

Abstract
Photobioreactors (PBRs) are very attractive for sunlight-driven production of biofuels and capturing of anthropogenic CO2. One major problem associated with PBRs however, is that the bacteria usually associated with microalgae in nonaxenic cultures can lead to biofouling and thereby affect algal productivity. Here, we report on a phylogenetic, metagenome, and functional analysis of a mixed-species bacterial biofilm associated with the microalgae Chlorella vulgaris and Scenedesmus obliquus in a PBR. The biofilm diversity and population dynamics were examined through 16S rRNA phylogeny. Overall, the diversity was rather limited, with approximately 30 bacterial species associated with the algae. The majority of the observed microorganisms were affiliated with Alphaproteobacteria, Betaproteobacteria, and Bacteroidetes. A combined approach of sequencing via GS FLX Titanium from Roche and HiSeq 2000 from Illumina resulted in the overall production of 350 Mbp of sequenced DNA, 165 Mbp of which was assembled in larger contigs with a maximum size of 0.2 Mbp. A KEGG pathway analysis suggested high metabolic diversity with respect to the use of polymers and aromatic and nonaromatic compounds. Genes associated with the biosynthesis of essential B vitamins were highly redundant and functional. Moreover, a relatively high number of predicted and functional lipase and esterase genes indicated that the alga-associated bacteria are possibly a major sink for lipids and fatty acids produced by the microalgae. This is the first metagenome study of microalga- and PBR-associated biofilm bacteria, and it gives new clues for improved biofuel production in PBRs.
INTRODUCTION
The increasing global CO2 concentrations and the resulting effects of global warming have led to the development of novel and alternative technologies for the production of non-fossil-based biofuels and methods of CO2 capture. Since photobioreactors (PBRs) allow the continuous cultivation of microalgae under relatively controlled conditions, they have been the focus of many research projects, often linked to large-scale biofuel production (1–3). The microalgae in the PBRs produce significant amounts of lipids that are the basis of biofuel production (1, 2, 4, 5). Unfortunately the technology is not yet sustainable (6), and certainly, more research is needed, not only to improve the technology, but also to better understand the complex biology and interactions of microalgae with their environment.
The term microalgae comprises a phylogenetically very heterogeneous group of prokaryotic and eukaryotic microorganisms. They all employ oxygenic photosynthesis and thereby convert atmospheric CO2 to biomass (7–9).
PBRs are usually vessels with varying volumes (3, 10, 11). Despite their advantages, however, PBRs suffer from several drawbacks, and biodiesel production with microalgae in PBRs faces many challenges (11, 12). One major problem associated with PBRs is linked to biofouling and the growth of microbial biofilms within the reactor. Bacterial growth in running PBRs results in disrupted flow, increased pressure, and reduced light intensities and ultimately affects algal productivity (13). While it is well known that microalgae are often associated with diverse bacteria in their natural aquatic ecosystems and in experimental cultures (14–16), only very few studies have addressed a detailed phylogenetic characterization of the associated microbes (17–19). Bacteria observed in these nonaxenic cultures belong to the classes Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria, but bacteria affiliated with the phylum Bacteroidetes are also often observed. Additionally, many uncultivated bacteria are noted. However, no archaeal species have been detected, and only one study has reported the presence of an uncultivated fungus (18). Two very recent studies have addressed a partial phylogenetic characterization of the alga-associated microbial community within PBRs (16, 20). The authors of these studies show that the microalga Chlorella vulgaris is associated mainly with the Alphaproteobacteria (i.e., the genus Sphinogomonas) and Dunaliella tertiolecta is associated with the Alphaproteobacteria and Gammaproteobacteria.
Microalgae are in general believed to be auxotrophic for vitamin B12, and a significant fraction of microalgal species appear to be auxotrophic for thiamine, while some are also auxotrophic for biotin (21–24). Within this framework, it is also possible that other bacterial factors or metabolic activities have a positive influence on algal growth (25). Only very few studies have suggested that there is actually a negative impact on the population dynamics of the algae (26).
Metagenome technologies have proven to be powerful tools for the analysis of complex microbial communities and have led to a tremendous increase of knowledge about the functions, protein families, biotechnology, and ecology of microbial communities within the last decade (27–29). Recent examples from metagenome studies have given us a first insight into the complex interactions of microorganisms with their environments and hosts (29–33). Surprisingly, only very few studies have focused on metagenomes associated with algae (34, 35), and to date, no study is available that has addressed aquatic microalgae and their bacterial communities in PBRs.
Therefore, here, we have analyzed the phylogenetic and metagenomic diversity of a mixed-species biofilm grown in a PBR. The data suggest that the microbial community is composed of approximately 30 different microbial species and is capable of supporting algal growth by the production of B vitamers (cobalamin [B12] and biotin [B7]) and possibly other cofactors not yet identified. Remarkably, our data also suggest that at least some of the bacteria within the PBR biofilm cannot be cultivated without the microalgae and that the bacteria encode the potential to affect biofuel production.
MATERIALS AND METHODS
Microorganisms used in this study and cultivation media.
Scenedesmus obliquus strain U169 and Chlorella vulgaris strain U126 were obtained from the collection of algae at the University of Hamburg, Hamburg, Germany (http://www.biologie.uni-hamburg.de/bzf/zeph/zephsvcke.htm). The defined medium for cultivation of algae was composed of 2 g liter−1 Flory Basis Fertilizer 1 (Euflor, Germany) and supplemented with 3.22 g liter−1 KNO3. Flory Basis Fertilizer 1 is solely based on mineral compounds and was not supplemented with any vitamins. The pH was adjusted to 7.0. The algae were cultivated at 17°C in liquid medium at a natural light intensity in polyethyleneterephthalate flat-panel photobioreactors. The culture medium circulated at 1 liter min−1 in the PBR system. The PBR was aerated with compressed air and flue gas obtained from a combined block heat and power station. Physical parameters and culture conditions were monitored continuously (WTW IQ Sensor Net; System 2020 XT, Germany). The light intensity was determined with a LI-190 sensor (Li-Cor, USA). For a detailed description of the reactor, see Fig. S1 in the supplemental material and reference 36.
Escherichia coli strains were grown at 37°C in lysogenic broth (LB) medium (1% tryptone, 0.5% yeast extract, 1% NaCl) (37) supplemented with appropriate antibiotics. Lactobacillus plantarum ATCC 8014 and Lactobacillus delbrueckii subsp. lactis DSM 20335 for B vitamin detection were obtained from the DSMZ, Braunschweig, Germany. Lactobacillus strains were grown on MRS medium (38) under anaerobic conditions.
Media for cultivation of individual bacterial isolates derived from the PBR biofilm community were prepared as follows. R2A medium was prepared as described previously (39), and M9, TSB, and NB media were prepared according to the method of Sambrook and Russell (37). To stimulate microbial growth, the media were in part supplemented with algal culture extracts ranging from 5% to 50% (vol/vol). The inoculated plates were incubated for 5 to 7 days at 22°C under aerobic and anaerobic conditions.
Scanning electron microscopy.
For scanning electron microscopy (SEM), biofilm samples were fixed in paraformaldehyde (1%) and glutaraldehyde (0.25%), dehydrated by ascending alcohol series, and dried at the critical point with Balzers CPD 030 Critical Point Dryer (Bal-Tec, Schalksmühle, Germany). After coating samples with gold using an SCD 050 sputter coater (Bal-Tec), scanning electron micrographs were taken with a Leo 1525 (Zeiss, Germany).
PBR biofilm DNA extraction and molecular technologies.
Total nucleic acids were extracted from the biofilm samples using a previously published enzymatic cell lysis protocol with some modifications (40). The samples were stirred (200 rpm) overnight in 10 ml of extraction buffer (100 mM Tris-HCl, pH 8.0, 100 mM sodium EDTA, pH 8.0, 1.5 M NaCl, 0.1% Tween 80) with the addition of 5 mg lysozyme and 0.5 mg proteinase K at 37°C. Subsequently, SDS (1 ml; 20%) was added and incubated at 65°C for 90 min. The sample was centrifuged for 10 min at room temperature and 6,000 × g. The pellet was resuspended in 10 ml extraction buffer, incubated for 10 min at 65°C, and centrifuged again. The supernatant was collected and mixed with 1 volume polyethylene glycol (PEG) 6000 (30%) and 1.6 M sodium chloride. After 2 h of incubation at room temperature, the mixture was centrifuged again for 20 min at 10,000 × g. The resulting pellet was resuspended in 2 ml TE buffer (10 mM Tris-HCI, 1 mM sodium EDTA, pH 8.0). For separating proteins and polysaccharides, 7.5 M potassium acetate was added to the mixture, achieving a final concentration of 0.75 M. After 5 min of incubation on ice, proteins and polysaccharides were centrifuged at 16,000 × g and 4°C for 30 min. The DNA was finally extracted with phenol-chloroform–isoamyl alcohol and precipitated overnight at −20°C after adding 0.7 volume isopropanol with 1/10 volume of 3 M sodium acetate. The isolated DNA was used for PCR amplifications, as well as metagenomic analyses.
For the phylogenetic characterization, 16S rRNA genes were amplified using the standard primers 27f and 1492r (41). The amplified genes were ligated into the pDrive cloning vector (Qiagen, Hilden, Germany) and transformed into chemically competent TOP10 E. coli cells (Invitrogen, Karlsruhe, Germany). The 16S rRNA gene was sequenced with automated ABI377 technology following the manufacturer's instructions.
The large-insert fosmid library was constructed according to the Copy Control fosmid library production kit manual (Epicentre Biotechnologies, Madison, WI, USA). A total of 14,976 fosmid clones harboring inserts with an average size of 35 kb were generated. The insert rate was approximately 98%.
Sequencing of metagenomic DNA.
For sequencing of metagenomic DNA on the GS FLX platform (Roche Applied Science), libraries were constructed by applying the GS Rapid Library Prep kit (Roche Applied Science) according to the manufacturer's protocol. A single library was sequenced in half a PicoTiter Plate on the GS FLX system using the titanium sequencing chemistry (Roche Applied Science, Mannheim, Germany). Raw data were processed by employing the analysis pipeline for whole-genome shotgun sequence reads and applying GS FLX System Software (version 2.3). Illumina sequencing was done using a single lane of a HiSeq 2000 paired-end run (2 sets of 100 cycles). De novo assembly of these short reads was performed using the Velvet assembly program version 1.2.08 (42).
For the sequences' functional characterization, we used the Integrated Microbial Genomes (IMG) pipeline. To further analyze the biological processes linked to the individual genes and open reading frames (ORFs), we mainly employed the KEGG (43), COG (44), and Pfam (45) databases, using a cutoff of 10−5.
Phylogenetic analysis of bacteria associated with algae.
16S rRNA genes were amplified using oligonucleotide primers (27f, 5′-AGA GTT GAT CMT GGC TCA G-3′, and 1492r, 5′-GGY TAC CTT GTT ACG ACT T-3′) (41). PCR mixtures contained 100 ng of template DNA per μl, 0.2 mM each of the four deoxynucleoside triphosphates, 1.5 mM MgCl2, 1 μM (each) primer, and 2.5 U of Taq DNA polymerase. The thermocycling conditions were 45 s of denaturation at 94°C, 45 s of primer annealing at 50°C, and 1 min 30 s of primer extension at 72°C. This cycle was repeated 30 times.
Denaturing gradient gel electrophoresis (DGGE) analyses were carried out with a DCode system (Bio-Rad, Munich, Germany) using denaturing gradients of 40 to 70% denaturants. For the amplification of the 16S rRNA gene fragments, the primers 341F-GC (5′-GCA CGG GGGG CCT ACG GGA GGC AGC AG-3′, containing a 30-bp GC-rich sequence at the 5′ end) and 907R (5′-CCG TCA ATT CCT TTR AGT TT-3′) were used (46, 47). The thermocycling conditions were 40 s of denaturation at 96°C, 40 s of primer annealing at 56°C, and 1 min of primer extension at 72°C. This cycle was repeated 30 times. DGGE bands were sampled for reamplification and sequencing by punching into individual bands with sterile pipette tips and were transferred to 1.5-ml microcentrifuge tubes that contained 50 μl of distilled water. The reamplified PCR products were cleaned up using the Gel/PCR DNA Fragments Extraction kit (Qiagen, Hilden, Germany) and finally sequenced using standard technologies.
For restriction fragment length polymorphism (RFLP) analysis, the amplified (27f and 1492r) 16S rRNA genes were cloned into pDrive and transformed in E. coli cells. The insert DNA of 100 clones of each sample (T0 to T5) was amplified using oligonucleotide primers (M13-20-f, 5′-GTA AAA CGA CGG CCA GT-3′, and M13-r, 5′-CAG GAA ACA GCT ATG ACC-3′). The thermocycling conditions were 45 s of denaturation at 94°C, 45 s of primer annealing at 57°C, and 1 min 45 s of primer extension at 72°C. This cycle was repeated 30 times. The fragments were digested by the restriction enzyme HpaII. Clones that showed different patterns were sequenced using standard technologies.
Furthermore, to investigate the composition of the microbial community in the photobioreactor, rRNA sequences of the small subunit (SSU) were extracted from the unassembled FLX 454 and Illumina data using SortMeRna (48). Low-quality (<20 Ns) and short (<80-bp) reads were removed prior to further analysis. To gain insight into the community composition, sequences were directly mapped onto the most recent Silva database (SSURef 111 NR) using Bowtie2 (49). The Silva taxonomy of the best hit in the database was affiliated to the respective query sequence. For deeper analysis of the bacterial community structure, the hypervariable regions V3 and V6 of the bacterial 16S rRNA genes were extracted using V-Xtractor (50) and were also mapped on the most recent Silva database.
EPS characterization.
Exopolymeric substances (EPS) from the biofilm sample were extracted according to the method of Wingender et al. (51) with minor modifications. For this purpose, the biofilm samples were carefully removed from the PBR surface, weighed, and subsequently suspended in a ratio of 1:16 (wt/vol) in 0.14 M NaCl solution and homogenized by stirring at room temperature for 60 min. The suspension was centrifuged for 30 min at 20,000 × g and 10°C. The supernatants were twice sterile filtered using membrane filters (cellulose acetate; pore size, 0.20 μm). The filtrates were either processed immediately or aliquoted and stored until use at −20°C.
The lipid content was measured by extraction with n-hexane (52). To 0.5 ml of the EPS sample, 0.5 ml n-hexane was added and homogenized for 30 min by shaking at room temperature following a centrifugation step at 12,000 × g for 90 s. The upper phase (hexane and lipids) was transferred to a preweighed glass tube while avoiding the interphase. The solution was dried, and the glass vessel was weighed again.
The total carbohydrate content was determined using a phenol-sulfuric acid method (53) with dextran (neutral polysaccharides) and alginate (acidic polysaccharides) standards. To 0.5 ml of sample, 0.5 ml of phenol solution was added and mixed. Following this, 2.5 ml of concentrated sulfuric acid (H2SO4) was added, and the sample was mixed again following an incubation step at room temperature (10 min) and incubation at 30°C for 15 min. After a further 5-min incubation at room temperature, the absorbance of the samples was measured spectrophotometrically as the optical density at 480 nm (OD480) for acidic polysaccharides (alginate) and the OD490 for neutral polysaccharides (dextran).
Uronic acids were determined according to the method of Filisetti-Cozzi and Carpita (54) with glucuronic acid as a standard. To 0.4 ml of each sample, 40 μl solution 1 (4 M sulfamic acid [H3SO3N], pH 1.6) was added and mixed. After this step, 2.4 ml of solution 2 [0.075 M Na2(B4O5(OH)4) × 8H2O dissolved in H2SO4] was added, mixed, and then incubated for 20 min at 100°C. The samples were cooled in an ice bath for 5 min. Then, 80 μl of solution 3 (0.15% [wt/vol] m-hydroxybiphenyl [C12H10O] dissolved in 0.5% [wt/vol] NaOH) was added. Finally, the samples were thoroughly mixed and incubated for 10 min at room temperature, and the absorbance was recorded as OD525.
The protein content was measured using a modified Lowry assay (55, 56).
Furthermore, the total DNA within the EPS solution was detected spectrophotometrically after extraction from the EPS using the UltraClean Microbial DNA Kit (Dianova, Hamburg, Germany).
Lipase and esterase activities were assayed using various p-nitrophenyl (pNP) substrates and following previously published protocols (57).
Vitamin B12 and biotin detection.
For the determination of cobalamin the Difco B12 assay medium was used, and vitamin B12 was extracted and assayed according to the protocol published by Denter and Bisping (58) employing the L. delbrueckii subsp. lactis DSM 20335 indicator strain.
To determine the biotin content of the sample, the culture supernatant was centrifuged and sterile filtered. The biotin detection was done as described previously using the Lactobacillus growth assay and L. plantarum strain ATCC 8014 (59).
Nucleotide sequence accession numbers.
This project has been assigned the GenBank BioProject number PRJNA197241. The sequences derived from Illumina and FLX454 sequencing were deposited in the NCBI Short Read Archive under the study accession number SRP021903. The genome assembly, together with predicted gene models and annotations, is available from www.jgi.doe.gov (DOE Joint Genome Institute) under the IMG Project ID 9992.
The 16S rRNA gene sequences obtained have been deposited in the GenBank database under accession numbers KC994651 to KC994663, KC994664 to KC994680, KC994681 to KC994881, and KC994882 to KC994889.
RESULTS AND DISCUSSION
Biofilm development and chemical analysis.
PBRs are commonly inoculated with nonaxenic cultures of eukaryotic microalgae (14–16). In the current study, we examined a microbial biofilm in a PBR, which was inoculated with an S. obliquus and C. vulgaris culture. We monitored biofilm development over a period of 12 weeks by analyzing a total of six time points (T0 to T5) (Fig. 1). As early as 14 days after the initial transfer of the starter culture into the reactor, extensive biofilm formation was observed. The biofilm was tightly attached to the surface of the reactor and spread over the entire reactor within a period of 10 to 12 weeks, thereby strongly affecting the overall photosynthesis rates of the algae. This strong biofilm development resulted in shutdown of the PBR. Microscopic examinations of biofilm samples using SEM suggested that the bacteria were tightly attached to the algae (Fig. 2A to D). Additionally, EPS and nanowire-like filamentous structures were visible in the SEM images (Fig. 2). It is well known that EPS in biofilms mainly consists of polysaccharides, proteins, nucleic acids, and lipids (60). The concentrations of the individual components, however, vary and depend to a large extent on the different microorganisms and their medium and environmental conditions (61–63). EPS has diverse functions during biofilm development: it provides mechanical stability, builds up a three-dimensional network of polymers, and plays a role in cell-cell communication and defense against biocides (60, 64). A detailed chemical analysis of the PBR biofilm exopolymeric matrix suggested that it consisted mainly of fatty acids (10.4 mg/g), acidic and neutral polysaccharides (11.6 mg/g), and proteins (3.8 mg/g). However, only small amounts of uronic acid (<1%) and only traces of DNA were observed (Fig. 3). Since microalgae are well known to produce and release lipids (2, 5), it is not surprising that the EPS consisted largely of fatty acids.
Fig 1.

Development of a microbial biofilm over 12 weeks in a flat-panel PBR. T1, after 2 weeks; T2, after 4 weeks; T3, after 6 weeks; T4, after 8 weeks; T5, after 12 weeks of incubation. No microbial biofilm was observed at T0 (data not shown). Shortly after inoculation of the PBR at T0, a light intensity of approximately 150 μmol/m2s was measured at the backside of the reactor. The light intensities with a fully developed biofilm were in the range of 10 μmol/m2s or lower (T5).
Fig 2.

Scanning electron micrographs of a network of microalgae and bacteria in a PBR. Scale bars are indicated in the images (REM Leo 1525; 5.00 kV). The arrows point to nanowire-like structures that were identified. The large round cells (5 to 6 μm) are C. vulgaris, and the lemon-shaped cells (8 to 9 μm) are S. obliquus.
Fig 3.

Contents of biopolymers of the extracellular polymeric substances from biofilm samples harvested at T5. The DNA content was <4 ng/mg wet weight. The data are mean values of at least three replicates. The error bars indicate simple standard deviations.
Population structure of the alga-associated bacterial community.
For phylogenetic analysis, DNA from the mature biofilm (T5), the different biofilm developmental stages (T1 to T4), and the nonaxenic starter culture (T0) was extracted. The quality of the isolated DNA was sufficiently high for the construction of a large-insert metagenome library and DNA-sequencing analysis using pyrosequencing and Illumina-based technologies.
We used different strategies to estimate the microbial diversity within the developing biofilm samples and the established biofilm. We amplified 16S rRNA genes using standard primers (27f and 1492r [41]) and cloned the amplified DNA fragments. Inserts of these clones were completely sequenced. After removal of potential chimeras and duplicates, we analyzed a total of 201 unique 16S rRNA gene sequences from the mature biofilm (T5) (KC994681 to KC994881). In order to identify unique phylotypes and to estimate the bacterial richness, a rarefaction analysis using QIIME (65) was performed. This analysis suggested that the 16S rRNA gene sequences represented 28 operational taxonomic units (OTUs) based on a >99% identity cutoff for bacterial 16S rRNA genes. Rarefaction curves reached saturation at distance levels of 20% (phylum level) and 3% (species level) (Fig. 4). This analysis also suggested that the overall diversity within the biofilm community was rather limited. In addition, a Shannon-Weaver index (66) of 3.01 at a genetic distance of 1% was observed. These results, together with abundance-based coverage estimators (ACE) (67) and Chao1 richness estimates (68), indicated that a significant fraction of the bacterial species diversity was assessed. The analyzed bacterial 16S rRNA gene sequences were mainly affiliated with members of the classes Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria (Fig. 5; see Table S1 in the supplemental material). Additionally, a significant number of 16S rRNA gene hits were associated with the phylum Bacteroidetes. Bacteroidetes are among the main colonizers of the guts of humans and animals, but they are also found in many other habitats and are regarded as specialists in the degradation of high-molecular-weight organic matter (34). Overall, the most abundant bacterial organisms were members of the families Sphingomonadaceae, Caulobacteraceae, Rhizobiaceae, Comamonadaceae, Xanthomonadaceae, Sphingobacteriaceae, and Flavobacteriaceae.
Fig 4.

Rarefaction curves indicating the observed numbers of OTUs within the 16S rRNA gene libraries derived from the PBR biofilm at T5 after filtering of the chloroplast sequences. The curves were calculated with QIIME (65). OTUs are shown at 1, 3, and 20% genetic-distance levels.
Fig 5.

Phylogenetic assignment of the 16S rRNA clone library derived from the PBR biofilm by using the reference sequences stored in the NCBI database. The 16S rRNA gene sequences were analyzed with Finch TV (Geospiza) and BioEdit (http://www.mbio.ncsu.edu/bioedit/bioedit.html).
Additional data from DGGE and RFLP analysis from the different time points during biofilm development and the nonaxenic starter culture supported these findings (see Fig. S2 and S3 in the supplemental material). The DGGE profiles and RFLP analysis, in combination with 16S rRNA gene sequences (DGGE, KC994651 to KC994663; RFLP, KC994664 to KC994680) derived from T0 to T5 further suggested that the phylogenetic structure of the biofilm population was rather stable over the entire time. The data also indicated that the microbes observed within the PBR biofilm were mostly already present in the starter culture used to inoculate the PBR at T0.
To further verify these data, we analyzed the DNA sequences of the mature PBR biofilm obtained by pyrosequencing and Illumina-based sequencing for the presence of hypervariable regions of rRNA gene fragments. Analysis of the V3 and V6 regions has been applied to study bacterial communities in diverse ecosystems (69–71). In total, 226 sequences from the FLX 454 (see Fig. S4A in the supplemental material) and 137,506 sequences from the Illumina (see Fig. S4C in the supplemental material) data sets were directly mapped onto the Silva database. For the analysis of the bacterial population, the hypervariable region V6 of the bacterial 16S rRNA gene was analyzed. Altogether, 41 sequences from the FLX 454 (see Fig. S4B in the supplemental material) and 558 sequences from the Illumina (see Fig. S4D in the supplemental material) data sets were included in the analysis.
In total, the results from the analysis (see Fig. S4 in the supplemental material) largely confirmed the results and the phylogenic composition estimated by using the 16S rRNA gene clone library.
However, it is notable that primer biases occurring during amplification of 16S rRNA genes (72) and the relatively high copy number of the microalgal plastid genomes, including 16S rRNA genes, resulted in slightly different species counts. The observed fungi, diatoms, and golden algae most likely are contaminants resulting from previous use of the PBRs.
Cultivation of microorganisms from the PBR.
Initial attempts to isolate bacteria associated with the algae by plating on different solid media (LB, tryptone-yeast [TY], Reasoner's 2A [R2A], TSB, and NB) under aerobic and anaerobic conditions, resulted in only two pure cultures. 16S rRNA gene sequencing suggested that the two isolates were affiliated with the genera Brevundimonas ( JQ661035.1; isolate A) and Paracoccus ( JQ404485.1; isolate B) (see Table S2 in the supplemental material). Members of these genera have been frequently isolated from soil and aquatic environments, but various clinical isolates are also known. Interestingly, tests in which we added various amounts of algal culture supernatant, including living cells, to the solid media did stimulate growth of another six bacterial organisms. These isolates were phylotyped by 16S rRNA gene sequencing (see Table S2 in the supplemental material). The 16S rRNA gene sequence of isolate C is most similar to that of Chryseobacterium taichungense YNB68 ( JQ071521.1). C. taichungense belongs to the Flavobacteriales and was only recently described as a novel species able to hydrolyze many chromogenic substrates (73). Isolate D could be associated with a Brevibacterium species that was not further characterized ( GQ199748.1). Brevibacteria are strictly aerobic Gram-positive microorganisms. Both isolates C and D were isolated on R2A agar plates supplemented with 50% (vol/vol) algal culture. The closest relative of isolate E is a member of the genus Roseomonas. The 16S rRNA gene sequence revealed a similarity of 78% to Roseomonas sp. ( HQ588850.1). Isolate E was cultivated in the presence of 25% (vol/vol) algal culture on NB agar plates. 16S rRNA analysis of isolate F indicated that this strain is associated with the yet-uncultured Cytophaga-Flavobacteria-Bacteroides (CFB) group bacterial clone S15D-MN13 ( AJ583211.1). The 16S rRNA gene sequence was 96% similar to the S15D-MN13 GenBank entry. Isolate G was similar to a hitherto uncultured gammaproteobacterium, clone L4 ( EU887990.1). 16S rRNA sequencing of isolate H suggested that the isolate is affiliated with Rhodococcus sp. strain 3/2 (EU041710.1). Isolates F to H were grown on NB medium supplemented with 50% (vol/vol) algal culture.
Altogether, these data suggest that only a minor fraction of the bacteria within the PBR are easily cultivable in laboratory media ( KC994882 to KC994889).
Analysis of the metabolic potential encoded by the PBR bacterial metagenome.
Today, our knowledge of the physiology and metabolism of complex microbial communities associated with eukaryotic microalgae is very limited. Therefore, we analyzed the partial metagenome sequences of the PBR biofilm microbial community with respect to possible gene functions. We established and assembled 164.7 Mbp of DNA sequences for the PBR biofilm community in contigs ranging from 165 bp to 203,650 bp (Table 1). Approximately 347,000 protein-coding genes could be assigned. Of all possible ORFs predicted, a total of 55,767 were similar to ORFs and genes in the KEGG database; 125,999 ORFs matched the COG database, and 35,935 ORFs could be analyzed by a MetaCyc pathway analysis (74). In addition, it should be noted that up to 1.5% of all ORFs were derived from eukaryotic DNA in the sample. Within this framework, it should be taken into account that most likely a small but significant number of ORFs are not correctly predicted due to overlapping genes/ORFs and noncorrected frame shifts. Based on the assumption that most of these 30 different bacterial species have genomes ranging from 3 to 8 Mbp, it is reasonable to speculate that the overall bacterial biofilm metagenome ranges from 90 Mbp to 240 Mbp. Therefore, the 164.7-Mbp assembled DNA represents a significant fraction of the overall bacterial biofilm metagenome. Although the available sequences do not allow a complete analysis of the physiological and metabolic functions within this bacterial community, the sequences give a good estimate of the biofilm genome structure and its metabolic potential.
Table 1.
Overall numbers of sequences and contigs generated for PBR biofilm analysis
| Parameter | Value |
|---|---|
| Reads FLX 454 (filtered) | |
| No. | 569,233 |
| Total length (bp) | 322,201,969 |
| Mean length (bp) | 566 |
| Range (bp) | 20–1,637 |
| GC mean (%) | 54 |
| Reads Illumina (filtered) | |
| No. | 217,902,594 |
| Total length (bp) | 21,136,551,618 |
| Mean length (bp) | 97 |
| Range (bp) | 97 |
| GC mean (%) | 51 |
| Contigs–assembly (velvet) | |
| No. | 165,176 |
| Total length (bp) | 164,776,921 |
| Mean length (bp) | 997 ± 2,780 |
| No. ≥400 bp | 86,050 |
| N50 size (bp) | 2,410 |
| Largest (bp) | 203,650 |
| GC mean (%) | 52 ± 10 |
The KEGG analysis suggested that the metabolic and catabolic potential of the microbes living in the PBR biofilm is highly diverse and flexible (Table 2). Genes for many of the classical pathways linked to the degradation of polysaccharides, proteins, and cellulose, but also for the degradation of aromatic compounds, were identified. With respect to the degradation of biopolymers, at least 64 amylases, 57 cellulases (mainly GH5 and M-family cellulose), 22 genes linked to the depolymerization of poly-β-hydroxybutyrate (PHB), and a high number of proteases (2,419) were observed.
Table 2.
Key features observed in the PBR biofilm metagenome using a KEGG-based analysis
| Trait | % of all hits |
|---|---|
| Amino acid metabolism | 16.6 |
| Polysaccharide degradation and metabolism | 16.1 |
| Energy metabolism | 8.1 |
| Vitamin and cofactor biosynthesis | 6.7 |
| Secondary-metabolite synthesis | 4.9 |
| Lipid metabolism | 4.8 |
| Transport mechanisms | 4.7 |
| Xenobiotic- and aromatic-compound degradation | 4.2 |
| Exopolysaccharide biosynthesis and modification | 1.9 |
| Cell appendages and motility | 1.8 |
| Secretion systems | 1.6 |
| Others | 14.2 |
| Total | 100 |
Many genes involved in lipid and fatty acid catabolism could be identified. Altogether, at least 1,137 genes and ORFs encoded esterolytic and/or lipolytic enzymes, suggesting a high hydrolytic activity for fatty acids within the analyzed microbial community. This observation was supported by data from a metagenome fosmid library screening. Altogether, 14,976 clones with an average insert size of 35 kbp were screened in tributyrin-containing indicator plates. This equals about 524 Mbp of screened DNA. From this screen, a surprisingly large number of lipolytic clones were identified. A total of 68 clones were identified in a first screen using the outlined screening procedures. Further tests with cell extracts of 16 of these clones confirmed that a wide range of fatty acids with various chain lengths were turned over by extracts of the fosmid clones (Fig. 6). The majority of the clones were active on the pNP-laurate (C12) but had almost negligible activities on the pNP-palmitate (C16) and pNP-stearate (C18) and thus most likely encoded esterases acting preferentially on shorter-chain fatty acids. However, very few clones also hydrolyzed the longer-chain fatty acid substrates and are perhaps more likely to encode true lipases (i.e., pFOS73A2, pFOS108D6, and pFOS131A4). Additional tests with culture supernatants supported this hypothesis. In these tests, we observed small but significant hydrolytic activities ranging from 0.0732 to 0.3373 milliunits per ml in the cell-free culture supernatant of the PBR. Lipolytic enzymes, including lipases (EC 3.1.1.3) and carboxylesterases (EC 3.1.1.1), are of general cell importance for membrane and fatty acid metabolism, and they are well known to be important during various pathogenic and nonpathogenic host-microbe interactions (75, 76). Thus, we speculate that the relatively high number of esterolytic and lipolytic genes is perhaps a result of the known production of lipids and fatty acids by the algae and thus has possibly resulted in an enrichment of highly lipolytic and esterolytic microbes. It is further notable that many short-chain fatty acid oxidoreductases and oxidoreductases without an assigned substrate (>600) were identified within the PBR metagenome. Some of these will most likely be important for the oxidation/reduction of the lipids derived from algae.
Fig 6.

Lipolytic and esterolytic activities of 16 PBR- and metagenome-derived fosmid clones. Activities were assayed in the presence of various pNP substrates (pNP-laurate, pNP-myristate, pNP-palmitate, and pNP-stearate) at a final concentration of 1 mM. Extinction was measured at 405 nm against an enzyme-free blank after 30 min of incubation at 37°C. The data are mean values of at least three replicates. The error bars indicate simple standard deviations.
The metagenome sequences further suggest that the bacteria associated within the PBR were mainly heterotrophs metabolizing a wide range of carbon and energy sources provided by the algae.
The microbial lifestyle in the PBR biofilm most likely included aerobic, as well as microaerobic, growth. Altogether, 1,844 cytochrome-encoding genes were counted, suggesting that highly diverse and branched electron transport chains are encoded by the bacteria living within the PBR biofilm. Branched electron transport chains are an adaptation to varying oxygen concentrations (77) and are observed in the majority of all bacterial genomes. In addition, all the genes needed for nitrate respiration were identified. The occurrence of these genes will certainly allow a wide range of metabolic activities under varying oxygen concentrations and could be an adaptation to life in biofilms that are known to include gradients of oxygen and often even have anoxic pockets (78, 79).
Biosynthesis genes of B-group vitamins.
Vitamin B12 is one of the most complex cofactors and an essential vitamer that is required for molecular rearrangements of hydrogen or methyl groups in prokaryotic and eukaryotic cells, and it is involved in the reduction of ribonucleotide triphosphate to 2′-deoxyribonucleotide triphosphate (80, 81). Higher eukaryotes (i.e., animals, humans, and protists) require B12 but do not synthesize the vitamer, and plants and fungi neither synthesize it nor use it. While plants have evolved a B12-independent methionine synthase, algae require the vitamer for the function of B12-dependent methionine synthase (22–24, 82, 83). Vitamin B12 is synthesized by bacteria and archaea, which have evolved two distinct routes for biosynthesis under aerobic and anaerobic conditions (22). More than 20 different steps are required for the biosynthesis of the corrinoid ring structure from uroporphyrinogen III (80, 81).
The KEGG-based analysis of the predicted genes and ORFs in the PBR biofilm metagenome suggested that all the genes with relevance to B12 vitamin biosynthesis were present, and many of them in multiple copies (see Fig. S5 in the supplemental material). Within this framework, it is notable that many algae are auxotrophic with respect to the production of thiamine and biotin, as well (21). Thiamine (vitamin B1) is essential for some of the key enzymes in the primary carbohydrate and branched-chain amino acid metabolism. Altogether, we have identified at least 218 genes in the biofilm metagenome that are involved in thiamine biosynthesis (see Fig. S6 in the supplemental material) (84, 85). Biotin is a cofactor of biotin-dependent carboxylase reactions, among them acetyl coenzyme A (CoA) carboxylase, which is essential for fatty acid synthesis (86, 87). For all major genes involved in biotin biosynthesis, multiple copies were detected (see Fig. S7 in the supplemental material). Based on the KEGG analysis, almost 6.7% of all ORFs and genes encoded metabolism of cofactors and vitamin biosynthesis. Additional tests in an experimental test reactor confirmed that small amounts of the two vitamers B12 and biotin could already be detected in the PBR culture supernatant 48 h after inoculation (Fig. 7 and 8). The pico- and nanomolar amounts observed were certainly sufficient to support algal growth.
Fig 7.
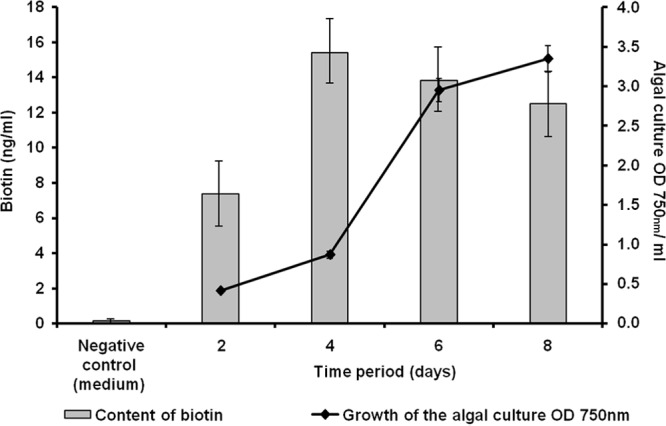
Biotin (ng/ml) detected in a PBR culture supernatant. Biotin was determined at different periods of growth of the algal culture. The data represent values of at least three replicates. The error bars indicate simple standard deviations.
Fig 8.
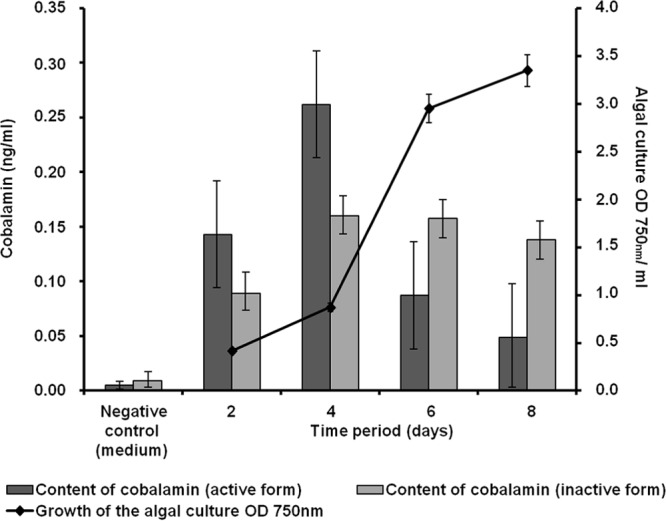
Content of cobalamin (ng/ml) in a model PBR. The cobalamin content (active and inactive forms) of the algal culture supernatant was determined at different time points during algal growth. The data represent values of at least three replicates. The error bars indicate simple standard deviations.
N-Acyl-homoserine lactone-dependent signaling plays a minor role in PBR biofilm formation.
Microbial biofilm formation depends to a large extent on the attachment of bacteria to surfaces, microcolony formation, and maturing of the biofilm structure (64, 88, 89). Within the PBR metagenome, many genes were identified that are essential to biofilm formation. Biofilm formation is regulated by a wide range of factors, including signaling molecules, such as autoinducer involved in quorum-sensing-dependent gene regulation. Interestingly, we have identified only a small number of genes that are involved in the synthesis of autoinducer I-type (N-acyl-homoserine lactone) molecules. N-Acyl-homoserine lactones are the key signaling molecules in the cell density-dependent system of gene regulation in many Gram-negative bacteria, and they are usually synthesized through a LuxI-like protein (90, 91). Employing the amino acid sequences of various LuxI homologues for Blastp searches, we could not detect more than a few homologues (<10) in the PBR metagenome. This finding is intriguing, and it might suggest that autoinducer I-dependent signaling may play only a minor role within the observed ecological niche.
Summary and conclusions.
Sequencing-based metagenome analyses in combination with function-based studies have in general significantly increased our knowledge of microbial communities and their phylogenetic makeup and genetic potential. Finally, these studies provided insight into the ecology of the analyzed communities and thereby enriched our understanding of biodiversity and biochemical functions. Many of these studies have been summarized previously (29, 92–94). Surprisingly, only a few studies have addressed metagenome technologies for the analysis of mixed-species biofilms in nonhuman interactions. The earliest studies dealt with the acid mine drainage biofilm (95) and a drinking water biofilm (96). More recently, studies have addressed microbial biofilm metagenomes of hydrothermal vent chimneys, biofilms on concrete water pipes, and an extremely acidophilic sulfur-oxidizing biofilm (97–99). Although not all studies have been listed here, it is evident that many different biotic and abiotic factors shape each of these communities and that each community clearly differs in its phylogenetic structure and genomic-information content.
In summary, our data give detailed insight into the microbial community and the metagenome of a PBR-associated microbial biofilm. Altogether, we observed 28 OTUs within the analyzed biofilm, and DGGE and RFLP analyses suggested that the community was rather stable over time. The sequence-based data and the laboratory measurement data clearly support earlier findings with respect to the bacterially produced B vitamins. Our data also suggest that bacterial growth depends in part on compounds released by the algae or bacteria within the community.
Finally, it is interesting that the bacteria in the PBR studied here encode many esterolytic and lipolytic enzymes. This finding was supported by extracting active lipolytic clones and by measuring the hydrolytic activities in the culture supernatants. Future work will have to use these data to further improve the efficiencies of PBRs with respect to CO2 fixation and downstream processing of biofuels and/or other valuable compounds produced and released by the algae.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the research and development project Technologies for Exploiting the Resource Microalgae (TERM) and the BMBF Project ChemBiofilm and was funded by the Federal Graduate School “C1-Chemistry in Resource and Energy Management” (C1-REM) (Hamburg, Germany).
Footnotes
Published ahead of print 2 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01641-13.
REFERENCES
- 1.Chisti Y. 2007. Biodiesel from microalgae. Biotechnol. Adv. 25:294–306 [DOI] [PubMed] [Google Scholar]
- 2.Williams PJLB, Laurens LML. 2010. Microalgae as biodiesel and biomass feedstocks: review and analysis of the biochemistry, energetics and economics. Energy Environ. Sci. 3:554–590 [Google Scholar]
- 3.Ugwu CU, Aoyagi H, Uchiyama H. 2008. Photobioreactors for mass cultivation of algae. Bioresour. Technol. 99:4021–4028 [DOI] [PubMed] [Google Scholar]
- 4.Gouveia L, Oliveira A. 2009. Microalgae as a raw material for biofuels production. J. Ind. Microbiol. 36:269–274 [DOI] [PubMed] [Google Scholar]
- 5.Abomohra AF, Wagner M, El-Sheekh M, Hanelt D. 2012. Lipid and total fatty acid productivity in photoautotrophic fresh water microalgae: screening studies towards biodiesel production. J. Appl. Phycol. 2012:1–6 [Google Scholar]
- 6.Waltz E. 2013. Algal biofuels questioned. Nat. Biotechnol. 31:12 [Google Scholar]
- 7.Andersen RA. 2004. Biology and systematics of heterokont and haptophyte algae. Am. J. Bot. 91:1508–1522 [DOI] [PubMed] [Google Scholar]
- 8.Falkowski PG, Katz ME, Knoll AH, Quigg A, Raven JA, Schofield O, Taylor FJ. 2004. The evolution of modern eukaryotic phytoplankton. Science 305:354–360 [DOI] [PubMed] [Google Scholar]
- 9.Falkowski PG, Raven JA. 2007. Aquatic photosynthesis, 2nd ed. Princeton University Press, Princeton, NJ [Google Scholar]
- 10.Lee YK. 2001. Microalgal mass culture systems and methods: their limitation and potential. J. Appl. Phycol. 13:307–315 [Google Scholar]
- 11.Lehr F, Posten C. 2009. Closed photo-bioreactors as tools for biofuel production. Curr. Opin. Biotechnol. 20:280–285 [DOI] [PubMed] [Google Scholar]
- 12.Scott SA, Davey MP, Dennis JS, Horst I, Howe CJ, Lea-Smith DJ, Smith AG. 2010. Biodiesel from algae: challenges and prospects. Curr. Opin. Biotechnol. 21:277–286 [DOI] [PubMed] [Google Scholar]
- 13.Carvalho AP, Meireles LA, Malcata FX. 2006. Microalgal reactors: a review of enclosed system designs and performances. Biotechnol. Prog. 22:1490–1506 [DOI] [PubMed] [Google Scholar]
- 14.Cole JJ. 1982. Interactions between bacteria and algae in aquatic ecosystems. Annu. Rev. Ecol. Syst. 13:291–314 [Google Scholar]
- 15.Lakaniemi AM, Hulatt CJ, Wakeman KD, Thomas DN, Puhakka JA. 2012. Eukaryotic and prokaryotic microbial communities during microalgal biomass production. Bioresour. Technol. 124:387–393 [DOI] [PubMed] [Google Scholar]
- 16.Lakaniemi AM, Intihar VM, Tuovinen OH, Puhakka JA. 2012. Growth of Chlorella vulgaris and associated bacteria in photobioreactors. Microb. Biotechnol. 5:69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otsuka S, Abe Y, Fukui R, Nishiyama M, Sendoo K. 2008. Presence of previously undescribed bacterial taxa in non-axenic Chlorella cultures. J. Gen. Appl. Microbiol. 54:187–193 [DOI] [PubMed] [Google Scholar]
- 18.Watanabe K, Takihana N, Aoyagi H, Hanada S, Watanabe Y, Ohmura N, Saiki H, Tanaka H. 2005. Symbiotic association in Chlorella culture. FEMS Microbiol. Ecol. 51:187–196 [DOI] [PubMed] [Google Scholar]
- 19.Ueda H, Otsuka S, Senoo K. 2009. Community composition of bacteria co-cultivated with microalgae in non-axenic algal cultures. Microbiol. Cult. Coll. 25:21–25 [Google Scholar]
- 20.Lakaniemi AM, Intihar VM, Tuovinen OH, Puhakka JA. 2012. Growth of Dunaliella tertiolecta and associated bacteria in photobioreactors. J. Ind. Microbiol. Biotechnol. 39:1357–1365 [DOI] [PubMed] [Google Scholar]
- 21.Croft MT, Warren MJ, Smith AG. 2006. Algae need their vitamins. Eukaryot. Cell 5:1175–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. 2002. The biosynthesis of adenosylcobalamin (vitamin B12). Nat. Prod. Rep. 19:390–412 [DOI] [PubMed] [Google Scholar]
- 23.Helliwell KE, Wheeler GL, Leptos KC, Goldstein RE, Smith AG. 2011. Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol. Biol. Evol. 28:2921–2933 [DOI] [PubMed] [Google Scholar]
- 24.Giovannoni SJ. 2012. Vitamins in the sea. Proc. Natl. Acad. Sci. U. S. A. 109:13888–13889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouget JL, Dakhama A, Lavoie MC, de la Noüe J. 1995. Algal growth enhancement by bacteria: is consumption of photosynthetic oxygen involved? FEMS Microbiol. Ecol. 18:35–43 [Google Scholar]
- 26.Berger PS, Rho J, Gunner H. 1979. Bacterial suppression of Chlorella by hydroxylamine production. Water Res. 13:267–273 [Google Scholar]
- 27.Handelsman J. 2004. Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68:669–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon C, Daniel R. 2011. Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. 77:1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streit WR, Schmitz RA. 2004. Metagenomics—the key to the uncultured microbes. Curr. Opin. Microbiol. 7:492–498 [DOI] [PubMed] [Google Scholar]
- 30.Simon C, Daniel R. 2009. Achievements and new knowledge unraveled by metagenomic approaches. Appl. Microbiol. Biotechnol. 85:265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremaroli V, Bäckhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249 [DOI] [PubMed] [Google Scholar]
- 32.Weinstock GM. 2012. Genomic approaches to studying the human microbiota. Nature 489:250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewin A, Wentzel A, Valla S. 2013. Metagenomics of microbial life in extreme temperature environments. Curr. Opin. Biotechnol. 24:516–525 [DOI] [PubMed] [Google Scholar]
- 34.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. 2011. Bacterial community assembly based on functional genes rather than species. Proc. Natl. Acad. Sci. U. S. A. 108:14288–14293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams TJ, Wilkins D, Long E, Evans F, Demaere MZ, Raftery MJ, Cavicchioli R. 2013. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15:1302–1317 [DOI] [PubMed] [Google Scholar]
- 36.Hindersin S, Leupold M, Kerner M, Hanelt D. 2013. Irradiance optimization of outdoor microalgal cultures using solar tracked photobioreactors. Bioprocess Biosyst. Eng. 36:345–355 [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38.De Man J, Rogosa M, Sharpe M. 1960. A medium for the cultivation of lactobacilli. J. Appl. Bacteriol. 23:130–135 [Google Scholar]
- 39.Reasoner DJ, Geldreich EE. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeates C, Gillings MR, Davison AD, Altavilla N, Veal DA. 1998. Methods for microbial DNA extraction from soil for PCR amplification. Biol. Proced. Online 1:40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175 In Stackebrandt E, Goodfellow MD. (ed), Nucleic acid techniques in bacterial systematics. John Wiley and Sons, New York, NY [Google Scholar]
- 42.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakaya A, Katayama T, Itoh M, Hiranuka K, Kawashima S, Moriya Y, Okuda S, Tanaka M, Tokimatsu T, Yamanishi Y, Yoshizawa AC, Kanehisa M, Goto S. 2013. KEGG OC: a large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res. 41:D353–D357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer ELL, Bateman A. 2008. The Pfam protein families database. Nucleic Acids Res. 36:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchholz-Cleven BEE, Rattunde B, Straub KL. 1997. Screening for genetic diversity of isolates of anaerobic Fe(II)-oxidizing bacteria using DGGE and whole-cell hybridization. Syst. Appl. Microbiol. 20:301–309 [Google Scholar]
- 47.Muyzer G, de Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopylova E, Noe L, Touzet H. 2012. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217 [DOI] [PubMed] [Google Scholar]
- 49.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartmann M, Howes CG, Abarenkov K, Mohn WW, Nilsson RH. 2010. V-Xtractor: an open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J. Microbiol. Methods 83:250–253 [DOI] [PubMed] [Google Scholar]
- 51.Wingender J, Strathmann M, Rode A, Leis A, Flemming HC. 2001. Isolation and biochemical characterization of extracellular polymeric substances from Pseudomonas aeruginosa. Methods Enzymol. 336:302–314 [DOI] [PubMed] [Google Scholar]
- 52.Hara A, Radin NS. 1978. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 90:420–426 [DOI] [PubMed] [Google Scholar]
- 53.Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. 1956. Colorimetric method for determination of sugars and related substances. Anal. Chem. 28:350–356 [Google Scholar]
- 54.Filisetti-Cozzi TM, Carpita NC. 1991. Measurement of uronic acids without interference from neutral sugars. Anal. Biochem. 197:157–162 [DOI] [PubMed] [Google Scholar]
- 55.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 56.Frølund B, Palmgren R, Keiding K, Nielsen PH. 1996. Extraction of extracellular polymers from activated sludge using a cation exchange resin. Water Res. 30:1749–1758 [Google Scholar]
- 57.Elend C, Schmeisser C, Leggewie C, Babiak P, Carballeira JD, Steele HL, Reymond JL, Jaeger KE, Streit WR. 2006. Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl. Environ. Microbiol. 72:3637–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Denter J, Bisping B. 1994. Formation of B-vitamins by bacteria during the soaking process of soybeans for tempe fermentation. Int. J. Food. Microbiol. 22:23–31 [DOI] [PubMed] [Google Scholar]
- 59.Entcheva P, Liebl W, Johann A, Hartsch T, Streit WR. 2001. Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Appl. Environ. Microbiol. 67:89–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat. Rev. Microbiol. 8:623–633 [DOI] [PubMed] [Google Scholar]
- 61.Branda SS, Vik S, Friedman L, Kolter R. 2005. Biofilms: the matrix revisited. Trends Microbiol. 13:20–26 [DOI] [PubMed] [Google Scholar]
- 62.Sutherland IW. 2001. The biofilm matrix: an immobilized but dynamic microbial environment. Trends Microbiol. 9:222–227 [DOI] [PubMed] [Google Scholar]
- 63.Flemming HC, Neu TR, Wozniak DJ. 2007. The EPS matrix: the house of biofilm cells. J. Bacteriol. 189:7945–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56:187–209 [DOI] [PubMed] [Google Scholar]
- 65.Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. 2011. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinformatics 10:10.7. 10.1002/0471250953.bi1007s36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shannon CE, Weaver W. 1949. The mathematical theory of communication, p 379–423 University of Illinois Press, Urbana, IL [Google Scholar]
- 67.Chao A, Lee SM. 1992. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 87:210–217 [Google Scholar]
- 68.Chao A. 1984. Nonparametric estimation of the number of classes in a population. Scan. J. Statist. 11:265–270 [Google Scholar]
- 69.Christen R. 2008. Global sequencing: a review of current molecular data and new methods available to assess microbial diversity. Microbes Environ. 23:253–268 [DOI] [PubMed] [Google Scholar]
- 70.Gloor GB, Hummelen R, Macklaim JM, Dickson RJ, Fernandes AD, MacPhee R, Reid G. 2010. Microbiome profiling by Illumina sequencing of combinatorial sequence-tagged PCR products. PLoS One 5:e15406. 10.1371/journal.pone.0015406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. 2008. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. 10.1371/journal.pgen.1000255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shen FT, Kämpfer P, Young CC, Lai WA, Arun AB. 2005. Chryseobacterium taichungense sp. nov., isolated from contaminated soil. Int. J. Syst. Evol. Microbiol. 55:1301–1304 [DOI] [PubMed] [Google Scholar]
- 74.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Pujar A, Shearer AG, Travers M, Weerasinghe D, Zhang P, Karp PD. 2012. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 40:D742–D753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arpigny JL, Jaeger KE. 1999. Bacterial lipolytic enzymes: classification and properties. Biochem. J. 343:177–183 [PMC free article] [PubMed] [Google Scholar]
- 76.Jaeger KE, Ransac S, Dijkstra BW, Colson C, van Heuvel M, Misset O. 1994. Bacterial lipases. FEMS Microbiol. Rev. 15:29–63 [DOI] [PubMed] [Google Scholar]
- 77.Anraku Y. 1988. Bacterial electron transport chains. Annu. Rev. Biochem. 57:101–132 [DOI] [PubMed] [Google Scholar]
- 78.Stewart PS, Franklin MJ. 2008. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 6:199–210 [DOI] [PubMed] [Google Scholar]
- 79.Williamson KS, Richards LA, Perez-Osorio AC, Pitts B, McInnerney K, Stewart P, Franklin MJ. 2012. Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of ribosome hibernation factors in the antibiotic-tolerant subpopulation and hypoxia-induced stress response in the metabolically active population. J. Bacteriol. 194:2062–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martens J, Barg H, Warren MJ, Jahn D. 2002. Microbial production of vitamin B12. Appl. Microbiol. Biotechnol. 58:275–285 [DOI] [PubMed] [Google Scholar]
- 81.Roth J, Lawrence J, Bobik T. 1996. Cobalamin (coenzyme B12): synthesis and biological significance. Annu. Rev. Microbiol. 50:137–181 [DOI] [PubMed] [Google Scholar]
- 82.Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG. 2005. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438:90–93 [DOI] [PubMed] [Google Scholar]
- 83.Duda J, Pedziwilk Z, Zodrow K. 1957. Studies on the vitamin B12 content of the leguminous plants. Acta Microbiol. Pol. 6:233–238 [PubMed] [Google Scholar]
- 84.Begley TP, Ealick SE, McLafferty FW. 2012. Thiamin biosynthesis: still yielding fascinating biological chemistry. Biochem. Soc. Trans. 40:555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jurgenson CT, Begley TP, Ealick SE. 2009. The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78:569–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Streit WR, Entcheva P. 2003. Biotin in microbes, the genes involved in its biosynthesis, its biochemical role and perspectives for biotechnological production. Appl. Microbiol. Biotechnol. 61:21–31 [DOI] [PubMed] [Google Scholar]
- 87.Lin S, Cronan JE. 2011. Closing in on complete pathways of biotin biosynthesis. Mol. Biosyst. 7:1811–1821 [DOI] [PubMed] [Google Scholar]
- 88.Monds RD, O'Toole GA. 2009. The developmental model of microbial biofilms: ten years of a paradigm up for review. Trends Microbiol. 17:73–87 [DOI] [PubMed] [Google Scholar]
- 89.Klausen M, Gjermansen M, Kreft JU, Tolker-Nielsen T. 2006. Dynamics of development and dispersal in sessile microbial communities: examples from Pseudomonas aeruginosa and Pseudomonas putida model biofilms. FEMS Microbiol. Lett. 261:1–11 [DOI] [PubMed] [Google Scholar]
- 90.LaSarre B, Federle MJ. 2013. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77:73–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galloway WR, Hodgkinson JT, Bowden SD, Welch M, Spring DR. 2011. Quorum sensing in Gram-negative bacteria: small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev. 111:28–67 [DOI] [PubMed] [Google Scholar]
- 92.Gilbert JA, Dupont CL. 2011. Microbial metagenomics: beyond the genome. Annu. Rev. Mar. Sci. 3:347–371 [DOI] [PubMed] [Google Scholar]
- 93.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, Bork P, Hugenholtz P, Rubin EM. 2005. Comparative metagenomics of microbial communities. Science 308:554–557 [DOI] [PubMed] [Google Scholar]
- 94.Simon C, Wiezer A, Strittmatter AW, Daniel R. 2009. Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Appl. Environ. Microbiol. 75:7519–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428:37–43 [DOI] [PubMed] [Google Scholar]
- 96.Schmeisser C, Stöckigt C, Raasch C, Wingender J, Timmis KN, Wenderoth DF, Flemming HC, Liesegang H, Schmitz RA, Jaeger KE, Streit WR. 2003. Metagenome survey of biofilms in drinking-water networks. Appl. Environ. Microbiol. 69:7298–7309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gomez-Alvarez V, Revetta RP, Santo Domingo JW. 2012. Metagenome analyses of corroded concrete wastewater pipe biofilms reveal a complex microbial system. BMC Microbiol. 12:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brazelton WJ, Baross JA. 2009. Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J. 3:1420–1424 [DOI] [PubMed] [Google Scholar]
- 99.Jones DS, Albrecht HL, Dawson KS, Schaperdoth I, Freeman KH, Pi Y, Pearson A, Macalady JL. 2012. Community genomic analysis of an extremely acidophilic sulfur-oxidizing biofilm. ISME J. 6:158–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.