

Abstract
Alishewanella species are expected to have high adaptability to diverse environments because they are isolated from different natural habitats. To investigate how the evolutionary history of Alishewanella species is reflected in their genomes, we performed comparative genomic and transcriptomic analyses of A. jeotgali, A. aestuarii, and A. agri, which were isolated from fermented seafood, tidal flat sediment, and soil, respectively. Genomic islands with variable GC contents indicated that invasion of prophage and transposition events occurred in A. jeotgali and A. agri but not in A. aestuarii. Habitat differentiation of A. agri from a marine environment to a terrestrial environment was proposed because the species-specific genes of A. agri were similar to those of soil bacteria, whereas those of A. jeotgali and A. aestuarii were more closely related to marine bacteria. Comparative transcriptomic analysis with pectin as a sole carbon source revealed different transcriptional responses in Alishewanella species, especially in oxidative stress-, methylglyoxal detoxification-, membrane maintenance-, and protease/chaperone activity-related genes. Transcriptomic and experimental data demonstrated that A. agri had a higher pectin degradation rate and more resistance to oxidative stress under pectin-amended conditions than the other 2 Alishewanella species. However, expression patterns of genes in the pectin metabolic pathway and of glyoxylate bypass genes were similar among all 3 Alishewanella species. Our comparative genomic and transcriptomic data revealed that Alishewanella species have evolved through horizontal gene transfer and habitat differentiation and that pectin degradation pathways in Alishewanella species are highly conserved, although stress responses of each Alishewanella species differed under pectin culture conditions.
INTRODUCTION
The diversity of Alishewanella species has begun to be explored only recently, since the establishment of this genus in 2000 (1). There are only 5 Alishewanella species with validly published names (1–5), 36 isolates from various environments, and 16S rRNA gene sequences of 27 uncultured Alishewanella strains, according to NCBI taxonomy. Their isolation sources include fermented foods, tidal flat sediments, plant leaf and root surfaces, soils, deserts in cold climates, sludge, permafrost soils, freshwater biofilms, metal tailings, guts of beetle larvae, lakes, wastewater, and heavy metal-resistant communities. These diverse isolation sources imply that Alishewanella species have a broad range of niches and high adaptability. Additionally, a predominance of Alishewanella species was reported for a culture-dependent analysis of a bacterial community from a lake (6).
Phylogenetic analysis of 16S rRNA genes from Alishewanella and taxonomically neighboring species showed that Alishewanella species have evolved from an ancestor dwelling in the marine environment (5). Isolation from different environments and geographic locations may indicate that Alishewanella species moved from marine habitats to diverse environments. Pioneering a new habitat may require acquisition of novel genetic and physiological characteristics. Therefore, strains isolated from different sources could be expected to possess distinctive biological characteristics reflecting the evolutionary process.
Habitat can determine the preference of the horizontally transferred genes between bacterial populations, and it has been hypothesized that transferred genes could render high adaptability for local interactions and adaptations. The best and simplest example is the acquisition of novel plasmids, chromosomal genes, or mobile genomic islands (GIs) and the acquisition of pathogen or antibiotic resistance (7). Another example from nonpathogenic species comparative genomic and metagenomic studies of Prochlorococcus spp., which identified that genetic variability between phylogenetically distinct bacterial groups occurred in genomic islands, laterally transferred via phages and differentially expressed (8).
The aims of this study were to identify the evolutionary history recorded in Alishewanella genomes and to determine the effects of evolutionary changes on phenotypes or transcriptional profiles. To address these aims, we first performed comparative genomic analysis using 3 Alishewanella species (A. jeotgali, A. aestuarii, and A. agri, isolated from fermented seafood, tidal flat sediment, and soil, respectively), characterized their genomic contents, and identified core and species-specific genes. Because Bacillus species with high average nucleotide similarity and highly conserved synteny have been reported to show apparently different phenotypes, we compared the transcriptional profiles of these 3 species to determine whether there were differences in core genes. Comparative transcriptomic analysis was conducted with cells grown on pectin as a sole carbon source because these 3 Alishewanella species are all able to utilize pectin as a carbon source. Research on pectin metabolism has been described for only a small number of bacterial species, and transcriptomic analysis of pectin metabolism has not been previously reported.
MATERIALS AND METHODS
Genome data.
The genomes of A. jeotgali, A. aestuarii, and A. agri were previously sequenced by our group and are publicly available under GenBank accession numbers AHTH00000000, ALAB00000000, and AKKU00000000, respectively (9–11). The locus tags of A. jeotgali, A. aestuarii, and A. agri genomes are AJE, AEST, and AGRI with 5-digit numbers, respectively.
Bioinformatics analysis.
Genetic information was acquired from the NCBI GenBank database. Metabolic pathways were determined by using the KEGG pathway database. Multiple-genome alignment was conducted via Mauve (12) and a Web version of the Artemis Comparison Tools (ACT) (13). To determine species-specific genes, a reciprocal BLASTP search was performed by using protein-coding sequences at cutoff E values of >1e−10. To determine the expression levels of pectin metabolic genes, previously experimentally proven genes from Escherichia coli, Bacillus, and Erwinia spp. were used as reference sequences. Genomic islands were determined by GC variation in genomic loci using GC-Profile online software (http://tubic.tju.edu.cn/GC-Profile/). Expression of the intergenic regions was manually checked by using ACT (13).
Antibiotic resistance test.
Antibiotic resistance was tested for ampicillin, kanamycin, gentamicin, tetracycline, chloramphenicol, rifampin, and norfloxacin. The 3 Alishewanella species were grown in nutrient broth, and approximately 105 CFU/ml was inoculated into fresh nutrient broth containing 1, 2, 5, 10, 20, 30, 40, or 50 μg/ml of antibiotics. Ampicillin was also tested at 100 μg/ml. Antibiotic resistance was determined after incubation of the cells at their optimal growth temperature (37°C for A. jeotgali and A. aestuarii and 30°C for A. agri) for 5 days. Resistance was defined as an optical density at 600 nm (OD600) reading of >0.1.
Plate assay for pectin methylesterase activity.
Pectin methylesterase (PME) activity was determined as previously described, with some modifications (14). Cells (10 μl) were deposited onto the surface of 1% pectin-containing minimal salt basal (MSB) medium (15). After 3 days of incubation, the diameters of colonies were measured. The plates were overlaid with 5 ml of a mixture containing 0.5% pectin, 50 mM potassium phosphate (pH 6.0), 0.1% Triton X-100, and 0.5% agarose. The overlaid plates were incubated at 30°C for 1 day, stained with 0.2% ruthenium red for 10 min, and destained with distilled water for 10 min. PME activity was determined as the diameter of the halo zone surrounding the colony.
Transcriptomic analysis via RNAseq.
For reference total RNA, A. jeotgali was grown in a batch culture with 10 mM succinate-containing MSB medium under shaking conditions (220 rpm). Total RNA of A. jeotgali cells grown on succinate was isolated from mid-exponential-phase cells (OD600 = 0.4). The amount of protein was measured in succinate-grown cells when the OD600 reached 0.4. The protein concentration was 553.3 ± 31.2 μg/ml, which was close to that of mid-exponential-phase cells growing on pectin. A. jeotgali, A. aestuarii, and A. agri were grown on apple pectin to the mid-exponential phase (protein concentration of 600 μg/ml, determined by a Bradford assay [16]). The growth temperatures were 37°C for A. jeotgali and A. aestuarii and 30°C for A. agri according to previous reports describing their optimal temperatures. Total RNA was isolated by using an RNeasy minikit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Total RNA (10 μg) from each sample was used as starting material to prepare sequencing libraries with TruSeq RNA sample preparation kits (Illumina, USA) according to the manufacturer's instructions. This kit contained rRNA reduction procedures using biotinylated probes. One lane per sample was used for sequencing with the Illumina Genome Analyzer IIx system to generate nondirectional, single-ended, 36-bp reads. Quality-filtered reads were mapped to the Alishewanella genomes as reference sequences by using CLC Genomics Workbench 4.0 (CLC Bio). The relative transcript abundance was computed by counting the reads per kilobase of exon model per million mapped sequence reads (RPKM) (17). RPKM is defined as the total matched reads/(mapped reads in millions × gene length in kilobases).
Measurement of cellular oxidation.
Intracellular superoxide anion generation was determined by using dihydrorhodamine (DHR) 123 (Sigma, USA) (18). The tested cells were cultivated with 10 mM succinate or 1% (wt/vol) apple pectin (the same conditions for the RNAseq analysis) in MSB medium at their optimal growth temperature. For measurement of oxidative stress in H2O2- and paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride)-treated cells, exponentially growing cells were treated with 0.5 or 1.0 mM H2O2 and paraquat for 10 min in MSB medium containing 10 mM succinate. Cells growing exponentially were washed twice and resuspended with phosphate-buffered saline (PBS). Cells were then treated with DHR 123 (2.5 μg/ml) and were incubated for 1 h under dark conditions. DHR 123-treated cells were washed and resuspended twice with PBS. The intracellular superoxide anion-mediated oxidation of DHR 123 was assayed via FACSverse flow cytometry (BD Biosciences, USA). The samples were analyzed by using a fluorescein isothiocyanate (FITC) argon ion laser for excitation. Fluorescence intensity was determined and analyzed by measuring 10,000 cell counts. BD FACSuite software was used for data analysis.
Statistical analysis.
Protein concentrations and PME activity were measured from triplicate experiments and were analyzed statistically by using analysis of variance (ANOVA) and t tests in Microsoft Excel.
Nucleotide sequence accession number.
RNAseq data were deposited in the Gene Expression Omnibus database under accession number GSE45511.
RESULTS AND DISCUSSION
General features of Alishewanella genomes.
The genome sequences of 3 Alishewanella species were previously reported by our laboratory (9–11). However, intensive analyses had not yet been conducted. A. jeotgali had the largest genome (3.8 Mb) among the 3 Alishewanella species. GC contents of the 3 genomes were all similar (∼51%). While rRNA genes are usually closely associated in terms of synteny, sometimes with tRNA, we could not determine the number of rRNAs as operons because rRNA genes were located at the ends of contig sequences. Therefore, only the copy numbers of 5S, 16S, and 23S rRNA genes are presented in Table 1. Genome data of A. aestuarii and A. agri did not contain 5S rRNA gene sequences because they were probably present in gapped genomic regions. COG analysis of the 3 genomes demonstrated that proteins related to amino acid transport and metabolism (COG abbreviation E) were the most abundant in the 3 Alishewanella genomes (Fig. 1). A previous report of the A. agri genome presumed that contig027 to contig029 could be plasmid sequences according to their sizes and paired-end sequences (11). However, more detailed analyses via reciprocal BLAST searches indicated that homologs of contig027 to contig029 were present in both the A. jeotgali and A. aestuarii genomes from AJE_03786 to AJE_04116 (73 kb) and AEST_28220 to AEST_28880, respectively. Moreover, no plasmid replication-related proteins were identified from A. agri contig027 to contig029. The extremely high level of coverage of genome sequences and our homology searches indicated the absence of a plasmid in A. aestuarii. The numbers of protein-coding sequences were 3,669, 3,380, and 3,223 in the A. jeotgali, A. aestuarii, and A. agri genomes, respectively. Functional predictions and COG analyses were relatively well characterized in A. aestuarii. The DNA coding density and average gene length were the highest in the A. agri genome. Average nucleotide identities were calculated to be 93.9%, 85.7%, and 85.4% for A. jeotgali versus A. aestuarii, A. aestuarii versus A. agri, and A. agri versus A. jeotgali, respectively.
Table 1.
General features of Alishewanella species genomes
| Parameter | Value for Alishewanella species |
||
|---|---|---|---|
| A. jeotgali | A. aestuarii | A. agri | |
| Isolation source | Fermented food | Tidal flat sediment | Landfill soil |
| Genome size (bp) | 3,844,563 | 3,588,054 | 3,491,709 |
| GC content (%) | 50.66 | 50.98 | 50.60 |
| Plasmid detected | None | None | None |
| No. of tRNA genes | 64 | 70 | 68 |
| No. of rRNA genes | 6 | 4 | 3 |
| 5S | 1 | 0b | 0b |
| 16S | 1 | 2 | 1 |
| 23S | 4 | 2 | 2 |
| No. of protein-coding sequences | 3,669 | 3,380 | 3223 |
| No. (%) of protein-coding sequences with predicted function | 1,728 (47.1) | 2,305 (68.2) | 1,625 (50.4) |
| No. (%) of protein-coding sequences with assigned COGa | 2,227 (60.7) | 2,370 (70.1) | 2,349 (72.9) |
| DNA coding density (%) | 88.9 | 90.6 | 91.6 |
| Average gene length (bp) | 931.6 | 957.4 | 1,008.3 |
The COG category “function unknown (S)” was not included.
The number of rRNA genes was counted from different contigs. Additional rRNA genes can be present in gapped sequences.
Fig 1.
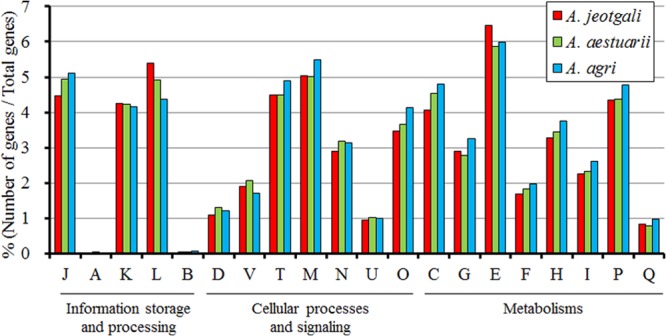
COG analysis of A. jeotgali, A. aestuarii, and A. agri genomes. COG abbreviations: J, translation, ribosomal structure, and biogenesis; A, RNA processing and modification; K, transcription; L, replication, recombination, and repair; B, chromatin structure and dynamics; D, cell cycle control, cell division, and chromosome partitioning; V, defense mechanisms; T, signal transduction mechanisms; M, cell wall/membrane/envelope biogenesis; N, cell motility; U, intracellular trafficking, secretion, and vesicular transport; O, posttranslational modification, protein turnover, and chaperones; C, energy production and conversion; G, carbohydrate transport and metabolism; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism.
Genomic islands may have imported potentially different functions or metabolic pathways.
Seven and four genomic islands (GIs) were determined by GC content deviations in A. jeotgali and A. agri, while no significant variation in GC content was found in A. aestuarii (Table 2; see also Fig. S1 in the supplemental material). The total lengths of GIs in A. jeotgali and A. agri were 204.8 and 136.3 kb, respectively, and the greatest GC content deviations were found in GI 2 (36.3%) and GI 10 (41.9%) from A. jeotgali and A. agri, respectively, compared to the average GC content (∼51%) (Tables 1 and 2). Functions of GIs were predicted based on the gene contents (Table 2). GI 1 was thought to be related to a restriction-modification system due to the presence of exonuclease (AJE_00370, AJE_00385, and AJE_00390), endonuclease (AJE_00400 and AJE_00510), a type I site-specific restriction-modification system (AJE_00425, AJE_00435, and AJE_00440 for R, M, and S subunits, respectively), and ATPase (AJE_00375 and AJE_00465). GI 3 contained many genes involved in flagellum biosynthesis and regulation. A similar gene organization was found for Pseudoalteromonas tunicata D2, Idiomarina baltica OS14, Pseudoalteromonas haloplanktis TAC125, Vibrio splendidus 12B01, the Alteromonas macleodii “deep ecotype,” and Shewanella sp. strain PV-4. Interestingly, a portion of gene contents from GIs 3, 5, and 11 was involved in flagellum biosynthesis, metal transport, and sugar metabolism. Pál et al. investigated the evolution of metabolic networks of E. coli over the past 100 million years and concluded that most changes were due to horizontal gene transfer and that the driving force of gene evolution was changing environments (19). In this context, horizontal gene transfer may have conferred important physiological features with relevance to the environmental conditions and contributed to speciation from a common ancestor of Alishewanella species. The genomic loci of 20 copies of transposases in the A. aestuarii genome were not associated with GC content variations throughout the A. aestuarii genome sequences. Because horizontally transferred genes do not necessarily have different GC contents from that of the rest of genome, analyses that are more detailed may be required for A. aestuarii (20).
Table 2.
Genomic islands determined by GC content variations in the A. jeotgali and A. agri genomesa
| GI | No. of contig(s) | Start position | Stop position | Size (bp) | No. of genes | GC content (%) | No. of hypothetical proteins | No. of transposase integrases | Predicted function(s) |
|---|---|---|---|---|---|---|---|---|---|
| Alishewanella jeotgali | |||||||||
| 1 | 2 | 13959 | 76263 | 62,305 | 48 | 44.6 | 22 | 4 | DNA restriction-modification system |
| 2 | 3 | 996 | 7581 | 6,586 | 3 | 36.3 | 1 | 1 | Not determinedb |
| 3 | 5 | 418566 | 467914 | 49,349 | 50 | 44.7 | 12 | 0 | Flagellum biosynthesis |
| 4 | 35–36 | 22575 | 12069 | 23,960 | 14 | 42.3 | 9 | 0 | Signal transduction, DNA manipulation |
| 5 | 36 | 12070 | 35419 | 23,350 | 26 | 54.7 | 2 | 2 | Metal transporter |
| 6 | 36–37 | 35420 | 16867 | 22,675 | 19 | 46.9 | 5 | 3 | Antibiotics resistance |
| 7 | 39–40 | 131943 | 17755 | 36,580 | 36 | 41 | 20 | 4 | DNA restriction-modification system |
| Alishewanella agri | |||||||||
| 8 | 1 | 142737 | 184463 | 41,727 | 32 | 43.1 | 7 | 1 | Cell wall synthesis |
| 9 | 1 | 377622 | 397599 | 19,978 | 29 | 45.1 | 18 | 0 | Mixed features |
| 10 | 3 | 19166 | 45148 | 25,983 | 21 | 41.9 | 6 | 3 | Prophage, metal transport, oxidative stress defense |
| 11 | 11 | 18059 | 66686 | 48,592 | 43 | 45 | 8 | 1 | Sugar metabolism |
GIs were not identified in the A. aestuarii genome.
The function of GI 3 was not predicted because there were not enough genes.
Gene contents in the low-GC-content regions implied the occurrence of horizontal gene transfer via prophage and transposases. The major features of typical phage islands were the presence of phage integrase, which performs integration of the phage elements, phage transcriptional regulators, helicase activity-possessing primase, and genes related to genome packaging, followed by many genes with unknown functions (21, 22). In accordance with previous reports, some of the low-GC-content genomic regions were associated with phage-related sequences. In A. jeotgali, AJE_12214, AJE_12313, and AJE_12348, located at the end of a low-GC-content region (bp 2673570 to 2710150; 36.6 kb), were phage integrase followed by a putative prophage repressor (AJE_12273) and many hypothetical proteins. In A. agri, AGRI_02408 and AGRI_02413, located at the end of a low-GC-content region (bp 487577 to 513560; 30.0 kb), were phage integrase family proteins followed by RNA-directed DNA polymerase (reverse transcriptase [AGRI_02393]).
Transposition of genetic materials also contributed to GC-content-variable regions in the Alishewanella genomes. These regions contained many copies of transposase genes and transposition-related sequences, such as the res subunit (AJE_10438) and cointegrate resolution protein T (AGRI_02333). All of the low-GC-content regions of the A. agri genome were associated with transposases. Transposition events produced pseudogenes via overlapping of a hypothetical protein AJE_01591 over tRNA-serine (bp 369529 to 370029) in A. jeotgali and hypothetical proteins AGRI_00850 and AGRI_01710 over tRNA-alanine (bp 184489 to 184653) and tRNA-serine (bp 378416 to 378838), respectively, in A. agri. A study of the distribution of pathogenicity islands (PIs) containing virulence factors such as adhesins, toxins, invasins, protein secretion systems, iron uptake systems, and others demonstrated that the GC content of PIs was often different from the average value of the chromosome and that their dispersion may be attributed to horizontal gene transfer in bacterial genomes, as evidenced by the presence of integrase determinants and other mobility loci (23). In some cases, tRNA genes (AJE_t00892 [tRNA-Met], AJE_t04154 [tRNA-Arg], AJE_t10470 [tRNA-Arg], and AJE_t12268 [tRNA-Leu]) were flanked by the ends of low-GC-content regions.
Prominent gene rearrangement in A. jeotgali shown in multiple-genome alignments.
Multiple-genome alignments were performed with concatenated contig sequences because the order of all contigs was confirmed by paired-end sequencing. Figure S2 in the supplemental material shows the alignment results using ACT (http://www.webact.org/). The genomes of A. aestuarii and A. agri seemed to be largely conserved, lacking severe gene rearrangements. However, homologous genes of A. aestuarii were found throughout scattered regions of the A. jeotgali genome, implying more intensive gene rearrangements than those observed in A. aestuarii and A. agri. Because gene rearrangement could occur in the course of genome evolution, A. jeotgali may have encountered a certain environment requiring flexible adaptation (24).
Species-specific genes were horizontally transferred from a bacterial population in an overlapping habitat.
Reciprocal BLASTP searches revealed 373, 386, and 238 coding sequences of low amino acid identity (E values of > 1e−10) to the other Alishewanella species A. jeotgali, A. aestuarii, and A. agri, respectively (see Fig. S3 in the supplemental material). We designated these proteins species-specific gene products and investigated their predicted functions to determine whether their presence provided any genetic or physiological characteristics for adapting to their different habits. Hypothetical proteins accounted for 36.2%, 85.2%, and 62.2% (135, 329, and 148 genes, respectively) of species-specific genes in A. jeotgali, A. aestuarii, and A. agri, respectively. The presence of multiple copies of transposase may mediate the introduction of new genes from other species or environmental DNA (9, 5, and 8 transposases in A. jeotgali, A. aestuarii, and A. agri, respectively). Another common feature of species-specific genes was the presence of phage-related sequences such as integrase, phage tape measure protein, coat protein, head subunit protein, and phage transcriptional regulator (20, 2, and 7 phage-related sequences in A. jeotgali, A. aestuarii, and A. agri, respectively). Prophage is known as a major contributor to microbial diversification (25) via genomic rearrangement, altering the fitness of the bacteria to survive (26), and transferring virulence factors (27). Species-specific genes, other than hypothetical protein, transposase, and prophage, were categorized with similar functions and are listed in Tables S1, S2, and S3 in the supplemental material.
We reasoned that species-specific genes were acquired from exogenous sources such as other bacterial populations in the vicinity. To confirm this hypothesis, we investigated the closest sequences of species-specific genes from GenBank based on a BLASTP search to identify whether there were more similar sequences from taxonomic groups other than the Alishewanella genus. All the species-specific genes found their closest matches in another bacterial group, as expected. E values obtained from the BLASTP results showed that species-specific genes from the 3 Alishewanella species had E values that were all <10−10 and that some genes coded for identical amino acid sequences, as shown in sequence identity and coverage (see Tables S1, S2, and S3 in the supplemental material). Next, we identified the bacterial species from which the closest sequence of species-specific genes originated. A. jeotgali- and A. aestuarii-specific genes seemed to originate from Alteromonas, Pseudoalteromonas, Rheinheimera, Vibrio, Shewanella, and Idiomarina species. Their previously described isolation sources were all aquatic environments such as lake and seawater. However, many A. agri-specific genes seemed to originate from terrestrial environment dwellers such as Pseudomonas, Geobacillus, Rhizobium, and Escherichia spp. This result implied that habitat differentiation may have driven horizontal gene transfer from soil bacteria to A. agri and that accumulation of exogenously acquired genes may have resulted from the genomic evolution of A. agri. We also examined whether A. jeotgali and A. aestuarii harbored the same predicted function as A. agri-specific genes. From this analysis, we categorized A. agri-specific genes into 2 groups. The first group contained genes encoding proteins with annotated functions found only in A. agri; for example, this group contained phytanoyl coenzyme A (CoA) dioxygenase (AGRI_00310) and an AbrB family transcriptional regulator (AGRI_01840), functions which A. jeotgali and A. aestuarii did not possess. The second group contained A. agri-specific genes having low sequence identities with those of A. jeotgali and A. aestuarii encoding proteins with the same function. For example, 3 Alishewanella species have multiple copies of glycosyltransferase; however, A. agri-specific glycosyltransferases (AGRI_00730, AGRI_00770, and AGRI_00785) are distinguished from other glycosyltransferases found in A. jeotgali, A. aestuarii, and A. agri by low sequence identity. BLASTP results and comparisons of the genes encoding proteins with the same predicted function suggested that A. agri acquired new or additional genes from other bacteria and that some portion of these horizontally transferred genes was from soil bacteria, implying that its habitat was moved from a marine environment to a soil environment.
Oxidative stress.
Many oxidative stress-related genes were present in Alishewanella species (see Table S4 in the supplemental material). Manganese superoxide dismutase and iron superoxide dismutase, which both catalyze superoxide radicals into hydrogen peroxide and oxygen, were identified in all 3 Alishewanella species; however, the presence of sodC, the Cu-Zn superoxide dismutase, was not predicted. Catalases that decompose hydrogen peroxide to water and oxygen were present, with 3 gene copies in A. jeotgali and A. agri and 2 gene copies in A. aestuarii, which lacked homologs for AJE_10739 and AGRI_12426. Glutathione reductase, glutathione peroxidase, glutaredoxin, and glutaredoxin-related protein were present in Alishewanella genomes.
Motility.
A previous study reported that A. jeotgali and A. aestuarii have motility and confirmed the presence of their single flagellum; A. agri did not have this phenotype (2, 3). We also confirmed their motility on agar plates (data not shown). Interestingly, RAST and PGAAP predicted 34.9-kb and 19.0-kb genomic loci, from AJE_03261 to AJE_03431 (35 genes) and from AJE_03546 to AJE_03646 (21 genes), related to flagellum biosynthesis and regulation in A. jeotgali, with homologs in A. aestuarii and A. agri having identical gene arrangements. Two copies of the flagellar motor protein MotA (AJE_03291 and AJE_14615) and 4 copies of MotB (AJE_00485, AJE_00495, AJE_03286, and AJE_14620) were identified. However, only AJE_03291 and AJE_03286 were most likely to be associated with other flagellar-related genes in the vicinity. An aspartic acid residue (Asp32 in E. coli), which was suggested to be essential for flagellar rotation via proton movement through the motor (28), was conserved in MotB proteins from the 3 Alishewanella species. Flagellum-specific chaperones required for exportation of structural components in a timely fashion were also located in other neighboring flagellar-related genes. The absence of motility in A. agri, despite the presence of all genetic components, remains to be investigated.
Antibiotic resistance in Alishewanella species.
Experimental data performed in this study demonstrated that the 3 Alishewanella species could grow at ampicillin, kanamycin, gentamicin, tetracycline, chloramphenicol, rifampin, and norfloxacin concentrations of 100, 10, 2, 20, 5, 50, and 50 μg/ml, respectively. Interestingly, there were no differences in antibiotic resistance among the species. Ampicillin is a beta-lactam antibiotic that inhibits the synthesis of the cell wall. Beta-lactamase is responsible for the inactivation of beta-lactam antibiotics via cleavage of the lactam ring structure. A. jeotgali harbored 6 beta-lactamases, while A. aestuarii and A. agri had 5 beta-lactamases (see Table S5 in the supplemental material). AmpG and AmpE permeases, which are required for the expression of beta-lactamase in P. aeruginosa PAO1 and E. coli, were also identified in Alishewanella genomes (see Table S5 in the supplemental material) (29, 30). Putative TetR family transcriptional regulators of the multidrug efflux pump (AJE_11219, AEST_24320, and AGRI_12981) were located upstream of one of the beta-lactamase genes in each species (AJE_11214, AEST_24330, and AGRI_12976, respectively). The macrolide-specific efflux pump and multidrug efflux pumps could also have important roles in antibiotic resistance.
Carbon metabolism.
Carbon source utilization by species was tested in MSB medium with 21 compounds (see Table S6 in the supplemental material). Interestingly, Alishewanella species could not utilize any aromatic compounds or hydrocarbons such as gentisate, benzoate, salicylate, naphthalene, toluene, paraffin, diesel, and hexadecane. Genes for glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway, and the Entner-Doudoroff pathway were conserved in the 3 Alishewanella species (see Table S7 in the supplemental material). Naphthalene monooxygenase, the first enzyme in the naphthalene degradation pathway, was absent in the 3 Alishewanella species. Salicylate hydroxylase, which converts salicylate into catechol, was not identified in any of the 3 Alishewanella species. Gentisate 1,2-dioxygenase, which catalyzes the conversion of gentisate (2,5-dihydroxybenzoate) into maleylpyruvate, was not identified in any of the 3 Alishewanella species. Alkane 1-monooxygenase, which oxidizes alkane chains and produces alcohol for further degradation, was not identified. Thus, these data suggested that Alishewanella species seem to be unable to degrade naphthalene, salicylate, gentisate, and aliphatic hydrocarbons because of the absence of key enzymes required for their metabolism. The β-ketoadipate pathway is responsible for downstream pathways of many aromatic compounds and consists of catechol and protocatechuate branches. All genes for the catechol branch of the β-ketoadipate pathway were present in A. jeotgali and A. agri but not in A. aestuarii (see Table S8 in the supplemental material). The protocatechuate branch of the β-ketoadipate pathway was absent in all 3 Alishewanella species. The absence of key enzymes for the degradation of aliphatic and aromatic hydrocarbons implied the preference for carbohydrates rather than hydrocarbon, in terms of carbon source utilization.
Pectin metabolism.
Pectin is a complex polysaccharide with a 1,4-linked α-d-galacturonic acid backbone. Pectin is a major component of the plant cell wall; hence, it is an abundant plant biomass in the natural environment (31), including coastal ocean and soil (32). Considering the preference for carbohydrates rather than hydrocarbons in carbon source utilization, the ability to utilize pectin could be quite an important feature in Alishewanella species. We provided apple pectin or citrus pectin as a sole carbon source and measured the growth of each species based on the increase in the total protein concentration rather than measuring the optical density because 1% pectin in MSB medium has its own turbidity. Growth curves for the 3 Alishewanella species showed that the concentration of protein increased faster and reached a higher level in 108 h when apple pectin was utilized (Fig. 2A). A. agri reached a higher protein concentration than A. jeotgali and A. aestuarii, regardless of the pectin source. The superior growth of A. agri on pectin was consistent with PME activity measured by ruthenium red staining (Fig. 2B). Interestingly, only A. agri showed PME activity on both apple and citrus pectin, whereas the PME activities of A. jeotgali and A. aestuarii were measurable only on citrus pectin-containing medium. The greater PME activity of A. agri could be one reason for its better growth on pectin, since PME deesterifies pectin to produce substrates for subsequent pectinolytic enzymes such as polygalacturonase.
Fig 2.

Characteristics of pectin utilization in Alishewanella species. (A) Growth curves of Alishewanella species on apple pectin (solid lines) and citrus pectin (dotted lines). Growth was determined by measuring the total amount of protein. (B) Pectin methylesterase (PME) activity was measured as the diameter of the halo zone after ruthenium red staining. A. jeotgali and A. aestuarii did not show PME activity on citrus pectin. (C) Gene arrangement of pectin catabolic genes. The functions of gene locus tags are summarized in Table S9 in the supplemental material. The asterisks in panel A indicate statistically significant differences, as measured by ANOVA. Bar graphs with different letters in panel B are statistically different by a t test (P < 0.05). TR, transcriptional regulator.
Pectin metabolic genes were predicted via genome annotation and BLASTP comparison using previously described genes from E. coli, Pectobacterium atrosepticum, B. subtilis, and Erwinia chrysanthemi. The arrangement of pectin metabolism genes is shown in Table S9 in the supplemental material and in Fig. 2C. Pectin methylesterase (pmeA) was located at the ends of pectin metabolic loci. Deesterified pectin was degraded by pectate lyase (PelAB) and produced d-galacturonate and 4-deoxy-5-keto-l-threo-hexuronate at a ratio of 4:1. The transport system for 4-deoxy-5-keto-l-threo-hexuronate was identified experimentally in E. chrysanthemi and annotated kdgT (33). However, a homolog of kdgT was not found in the Alishewanella genomes. Downstream catabolic pathways are mediated by 4-deoxy-5-keto-uronate isomerase (kduI) and 2-dehydro-3-deoxy-d-gluconate-5-dehydrogenase (kduD), which produce 3-deoxy-d-glycero-hex-2,5-diulosonate and 2-keto-3-deoxy-d-gluconate (KDG), respectively. The transport system for d-galacturonic acid was experimentally proven and termed exuT (34); however, a homolog of exuT was not identified in the Alishewanella genomes. Instead, pectin metabolism loci of Alishewanella contained the tripartite ATP-independent transport system (TRAP) transporter family. Similar arrangements of TRAP transporter genes and pectin metabolic genes were found in Pasteurellales, Vibrionales, Alteromonadales, Oceanospirillales, and Photorhabdus luminescens TTO1 from the Enterobacteriales (35). d-Galacturonic acid is further converted to uronate isomerase (uxaC), d-altronate hydrolase (uxaA), and d-altronate oxidoreductase (uxaB) and produces 2-keto-3-deoxy-d-gluconate (KDG). 2-Keto-3-deoxy-d-gluconate kinase (kdgK) converts KDG into 2-keto-3-deoxygluconate-6-phosphate (KDPG), and 2-keto-3-deoxy-6-phosphogluconate aldolase (kdgA) converts KDPG into d-glyceraldehyde-3-phosphate and pyruvate. Alishewanella species do not have a catabolic pathway for d-glucuronate due to the absence of d-mannoate hydrolase (uxuA) and d-mannoate oxidoreductase (uxuB). Hexuronate metabolism in E. coli is regulated by 2 GntR family transcriptional regulators, uxuR and exuR. uxuR is known to control d-glucuronate metabolism, while exuR controls genes involved in the metabolism of d-galacturonate and d-glucuronate (35). Two transcriptional regulators were found in the Alishewanella genomes (LacI and IclR family), located upstream of pelB. The IclR family transcriptional regulator was a homolog of KdgR, the transcriptional regulator of KDG metabolism in E. coli. KdgR is known to regulate pectin metabolism in Enterobacteriales (35). Based on our comparative genomics data, we constructed the pectin metabolic pathway in Alishewanella species. Briefly, pectin was deesterified by pectin methylesterase and produced pectate. Pectate lyase depolymerized pectate and produced d-galacturonate and 5-keto-4-deoxy-hexuronate. These 2 products were metabolized by uxaCAB and kduID, respectively, and resulted in the formation of a common metabolite, KDG. kdgK and kdgA were responsible for the downstream pathway, and the 2 final products, d-glyceraldehyde-3-phosphate and pyruvate, were substrates for glycolysis and the TCA cycle.
Transcriptomic analysis of Alishewanella species utilizing pectin as a sole carbon source.
Next, we performed transcriptomic analysis of the 3 Alishewanella species grown on apple pectin as a sole carbon source because (i) pectin metabolism was a conserved characteristic in the 3 Alishewanella species, despite the evidence for genetic recombination, e.g., horizontal gene transfer from other bacterial populations in their habitat, incorporation of prophage, and loss of the β-ketoadipate pathway in A. aestuarii; (ii) pectin could be an important carbon source for carbohydrate-preferring Alishewanella in the natural environment; and (iii) pectin metabolism (or d-galacturonate metabolism) has been investigated in only a limited number of bacterial species, including E. coli, B. subtilis, and E. chrysanthemi. Transcriptomic studies related to pectin metabolism have not yet been reported.
Transcriptomic analysis was performed via the RNAseq technique. The expression levels of genes from A. jeotgali grown on succinate (here called AJE_Succinate) were used as references, and RNAseq results from A. jeotgali grown on apple pectin (here called AJE_Pectin) were compared. RPKM values of A. aestuarii and A. agri grown on apple pectin (here called AEST_Pectin and AGRI_Pectin, respectively) were compared to those of AJE_Pectin.
The average RPKM values of AJE_Succinate, AJE_Pectin, AEST_Pectin, and AGRI_Pectin were 350.6, 357.0, 322.3, and 327.9, respectively (see Fig. S4 in the supplemental material). Median RPKM values were 250.6, 214.7, 180.7, and 236.9, respectively. Genes with RPKM values of >600 represented 1.7%, 4.6%, 5.2%, and 8.4% of the total coding sequences of AJE_Succinate, AJE_Pectin, AEST_Pectin, and AGRI_Pectin, respectively. RPKM values of the samples are represented as a color gradient ranging from 0 to 600 in Fig. 4 and 5. Figure S4 in the supplemental material shows that most genes from A. jeotgali and A. aestuarii were expressed with RPKM values of 200 to 300, while genes from A. agri were distributed across a wider range (RPKM values of 200 to 500).
Fig 4.

Expression of glycolysis, TCA cycle, pentose phosphate pathway, and Entner-Doudoroff pathway genes in succinate-grown A. jeotgali and pectin-grown A. jeotgali, A. aestuarii, and A. agri cells. Gene expression levels are represented by a color gradient based on the RPKM value. Glucose-6P, glucose-6-phosphate; FAD, flavin adenine dinucleotide; FADH2, reduced flavin adenine dinucleotide.
Fig 5.
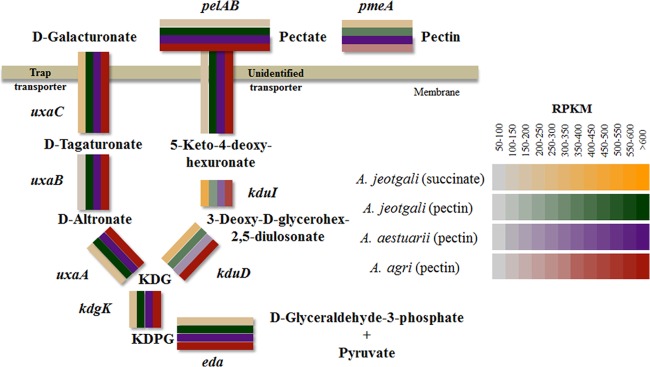
Expression levels of pectin metabolism pathway genes in succinate-grown A. jeotgali and pectin-grown A. jeotgali, A. aestuarii, and A. agri cells. Gene expression levels are represented by a color gradient based on the RPKM value.
The RPKM values of AJE_Succinate and AJE_Pectin were represented as an xy plot (Fig. 3). Several genes seemed to be expressed highly under only 1 condition, either pectin or succinate. Interestingly, many transposases and reverse transcriptases were highly expressed only under succinate conditions. Several functional genes were highly expressed only under pectin conditions. These genes included diacylglycerol kinase (dgkA [AJE_06506]), peptidase M28 (yfbL [AJE_07241]), 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (folK [AJE_13045]), general secretory pathway protein E (gspE [AJE_09889]), 6-phosphogluconate dehydrogenase (gnd [AJE_16359]), Na+/H+ antiporter (nhaC [AJE_11224]), and signal transduction histidine kinase (baeS [AJE_12378]). To understand cellular processes in AJE_Pectin, fold changes in RPKM values of AJE_Pectin/AJE_Succinate were represented by COG categories. Figure S5 in the supplemental material shows that most genes with predicted COG categories were upregulated in AJE_Pectin. Therefore, we classified upregulated genes (>2-fold) by their functions and associated metabolic pathways or biological processes to reconstruct important cellular processes of AJE_Pectin at the transcriptional level. The following subsections provide descriptions of selected features from our transcriptomic results. Table S10 in the supplemental material summarizes the upregulated genes (>2-fold) with functional categories and RPKM values of homologs from A. aestuarii and A. agri.
Fig 3.
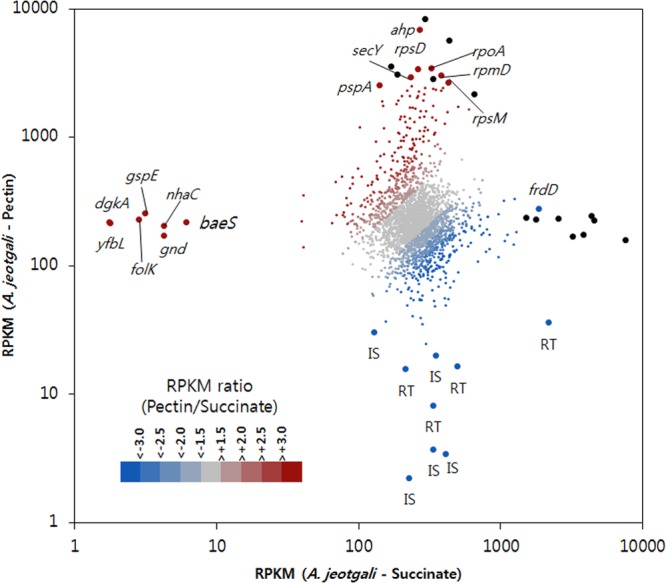
xy plot of RPKM values from A. jeotgali cells grown on pectin and succinate. A dot indicates a gene, and its x and y coordinates are the RPKM from succinate- and pectin-grown A. jeotgali cells. Fold changes (RPKM ratio of pectin/succinate) are represented with a color gradient. Gene symbols are marked next to the selected dot. Black dots indicate hypothetical proteins or proteins annotated with only general function. IS, transposase; RT, reverse transcriptase.
Glyoxylate bypass.
We investigated the expression of glycolysis, TCA cycle, pentose phosphate pathway, and Entner-Doudoroff pathway genes because succinate and downstream metabolites of pectin are substrates for these general metabolic pathways (Fig. 4). While most genes in AJE_Pectin, AEST_Pectin, and AGRI_Pectin were not expressed at levels significantly different from those in AJE_Succinate, isocitrate lyase (aceA [AJE_04350]) and malate synthase (aceB [AJE_04360]), involved in glyoxylate bypass, were the most significantly upregulated genes. Homologs of aceA and aceB from A. aestuarii and A. agri also exhibited very high RPKM values. The Leloir pathway, which converts galactose into glucose, was not responsible for the upregulation of glyoxylate bypass because Alishewanella genomes do not contain homologs of galactokinase and galactose-1-phosphate uridylyltransferase. Glyoxylate bypass is considered necessary for efficient utilization of carbon sources because it bypasses the CO2-generating steps of the TCA cycle and allows cells to synthesize the building blocks of cellular components when grown on a simple carbon source such as acetate. Generally, glyoxylate bypass is upregulated when acetyl-CoA is a direct product of a metabolic pathway (36). However, glyoxylate bypass was induced by oxidative stress in several bacterial species. For example, E. coli experiencing superoxide stress upregulated glyoxylate bypass (37). Pseudomonas sp. strain TLC6-6.5-4 also upregulated its glyoxylate bypass pathway when exposed to copper stress, which caused oxidative stress (38). Proteomic and metabolomic data from Bacillus licheniformis showed the induction of glyoxylate bypass under conditions of peroxide stress (39). Therefore, we speculated that oxidative stress induces glyoxylate bypass in A. jeotgali grown on pectin, because A. jeotgali and A. aestuarii were exposed to oxidative stress when they were grown on pectin (as discussed below).
After confirming that oxidative stress was generated by hydrogen peroxide and paraquat using flow cytometry (see Fig. S6 in the supplemental material), we measured the expression levels of genes associated with the TCA cycle and glyoxylate bypass by using identical conditions to those shown in Fig. S6 in the supplemental material. Quantitative reverse transcription-PCR (qRT-PCR) results showed the relative upregulation of isocitrate lyase (aceA) and malate synthase (aceB) expression, while most other genes were downregulated (see Fig. S7 in the supplemental material). Therefore, we suggest that oxidative stress induced glyoxylate bypass in Alishewanella species and that oxidative stress generated during pectin utilization is responsible for highly expressed glyoxylate bypass genes. Changes in the flux of metabolites in central carbon metabolism under conditions of oxidative stress (37) or malate accumulation, producing more NADPH to cope with oxidative stress, could partially explain the upregulation of glyoxylate bypass under oxidative stress conditions (40).
Pectin metabolism.
Based on the reconstructed pectin metabolic pathway, the expression of pectin metabolism-related genes was investigated. As shown in Fig. 5, pectin metabolic genes were highly expressed (RPKM values of >600) in pectin-utilizing cells, while succinate-utilizing A. jeotgali cells showed relatively lower expression levels of these genes. Their expression levels were higher than the top 10% of the total coding sequences. This result was expected because we supplied apple pectin as a sole carbon source. It is worthwhile to mention that the TRAP transporter system located in the pectin metabolic loci was also highly expressed. Because of the involvement of the TRAP transporter in d-galacturonic acid metabolism in several Proteobacteria, as shown by a comparative genomic study (35), we determined the expression level of the TRAP transporter in 3 Alishewanella species during the utilization of polygalacturonic acid using qRT-PCR. Figure S8 in the supplemental material shows the upregulation of the TRAP transporter in cells grown on polygalacturonic acid, suggesting their involvement in pectin metabolism. 2-Deoxy-d-gluconate 3-dehydrogenase (kduD) and 5-keto-4-deoxyuronate isomerase (kduI) did not show significantly different expression levels. Unsaturated rhamnogalacturonyl hydrolase (rhiN [AJE_04916]) exhibited a 3.4-fold increase in transcription. Unsaturated rhamnogalacturonyl belongs to glycosyl hydrolase family 88, and hydrolase catalyzes the hydrolysis of unsaturated rhamnogalacturonan disaccharide to yield unsaturated d-galacturonic acid and l-rhamnose. Rhamnogalacturonan is a form of pectin and contains α-l-rhamnose in the polygalacturonate chain. In some stretches, every second monomer is l-rhamnose, and this sugar can form side chains with galactan, arabinan, or arabinogalactan. Therefore, cells may require several different kinds of pectinolytic enzymes to utilize heteropolymers of pectin.
Transcription and translation.
We identified the upregulation of genes related to transcription and translation, such as RNA polymerase, sigma factor, ribosomal protein, and initiation factor. Figure 3 shows that the RPKM values of the RNA polymerase subunit (rpoA) and many ribosomal proteins (rpsD, rpmD, and rpsM) were extraordinarily high. In contrast, their RPKM values were about average when succinate was utilized as a carbon source. To exclude the possibility of early exhaustion of succinate, which would subsequently shut down translation, we compared the growth curves of A. jeotgali cells grown with 6, 8, or 10 mM succinate (see Fig. S9 in the supplemental material). The mean generation times were 0.94 ± 0.03 h−1, 1.02 ± 0.01 h−1, and 1.07 ± 0.03 h−1 for 6, 8, and 10 mM succinate, respectively. The maximum optical densities were 0.599 ± 0.04, 1.397 ± 0.73, and 1.411 ± 0.82 for 6, 8, and 10 mM succinate, respectively. A. jeotgali grew at a similar rate and reached a maximum optical density in 8 mM succinate. The growth curves showed that MSB medium containing 10 mM succinate, from which total RNA was isolated, provided enough substrate, and there were no artifacts related to a shortage of the carbon source.
Transporters.
Several ion transporters, including Na+/H+ antiporters (nhaB [AJE_04480] and nhaC [AJE_11224]), a monovalent cation/proton antiporter (mnhG [AJE_06366]), an ion transport protein (AJE_09454), a Ca2+/Na+ antiporter (AJE_11724), sulfate/thiosulfate transporter subunits (cysA [AJE_17435] and cysW [AJE_17430]), and a formate/nitrite transporter (focA [AJE_09122]), were upregulated. The carboxyl groups and methyl esters of pectin molecules have the ability to bind oppositely charged ions, and different ion concentrations in the medium could be achieved, resulting in the differential expression of ion transport genes (41).
Oxidative stress.
Many oxidative stress-related genes, including catalase (katG [AJE_01354]), alkyl hydroperoxide reductase (ahpC [AJE_08402] and ahpF [AJE_08407]), superoxide dismutase (sod [AJE_14235]), and peptide methionine sulfoxide reductase (mrsA [AJE_14655]) genes, were upregulated by >2-fold. The Fe(II)-trafficking protein (yggX [AJE_04240]) is known to protect DNA from iron-mediated oxidative damage (42) and was upregulated by 2.1-fold. The generation of oxidative stress by pectin-utilizing Alishewanella species was investigated by using DHR 123 (18). Nonfluorescent DHR 123 is oxidized by hydrogen peroxide, and the oxidation of DHR 123 is catalyzed by peroxidase activity. The fluorescent product, rhodamine 123, cannot cross the cell membrane and is retained in the cell. DHR 123 fluorescence was detected and visualized by flow cytometry analysis. Figure S10 in the supplemental material shows that A. jeotgali and A. aestuarii exhibited higher levels of oxidative stress when they were grown on pectin as a sole carbon source. However, interestingly, the fluorescent population of A. agri did not differ under the 2 conditions. The RPKM values of oxidative stress-related genes in A. agri were not as high as those in A. jeotgali and A. aestuarii. Therefore, A. agri seemed able to cope with oxidative stress via more efficient mechanisms or was more tolerant to oxidative stress than the other species. The reduced oxidative stress in A. agri could be the reason for its superior growth on pectin compared to A. jeotgali and A. aestuarii. Notably, we could not link oxidative stress with highly expressed glyoxylate bypass during pectin utilization in A. agri, unlike in the other 2 Alishewanella species. This can be explained by the rapid removal of reactive oxygen species before detection by DHR 123 or because the amount of oxidative stress was not high enough to be detected. Neither possibility was tested in this study.
Many upregulated proteases and chaperones (clp, dnaJ, dnaK, hscA, groEL, sugE, and lon) are expected to degrade damaged or misfolded proteins and assist in the proper folding of the peptide chain. Peptide methionine sulfoxide reductase (mrsA [AJE_14655]) was upregulated by 2.1-fold and can reduce methionine that had been oxidized by reactive oxygen species (thereby forming methionine sulfoxide) back to methionine. Relatively lower RPKM values of proteases, chaperones, and methionine sulfoxide reductase in AGRI_Pectin were confirmed.
Methylglyoxal detoxification.
Methylglyoxal is a metabolic by-product generated naturally. It is highly toxic due to its chemical reaction with biomolecules such as proteins and nucleic acids. Lactoylglutathione lyase, also known as glyoxalase I (gloA [AJE_16619]), detoxifies methylglyoxal and was upregulated by 2.7-fold.
Membrane integrity.
The lipids of the outer membrane are asymmetrically distributed, with lipopolysaccharides (LPSs) on the outside and phospholipids (PLs) on the inside (43). PLs are exposed to the surface of the outer membrane only in stressed cells (43). The ABC transporter membrane protein (AJE_06271) is a homolog of mlaE of E. coli and is known to maintain lipid asymmetry in the outer membrane (44). mlaE was upregulated by 4.7-fold in AJE_Pectin. Membrane integrity was not measured; however, the upregulation of mlaE indicated that the membrane integrity of A. jeotgali may be compromised under pectin-utilizing conditions. Upregulation of the fatty acid biosynthesis pathway (lpxC, fabZ, and fabD) may be required for rebuilding of the damaged cell membrane.
Motility.
The upregulation of several motility genes was observed in AJE_Pectin. These genes included genes encoding chemotaxis protein, flagellar motor switch protein, flagellar M-ring protein, regulator of flagellar synthesis, and type IV pilus assembly pilin (motA, spoA, fliF, flgM, and pilE, respectively). Motility tests were performed with succinate- and pectin-containing motility agar plates; however, no differences in motility were observed (data not shown). A motile phenotype was not observed for A. agri, although motility-related genes were expressed at approximately average RPKM values for A. agri. We could not explain a nonmotile phenotype of A. agri from RNAseq data.
Gene expression of homologs between A. aestuarii and A. agri.
The expression levels of homologous genes from the 3 different species (see Table S10 in the supplemental material) were not similar; that is, some genes were highly expressed in only 1 or 2 of the Alishewanella species, while others were highly expressed in all 3 species. This pattern prompted us to identify whether there was any correlation in gene expression levels between species. The Spearman's rank correlation value calculated from RPKM values of 2,671 core genes from A. aestuarii and A. agri was only 0.018, indicating that high RPKM values in homologous genes did not guarantee high gene expression levels in another species (see Fig. S11 in the supplemental material). This result was quite interesting, because we performed transcriptomic analysis under identical conditions and confirmed similar expression levels of genes involved in several cellular functions such as glyoxylate bypass, the pectin metabolic pathway, and transcription- and translation-related genes.
Conclusion.
The Alishewanella genus provides a good opportunity to investigate bacterial evolutionary history, because while these species appeared to be isolated within different environments, they still contain many conserved metabolic pathways, such as the pectin metabolism pathway. There is no golden rule to determine habitat differentiation of bacterial species; however, we suggest that genomic evolution and habitat differentiation of Alishewanella species were relatively recent events based on our results showing relatively conserved genomic synteny, large deviations in the GC content of GIs with potential metabolic functions, and horizontally transferred soil bacterium-associated A. agri genes. Comparative transcriptomic analyses were conducted with pectin-metabolizing A. jeotgali and are depicted in Fig. 6. Comparisons of gene expression patterns in the 3 Alishewanella species showed different aspects of evolution (or adaptation), revealing different transcriptional profiles of homologous genes. Therefore, we speculate that the 3 Alishewanella species dealt with an identical situation by using different sets of genes. In addition to evolutionary context, comparative transcriptomic studies of pectin metabolism in the 3 different species have not been performed previously, and the relationship of pectin metabolism to diverse stress conditions has not been described in many previous studies. Therefore, this study provides valuable information for pectin metabolism in terms of global cellular processes as well as bacterial evolution.
Fig 6.

Upregulated cellular processes of A. jeotgali. Fold changes were calculated as the RPKM values of pectin-utilizing cells/succinate-utilizing cells. See the text for relevant genes and descriptions. RNAP, RNA polymerase.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the Next-Generation BioGreen 21 Program (PJ0082082013), Rural Development Administration, Republic of Korea.
Footnotes
Published ahead of print 9 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02350-13.
REFERENCES
- 1.Vogel BF, Venkateswaran K, Christensen H, Falsen E, Christiansen G, Gram L. 2000. Polyphasic taxonomic approach in the description of Alishewanella fetalis gen. nov., sp. nov., isolated from a human foetus. Int. J. Syst. Evol. Microbiol. 50:1133–1142 [DOI] [PubMed] [Google Scholar]
- 2.Kim MS, Roh SW, Nam YD, Chang HW, Kim KH, Jung MJ, Choi JH, Park EJ, Bae JW. 2009. Alishewanella jeotgali sp. nov., isolated from traditional fermented food, and emended description of the genus Alishewanella. Int. J. Syst. Evol. Microbiol. 59:2313–2316 [DOI] [PubMed] [Google Scholar]
- 3.Roh SW, Nam YD, Chang HW, Kim KH, Kim MS, Oh HM, Bae JW. 2009. Alishewanella aestuarii sp. nov., isolated from tidal flat sediment, and emended description of the genus Alishewanella. Int. J. Syst. Evol. Microbiol. 59:421–424 [DOI] [PubMed] [Google Scholar]
- 4.Kim MS, Jo SK, Roh SW, Bae JW. 2010. Alishewanella agri sp. nov., isolated from landfill soil. Int. J. Syst. Evol. Microbiol. 60:2199–2203 [DOI] [PubMed] [Google Scholar]
- 5.Tarhriz V, Nematzadeh G, Mohammadzadeh F, Rahimi E, Hejazi MS. 2011. Isolation and characterization of some aquatic bacteria from Qurugöl Lake in Azerbaijan under aerobic conditions. Adv. Environ. Biol. 5:3173–3178 [Google Scholar]
- 6.Polz MF, Alm EJ, Hanage WP. 2013. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 29:170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobrindt U, Hochhut B, Hentschel U, Hacker J. 2004. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2:414–424 [DOI] [PubMed] [Google Scholar]
- 8.Coleman ML, Sullivan MB, Martiny AC, Steglich C, Barry K, Delong EF, Chisholm SW. 2006. Genomic islands and the ecology and evolution of Prochlorococcus. Science 311:1768–1770 [DOI] [PubMed] [Google Scholar]
- 9.Jung J, Choi S, Chun J, Park W. 2012. Genome sequence of pectin-degrading Alishewanella aestuarii strain B11T, isolated from tidal flat sediment. J. Bacteriol. 194:5476. 10.1128/JB.01255-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung J, Chun J, Park W. 2012. Genome sequence of extracellular-protease-producing Alishewanella jeotgali isolated from traditional Korean fermented food. J. Bacteriol. 194:2097. 10.1128/JB.00153-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Jung J, Sung JS, Chun J, Park W. 2012. Genome sequencing of pectin-degrading Alishewanella agri, isolated from landfill soil. J. Bacteriol. 194:5135–5136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss, and rearrangement. PLoS One 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. 2005. ACT: the Artemis comparison tool. Bioinformatics 21:3422–3423 [DOI] [PubMed] [Google Scholar]
- 14.Hsiao YM, Liu YF, Huang YL, Lee PY. 2011. Transcriptional analysis of pmeA gene encoding a pectin methylesterase in Xanthomonas campestris pv. campestris. Res. Microbiol. 162:270–278 [DOI] [PubMed] [Google Scholar]
- 15.Stanier RY, Palleroni NJ, Doudoroff M. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159–271 [DOI] [PubMed] [Google Scholar]
- 16.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 17.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621–628 [DOI] [PubMed] [Google Scholar]
- 18.Henderson LM, Chappell JB. 1993. Dihydrorhodamine 123: a fluorescent probe for superoxide generation? Eur. J. Biochem. 217:973–980 [DOI] [PubMed] [Google Scholar]
- 19.Pál C, Papp B, Lercher MJ. 2005. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 37:1372–1375 [DOI] [PubMed] [Google Scholar]
- 20.Jung J, Madsen EL, Jeon CO, Park W. 2011. Comparative genomic analysis of Acinetobacter oleivorans DR1 to determine strain-specific genomic regions and gentisate biodegradation. Appl. Environ. Microbiol. 77:7418–7424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novick RP, Christie GE, Penadés JR. 2010. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8:541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kung VL, Ozer EA, Hauser AR. 2010. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74:621–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hacker J, Kaper JB. 2000. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54:641–679 [DOI] [PubMed] [Google Scholar]
- 24.Arber W. 2000. Genetic variation: molecular mechanisms and impact on microbial evolution. FEMS Microbiol. Rev. 24:1–7 [DOI] [PubMed] [Google Scholar]
- 25.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. U. S. A. 96:2192–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brüssow H, Canchaya C, Hardt WD. 2004. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68:560–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner PL, Waldor MK. 2002. Bacteriophage control of bacterial virulence. Infect. Immun. 70:3985–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blair DF. 2003. Flagellar movement driven by proton translocation. FEBS Lett. 545:86–95 [DOI] [PubMed] [Google Scholar]
- 29.Kong KF, Aguila A, Schneper L, Mathee K. 2010. Pseudomonas aeruginosa β-lactamase induction requires two permeases, AmpG and AmpP. BMC Microbiol. 10:328. 10.1186/1471-2180-10-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balasubramanian D, Schneper L, Merighi M, Smith R, Narasimhan G, Lory S, Mathee K. 2012. The regulatory repertoire of Pseudomonas aeruginosa AmpC β-lactamase regulator AmpR includes virulence genes. PLoS One 7:e34067. 10.1371/journal.pone.0034067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridley BL, O'Neill MA, Mohnen D. 2001. Pectin: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57:929–967 [DOI] [PubMed] [Google Scholar]
- 32.Khotimchenko Y, Khozhaenko E, Kovalev V, Khotimchenko M. 2012. Cerium binding activity of pectins isolated from the seagrasses Zostera marina and Phyllospadix iwatensis. Mar. Drugs 10:834–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Condemine G, Robert-Baudouy J. 1987. 2-Keto-3-deoxygluconate transport system in Erwinia chrysanthemi. J. Bacteriol. 169:1972–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mata-Gilsinger M, Ritzenthaler P. 1983. Physical mapping of the exuT and uxaC operators by use of exu plasmids and generation of deletion mutants in vitro. J. Bacteriol. 155:973–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suvorova IA, Tutukina MN, Ravcheev DA, Rodionov DA, Ozoline ON, Gelfand MS. 2011. Comparative genomic analysis of the hexuronate metabolism genes and their regulation in gammaproteobacteria. J. Bacteriol. 193:3956–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakurai K, Arai H, Ishii M, Igarashi Y. 2011. Transcriptome response to different carbon sources in Acetobacter aceti. Microbiology 157:899–910 [DOI] [PubMed] [Google Scholar]
- 37.Rui B, Shen T, Zhou H, Liu J, Chen J, Pan X, Liu H, Wu J, Zheng H, Shi Y. 2010. A systematic investigation of Escherichia coli central carbon metabolism in response to superoxide stress. BMC Syst. Biol. 4:122. 10.1186/1752-0509-4-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li K, Pidatala RR, Ramakrishna W. 2012. Mutational, proteomic and metabolomic analysis of a plant growth promoting copper-resistant Pseudomonas spp. FEMS Microbiol. Lett. 335:140–148 [DOI] [PubMed] [Google Scholar]
- 39.Schroeter R, Voigt B, Jürgen B, Methling K, Pöther DC, Schäfer H, Albrecht D, Mostertz J, Mäder U, Evers S, Maurer KH, Lalk M, Mascher T, Hecker M, Schweder T. 2011. The peroxide stress response of Bacillus licheniformis. Proteomics 11:2851–2866 [DOI] [PubMed] [Google Scholar]
- 40.Singh R, Mailloux RJ, Puiseux-Dao S, Appanna VD. 2007. Oxidative stress evokes a metabolic adaptation that favors increased NADPH synthesis and decreased NADH production in Pseudomonas fluorescens. J. Bacteriol. 189:6665–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manunza B, Deiana S, Pintore M, Gessa C. 1998. Interaction of Ca2+ and Na+ ions with polygalacturonate chains: a molecular dynamics study. Glycoconj. J. 15:297–300 [DOI] [PubMed] [Google Scholar]
- 42.Osborne MJ, Siddiqui N, Landgraf D, Pomposiello PJ, Gehring K. 2005. The solution structure of the oxidative stress-related protein YggX from Escherichia coli. Protein Sci. 14:1673–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Preiss J, Ashwell G. 1963. Polygalacturonic acid metabolism in bacteria. II. Formation and metabolism of 3-deoxy-d-glycero-2,5-hexodiulosonic acid. J. Biol. Chem. 238:1577–1583 [PubMed] [Google Scholar]
- 44.Malinverni JC, Silhavy TJ. 2009. An ABC transport system that maintains lipid asymmetry in the gram-negative outer membrane. Proc. Natl. Acad. Sci. U. S. A. 106:8009–8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.