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrì MT, Cozzolino M. SOD1 and mitochondria in ALS: A dangerous liaison. Journal of Bioenergetics and Biomembranes. 2011 Dec;43(6):593–9. doi: 10.1007/s10863-011-9394-z. [DOI] [PubMed] [Google Scholar]
- 21.Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathologica. 2012 Sep;124(3):339–52. doi: 10.1007/s00401-012-1022-4. [DOI] [PubMed] [Google Scholar]
- 22.Weisiger RA, Fridovich I. Mitochondrial superoxide dismutase. Journal of Biological Chemistry. 1973 Jul 10;248(13):4793–6. [PubMed] [Google Scholar]
- 23.Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical Journal. 2009 Feb 1;417(1):1. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridovich I. Oxygen toxicity: A radical explanation. Journal of Experimental Biology. 1998;201(8):1203–9. doi: 10.1242/jeb.201.8.1203. [DOI] [PubMed] [Google Scholar]
- 25.Pacifici RE, Davies KJ. Protein, lipid and DNA repair systems in oxidative stress: The free-radical theory of aging revisited. Gerontology. 1991;37(1-3):166–80. doi: 10.1159/000213257. [DOI] [PubMed] [Google Scholar]
- 26.Gupta A, Hasan M, Chander R, Kapoor NK. Age-related elevation of lipid peroxidation products: Diminution of superoxide dismutase activity in the central nervous system of rats. Gerontology. 1991;37(6):305–9. doi: 10.1159/000213277. [DOI] [PubMed] [Google Scholar]
- 27.Daly MJ. Death by protein damage in irradiated cells. DNA Repair. 2012 Feb;11(1):12–21. doi: 10.1016/j.dnarep.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 28.Grimm S, Hoehn A, Davies KJ, Grune T. Protein oxidative modifications in the ageing brain: Consequence for the onset of neurodegenerative disease. Free Radical Research. 2011 Feb;45(1):73–88. doi: 10.3109/10715762.2010.512040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grune T, Davies KJA. The proteasomal system and HNE-modified proteins. Molecular Aspects of Medicine. 2003 Aug;24(4-5):195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 30.Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. The Lancet Neurology. 2007 Aug;6(8):734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 31.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984 Jul;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 32.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nature Reviews Neurology. 2011 Apr 8;7(3):137–52. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry G, Nunomura A, Hirai K, Zhu X, Pérez M, Avila J, et al. Is oxidative damage the fundamental pathogenic mechanism of alzheimer’s and other neurodegenerative diseases? Free Radic Biol Med. 2002 Dec 1;33(11):1475–9. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- 34.Leuner K, Schütt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxidants & Redox Signaling. 2012 Jul 15;16(12):1421–33. doi: 10.1089/ars.2011.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002 Jun 1;32(11):1050–60. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 36.Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with alzheimer’s disease: Insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8(11-12):2021–37. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- 37.Gustaw-Rothenberg K, Lerner A, Bonda DJ, Lee HG, Zhu X, Perry G, Smith MA. Biomarkers in alzheimer’s disease: Past, present and future. Biomark Med. 2010 Mar;4(1):15–26. doi: 10.2217/bmm.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butterfield DA, Bader Lange ML, Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochimica Et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2010 Aug;1801(8):924–9. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Leo ME, Borrello S, Passantino M, Palazzotti B, Mordente A, Daniele A, et al. Oxidative stress and overexpression of manganese superoxide dismutase in patients with Alzheimer’s disease. Neuroscience Letters. 1998 Jul 10;250(3):173–6. doi: 10.1016/s0304-3940(98)00469-8. [DOI] [PubMed] [Google Scholar]
- 40.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006 Oct 19;443(7113):787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 41.Cha M-Y, Han S-H, Son SM, Hong H-S, Choi Y-J, Byun J, Mook-Jung I. Mitochondria-Specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE. 2012 May 13;7(4):e34929. doi: 10.1371/journal.pone.0034929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. Journal of Neurochemistry. 2002 Feb;80(1):91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 43.Coskun P, Wyrembak J, Schriner SE, Chen H-W, Marciniack C, Laferla F, Wallace DC. A mitochondrial etiology of alzheimer and parkinson disease. Biochimica Et Biophysica Acta. 2012 Jun;1820(5):553–64. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010 Jun;51(5):440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 45.Choi SW, Gerencser AA, Ng R, Flynn JM, Melov S, Danielson SR, et al. No consistent bioenergetic defects in presynaptic nerve terminals isolated from mouse models of Alzheimer’s disease. J Neurosci. 2012 Nov 21;32(47):16775–84. doi: 10.1523/JNEUROSCI.2414-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, et al. Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE. 2007 Jul 20;2(6):e536. doi: 10.1371/journal.pone.0000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. Journal of Neurochemistry. 2004 Jul;89(5):1308–12. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 48.Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, et al. Reduction in mitochondrial superoxide dismutase modulates alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006 May 10;26(19):5167–79. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, et al. Amyloid - induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011 May 13;31(15):5589–95. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dickson DW. Parkinson’s disease and parkinsonism: Neuropathology. Journal of Neuropathology & Experimental Neurology. 1996 [Google Scholar]
- 51.Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, et al. Familial aggregation of Parkinson’s disease: A population-based case-control study in europe. EUROPARKINSON study group. Neurology. 1999 Jul 10;52(9):1876–82. doi: 10.1212/wnl.52.9.1876. [DOI] [PubMed] [Google Scholar]
- 52.Puschmann A. Monogenic parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013 Apr;19(4):407–15. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 53.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MMK, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science (New York, NY) 2004 Jun 21;304(5674):1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 54.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (New York, NY) 2003 Feb 10;299(5604):256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 55.Shimizu N, Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, et al. Access : Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism : Nature. Nature. 1998 May 9;392(6676):605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 56.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding omi/htra2 in Parkinson’s disease. Human Molecular Genetics. 2005 Aug 1;14(15):2099–111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 57.Radunovic A, Porto WG, Zeman S, Leigh PN. Increased mitochondrial superoxide dismutase activity in Parkinson’s disease but not amyotrophic lateral sclerosis motor cortex. Neuroscience Letters. 1997 Dec 19;239(2-3):105–8. doi: 10.1016/s0304-3940(97)00905-1. [DOI] [PubMed] [Google Scholar]
- 58.Ferrer I, Perez E, Dalfo E, Barrachina M. Abnormal levels of prohibitin and ATP synthase in the substantia nigra and frontal cortex in Parkinson’s disease. Neuroscience Letters. 2007 Apr 30;415(3):205–9. doi: 10.1016/j.neulet.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 59.Ihara Y, Chuda M, Kuroda S, Hayabara T. Hydroxyl radical and superoxide dismutase in blood of patients with Parkinson’s disease: Relationship to clinical data. Journal of the Neurological Sciences. 1999 Nov 30;170(2):90–5. doi: 10.1016/s0022-510x(99)00192-6. [DOI] [PubMed] [Google Scholar]
- 60.Marttila RJ, Lorentz H, Rinne UK. Oxygen toxicity protecting enzymes in Parkinson’s disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. Journal of the Neurological Sciences. 1988 Sep;86(2-3):321–31. doi: 10.1016/0022-510x(88)90108-6. [DOI] [PubMed] [Google Scholar]
- 61.Surmeier DJ, Guzman JN, Sanchez-Padilla J, Schumacker PT. The role of calcium and mitochondrial oxidant stress in the loss of substantia nigra pars compacta dopaminergic neurons in parkinson’s disease. Neuroscience. 2011 Dec 15;198:221–31. doi: 10.1016/j.neuroscience.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Varcin M, Bentea E, Michotte Y, Sarre S. Oxidative stress in genetic mouse models of parkinson’s disease. Oxid Med Cell Longev. 2012;2012:624925. doi: 10.1155/2012/624925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, et al. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochemical Research. 2011 Aug;36(8):1452–63. doi: 10.1007/s11064-011-0471-9. [DOI] [PubMed] [Google Scholar]
- 64.Jenner P. Oxidative stress and parkinson’s disease. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical Neurology. Vol. 83. 2007. pp. 507–20. [DOI] [PubMed] [Google Scholar]
- 65.Fukuda T. Neurotoxicity of MPTP. Neuropathology. 2001 Dec;21(4):323–32. doi: 10.1046/j.1440-1789.2001.00402.x. [DOI] [PubMed] [Google Scholar]
- 66.Tipton KF, Singer TP. Advances in Our Understanding of the Mechanisms of the Neurotoxicity of MPTP and Related Compounds. Journal of Neurochemistry. 1993 Oct;61(4):1191–206. doi: 10.1111/j.1471-4159.1993.tb13610.x. [DOI] [PubMed] [Google Scholar]
- 67.Choi SW, Gerencser AA, Lee DW, Rajagopalan S, Nicholls DG, Andersen JK, Brand MD. Intrinsic bioenergetic properties and stress sensitivity of dopaminergic synaptosomes. J Neurosci. 2011 Apr 23;31(12):4524–34. doi: 10.1523/JNEUROSCI.5817-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in parkinson’s disease: Molecular mechanisms and pathophysiological consequences. The EMBO Journal. 2012 Jul 18;31(14):3038–62. doi: 10.1038/emboj.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fukui M, Zhu BT. Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radical Biology and Medicine. 2010 Apr 15;48(6):821–30. doi: 10.1016/j.freeradbiomed.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaur D, Andersen J. Does cellular iron dysregulation play a causative role in parkinson’s disease? Ageing Research Reviews. 2004 Jul;3(3):327–43. doi: 10.1016/j.arr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 71.Yamaguchi H, Shen J. Absence of dopaminergic neuronal degeneration and oxidative damage in aged DJ-1-deficient mice. Molecular Neurodegeneration. 2007;2:10. doi: 10.1186/1750-1326-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proceedings of the National Academy of Sciences of the United States of America. 2007 Sep 11;104(37):14807–12. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun SY, An CN, Pu XP. DJ-1 protein protects dopaminergic neurons against 6-OHDA/mg-132-induced neurotoxicity in rats. Brain Research Bulletin. 2012 Sep 1;88(6):609–16. doi: 10.1016/j.brainresbull.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 74.Koh H, Kim H, Kim MJ, Park J, Lee HJ, Chung J. Silent information regulator 2 (sir2) and forkhead box O (FOXO) complement mitochondrial dysfunction and dopaminergic neuron loss in drosophila pten-induced kinase 1 (PINK1) null mutant. Journal of Biological Chemistry. 2012 May 13;287(16):12750–8. doi: 10.1074/jbc.M111.337907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron. 2010 Jul 29;67(2):181–98. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moro MA, Almeida A, Bolaños JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005 Nov 15;39(10):1291–304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 77.Kristián T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium. 2004;36(3-4):221–33. doi: 10.1016/j.ceca.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 78.Jaffer H, Morris VB, Stewart D, Labhasetwar V. Advances in stroke therapy. Drug Delivery and Translational Research. 2011 Dec 1;1(6):409–19. doi: 10.1007/s13346-011-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005 Aug 15;39(4):429–43. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 80.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, et al. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants & Redox Signaling. 2011 May 15;14(8):1505–17. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kelly PJ, Morrow JD, Ning M, Koroshetz W, Lo EH, Terry E, et al. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: The biomarker evaluation for antioxidant therapies in stroke (beat-stroke) study. Stroke. 2008 Feb;39(1):100–4. doi: 10.1161/STROKEAHA.107.488189. [DOI] [PubMed] [Google Scholar]
- 82.Ciancarelli I, Di Massimo C, De Amicis D, Carolei A, Tozzi Ciancarelli MG. Evidence of redox unbalance in post-acute ischemic stroke patients. Curr Neurovasc Res. 2012 Jun;9(2):85–90. doi: 10.2174/156720212800410885. [DOI] [PubMed] [Google Scholar]
- 83.Gariballa SE, Hutchin TP, Sinclair AJ. Antioxidant capacity after acute ischaemic stroke. QJM : Monthly Journal of the Association of Physicians. 2002 Oct;95(10):685–90. doi: 10.1093/qjmed/95.10.685. [DOI] [PubMed] [Google Scholar]
- 84.Connell BJ, Saleh M, Khan BV, Saleh TM. Lipoic acid protects against reperfusion injury in the early stages of cerebral ischemia. Brain Research. 2011 Mar 23;1375:128–36. doi: 10.1016/j.brainres.2010.12.045. [DOI] [PubMed] [Google Scholar]
- 85.Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009 Jun 27;29(21):7003–14. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buga A-M, Scholz CJ, Kumar S, Herndon JG, Alexandru D, Cojocaru GR, et al. Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PLoS ONE. 2012 Dec 12;7(12):e50985. doi: 10.1371/journal.pone.0050985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Popa-Wagner A, Carmichael ST, Kokaia Z, Kessler C, Walker LC. The response of the aged brain to stroke: Too much, too soon? Curr Neurovasc Res. 2007 Aug;4(3):216–27. doi: 10.2174/156720207781387213. [DOI] [PubMed] [Google Scholar]
- 88.Nilupul Perera M, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC, Donnan GA. Inflammation following stroke. Journal of Clinical Neuroscience. 2006 Feb;13(1):1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 89.Pannese E. Morphological changes in nerve cells during normal aging. Brain Structure & Function. 2011 Jul;216(2):85–9. doi: 10.1007/s00429-011-0308-y. [DOI] [PubMed] [Google Scholar]
- 90.Ashford JW, Atwood CS, Blass JP, Bowen RL, Finch CE, Iqbal K, et al. What is aging? What is its role in Alzheimer’s disease? What can we do about it? Journal of Alzheimer’s Disease. 2005 Jul;7(3):247–53. doi: 10.3233/jad-2005-7308. discussion 255-62. [DOI] [PubMed] [Google Scholar]
- 91.de Groot LCPMG, Verheijden MW, de Henauw S, Schroll M, van Staveren WA, Investigators S. Lifestyle, nutritional status, health, and mortality in elderly people across europe: A review of the longitudinal results of the SENECA study. The Journals of Gerontology. 2004 Dec;59(12):1277–84. doi: 10.1093/gerona/59.12.1277. [DOI] [PubMed] [Google Scholar]
- 92.Lores-Arnaiz S, Bustamante J. Age-related alterations in mitochondrial physiological parameters and nitric oxide production in synaptic and non-synaptic brain cortex mitochondria. Neuroscience. 2011 Aug 11;188:117–24. doi: 10.1016/j.neuroscience.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 93.Flynn JM, Choi SW, Day NU, Gerencser AA, Hubbard A, Melov S. Impaired spare respiratory capacity in cortical synaptosomes from Sod2 null mice. Free Radic Biol Med. 2011 Apr 1;50(7):866–73. doi: 10.1016/j.freeradbiomed.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxidants & Redox Signaling. 2011 Jun 15;14(10):2013–54. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fukui K, Onodera K, Shinkai T, Suzuki S, Urano S. Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Annals of the New York Academy of Sciences. 2001 May;928:168–75. doi: 10.1111/j.1749-6632.2001.tb05646.x. [DOI] [PubMed] [Google Scholar]
- 96.Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proceedings of the National Academy of Sciences of the United States of America. 1996 Jun 14;93(10):4765–9. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berr C, Balansard B, Arnaud J, Roussel AM, Alperovitch A. Cognitive decline is associated with systemic oxidative stress: The EVA study. Etude du vieillissement arteriel. J Am Geriatr Soc. 2000 Oct;48(10):1285–91. doi: 10.1111/j.1532-5415.2000.tb02603.x. [DOI] [PubMed] [Google Scholar]
- 98.Perkins AJ, Hendrie HC, Callahan CM, Gao S, Unverzagt FW, Xu Y, et al. Association of antioxidants with memory in a multiethnic elderly sample using the third national health and nutrition examination survey. American Journal of Epidemiology. 1999 Jul 1;150(1):37–44. doi: 10.1093/oxfordjournals.aje.a009915. [DOI] [PubMed] [Google Scholar]
- 99.Polidori MC, De Spirt S, Stahl W, Pientka L. Conflict of evidence: Carotenoids and other micronutrients in the prevention and treatment of cognitive impairment. BioFactors. 2012;38(2):167–71. doi: 10.1002/biof.1001. [DOI] [PubMed] [Google Scholar]
- 100.Clausen A, Doctrow S, Baudry M. Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol Aging. 2010 Apr;31(3):425–33. doi: 10.1016/j.neurobiolaging.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Etgen T, Sander D, Bickel H, Forstl H. Mild cognitive impairment and dementia: The importance of modifiable risk factors. Deutsches Arzteblatt International. 2011 Nov;108(44):743–50. doi: 10.3238/arztebl.2011.0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Winker R, Lukas I, Perkmann T, Haslacher H, Ponocny E, Lehrner J, et al. Cognitive function in elderly marathon runners: Cross-sectional data from the marathon trial (APSOEM) Wiener Klinische Wochenschrift. 2010 Dec;122(23-24):704–16. doi: 10.1007/s00508-010-1485-z. [DOI] [PubMed] [Google Scholar]
- 103.Radak Z, Chung HY, Koltai E, Taylor AW, Goto S. Exercise, oxidative stress and hormesis. Ageing Research Reviews. 2008 Feb;7(1):34–42. doi: 10.1016/j.arr.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 104.Radak Z, Chung HY, Goto S. Exercise and hormesis: Oxidative stress-related adaptation for successful aging. Biogerontology. 2005;6(1):71–5. doi: 10.1007/s10522-004-7386-7. [DOI] [PubMed] [Google Scholar]
- 105.Baker LD, Bayer-Carter JL, Skinner J, Montine TJ, Cholerton BA, Callaghan M, et al. High-intensity physical activity modulates diet effects on cerebrospinal amyloid-beta levels in normal aging and mild cognitive impairment. J Alzheimers Dis. 2012;28(1):137–46. doi: 10.3233/JAD-2011-111076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proceedings of the National Academy of Sciences of the United States of America. 2001 Feb 16;98(2):676–82. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics. 1995 Dec 1;11(4):376–81. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 108.Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nature Genetics. 1998 Mar;18(2):159–63. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 109.Melov S, Coskun P, Wallace D. Mouse models of mitochondrial disease, oxidative stress, and senescence. Mutation Research. 1999;434(3):233–42. doi: 10.1016/s0921-8777(99)00031-2. [DOI] [PubMed] [Google Scholar]
- 110.Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, et al. Lifespan extension and rescue of spongiform encephalopathy in Superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. Journal of Neuroscience. 2001 Nov 1;21(21):8348–53. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: Neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. Journal of Neurochemistry. 2004 Mar;88(3):657–67. doi: 10.1046/j.1471-4159.2003.02195.x. [DOI] [PubMed] [Google Scholar]
- 112.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999 Jun 1;19(9):3414–22. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, et al. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Experimental Neurology. 2001 Feb;167(1):189–95. doi: 10.1006/exnr.2000.7525. [DOI] [PubMed] [Google Scholar]
- 114.Huang TT, Carlson EJ, Kozy HM, Mantha S, Goodman SI, Ursell PC, Epstein CJ. Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free Radical Biology and Medicine. 2001 Nov 1;31(9):1101–10. doi: 10.1016/s0891-5849(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 115.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun A, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96(3):846–51. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, et al. Life-long reduction in mnsod activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiological Genomics. 2003 Dec 16;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 117.Hu D, Cao P, Thiels E, Chu CT, Wu GY, Oury TD, Klann E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol Learn Mem. 2007 Apr;87(3):372–84. doi: 10.1016/j.nlm.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, et al. Overexpression of mn superoxide dismutase does not increase life span in mice. The Journals of Gerontology. 2009 Nov;64(11):1114–25. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bitner BR, Perez-Torres CJ, Hu L, Inoue T, Pautler RG. Improvements in a mouse model of Alzheimer’s disease through SOD2 overexpression are due to functional and not structural alterations. Magn Reson Insights. 2012 Apr 29;5:1–6. doi: 10.4137/MRI.S9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science. 1995 Jul 14;269(5221):230–4. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 121.Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Annals of the New York Academy of Sciences. 2008 Dec;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- 122.Weyer G, Babej-Dolle RM, Hadler D, Hofmann S, Herrmann WM. A controlled study of 2 doses of idebenone in the treatment of alzheimer’s disease. Neuropsychobiology. 1997;36(2):73–82. doi: 10.1159/000119366. [DOI] [PubMed] [Google Scholar]
- 123.Gutzmann H, Hadler D. Sustained efficacy and safety of idebenone in the treatment of Alzheimer’s disease: Update on a 2-year double-blind multicentre study. Journal of Neural Transmission Supplementum. 1998;54:301–10. doi: 10.1007/978-3-7091-7508-8_30. [DOI] [PubMed] [Google Scholar]
- 124.Thal LJ, Grundman M, Berg J, Ernstrom K, Margolin R, Pfeiffer E, et al. Idebenone treatment fails to slow cognitive decline in alzheimer’s disease. Neurology. 2003 Dec 9;61(11):1498–502. doi: 10.1212/01.wnl.0000096376.03678.c1. [DOI] [PubMed] [Google Scholar]
- 125.Praticò D. Evidence of oxidative stress in alzheimer’s disease brain and antioxidant therapy. Annals of the New York Academy of Sciences. 2008 Dec 8;1147(1):70–8. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 126.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med. 1997 Apr 24;336(17):1216–22. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 127.Lew MF. The evidence for disease modification in Parkinson’s disease. Int J Neurosci. 2011;121(Suppl 2):18–26. doi: 10.3109/00207454.2011.620194. [DOI] [PubMed] [Google Scholar]
- 128.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant mitoq as a disease-modifying therapy in Parkinson’s disease. Movement Disorders. 2010 Aug 15;25(11):1670–4. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 129.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, Breteler MM. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA : The Journal of the American Medical Association. 2002 Jul 26;287(24):3223–9. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 130.Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, et al. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA : The Journal of the American Medical Association. 2002 Jul 26;287(24):3230–7. doi: 10.1001/jama.287.24.3230. [DOI] [PubMed] [Google Scholar]
- 131.Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2012 Nov 14;11 doi: 10.1002/14651858.CD002854.pub3. CD002854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Blacker D. Neither vitamin E nor donepezil delays progression from amnestic mild cognitive impairment to Alzheimer’s disease in the long term. Evid Based Ment Health. 2006 Feb;9(1):20. doi: 10.1136/ebmh.9.1.20. [DOI] [PubMed] [Google Scholar]
- 133.Pautler, Bitner BR, Perez-Torres CJ, Hu L, Inoue T, Pautler Improvements in a mouse model of alzheimer’s disease through SOD2 overexpression are due to functional and not structural alterations. Magn Reson Insights. 2012 Apr;29(5):1–6. doi: 10.4137/MRI.S9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Misawa H, Nakata K, Matsuura J, Moriwaki Y, Kawashima K, Shimizu T, et al. Conditional knockout of mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiology of Disease. 2006 Jul;23(1):169–77. doi: 10.1016/j.nbd.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 135.Oh SS, Sullivan KA, Wilkinson JE, Backus C, Hayes JM, Sakowski SA, Feldman EL. Neurodegeneration and early lethality in superoxide dismutase 2-deficient mice: A comprehensive analysis of the central and peripheral nervous systems. Neuroscience. 2012 Jul 14;212:201–13. doi: 10.1016/j.neuroscience.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]