

Abstract
Infections with the opportunistic yeast Candida glabrata have increased dramatically in recent years. Antifungal therapy of yeast infections commonly employs azoles, such as fluconazole (FLC), but C. glabrata frequently develops resistance to these inhibitors of ergosterol biosynthesis. The pyrimidine analog flucytosine (5-fluorocytosine [5FC]) is highly active versus C. glabrata but is now rarely used clinically due to similar concerns over resistance and, a related concern, the toxicity associated with high doses used to counter resistance. Azole-5FC combination therapy would potentially address these concerns; however, previous studies suggest that 5FC may antagonize azole activity versus C. glabrata. Here, we report that 5FC at subinhibitory concentrations antagonized the activity of FLC 4- to 16-fold versus 8 of 8 C. glabrata isolates tested. 5FC antagonized the activity of other azoles similarly but had only indifferent effects in combination with unrelated antifungals. Since azole resistance in C. glabrata results from transcription factor Pdr1-dependent upregulation of the multidrug transporter gene CDR1, we reasoned that 5FC antagonism might be similarly mediated. Indeed, 5FC-FLC antagonism was abrogated in pdr1Δ and cdr1Δ strains. In further support of this hypothesis, 5FC exposure induced CDR1 expression 6-fold, and this upregulation was Pdr1 dependent. In contrast to azoles, 5FC is not a Cdr1 substrate and so its activation of Pdr1 was unexpected. We observed, however, that 5FC exposure readily induced petite mutants, which exhibit Pdr1-dependent CDR1 upregulation. Thus, mitochondrial dysfunction resulting in Pdr1 activation is the likely basis for 5FC antagonism of azole activity versus C. glabrata.
INTRODUCTION
Candida albicans and related yeasts are typically present in low numbers among the normal mucosal flora, but antibiotic exposure or various immunologic abnormalities can lead to its overgrowth, resulting in oral or vaginal thrush. Immunocompromised individuals are also at risk for life-threatening invasive candidiasis; indeed, Candida species are the fourth-most-common cause of nosocomial bloodstream infection (1, 2). These infections are generally treated with oral or intravenous fluconazole (FLC), a triazole that, like the imidazoles that preceded it, inhibits the heme-dependent enzyme sterol 14α-demethylase in the ergosterol biosynthesis pathway. Additionally, more-recently introduced triazoles include itraconazole, voriconazole, and posaconazole.
In recent years, Candida glabrata has emerged as the second-most-common agent of candidiasis, trailing only C. albicans (2). Its emergence parallels the introduction and wide-spread clinical use of triazoles. In contrast to C. albicans, which typically exhibits an FLC MIC of ≤0.5 μg/ml, C. glabrata exhibits intrinsically low FLC susceptibility, with MICs of 8 to 16 μg/ml (3). Furthermore, under selective pressure, C. glabrata readily mutates to FLC resistance, with MICs of ≥64 μg/ml. Nearly all such mutants characterized to date have gain-of-function mutations in transcription factor Pdr1 that result in upregulated expression of multidrug transporter genes, particularly CDR1 (4–7). In light of C. glabrata's poor response to azoles, echinocandins are now recommended as first-line agents for C. glabrata infection (8). However, echinocandins must be administered intravenously, as is also the case with amphotericin B, which greatly restricts and complicates their use.
The pyrimidine analog 5-fluorocytosine (flucytosine [5FC]) can be administered orally or intravenously and exhibits high activity versus C. glabrata, with MICs of ≤0.06 μg/ml (9). However, 5FC is now rarely used clinically, primarily due to concerns over resistance associated with monotherapy and, a related concern, toxicity associated with the high doses often employed to overcome resistance (10). 5FC resistance in C. glabrata has been associated with loss-of-function mutations in cytosine permease (Fcy2L), cytosine deaminase (Fcy1), or uracil phosphoribosyltransferase (Fur1) (11).
In theory, 5FC-azole combination therapy represents a promising alternative to 5FC or FLC monotherapy of C. glabrata infection, since it would reduce the likelihood of resistance. Combinations are typically additive in their activity, or in some cases synergistic, providing further rationale for their use. For example, the combination 5FC-amphotericin B was reported to be synergistic versus C. glabrata (12). Since uptake is a major determinant of 5FC susceptibility (as evidenced by the resistance conferred by cytosine permease mutation), a likely explanation for this synergism is increased membrane permeability in amphotericin B-treated cells (13).
Conversely, antifungal combinations may be antagonistic. Indeed, 5FC has been reported to antagonize the activity of three different azoles versus C. glabrata, including FLC and the imidazoles miconazole and ketoconazole (12, 14, 15). This antagonism argues against the clinical use of 5FC-azole combination therapy. On the other hand, only additive activity was reported for 5FC in combination with voriconazole and itraconazole (12, 16). The basis for these different results is unclear.
We begin here by showing that 5FC antagonized the activity of all four triazoles versus C. glabrata, although the converse was not observed; i.e., triazoles did not antagonize 5FC activity. Since 5FC and azoles have distinct mechanisms of both action and resistance, elucidating the mechanism behind 5FC-azole antagonism was of considerable interest. Here, we demonstrate that 5FC activity, as expected, was not affected by pdr1 and cdr1 mutations, which strongly affect azole susceptibility. On the other hand, 5FC-FLC antagonism was dependent on both of these genes. Consistent with this, 5FC treatment induced Pdr1-dependent CDR1 upregulation. This is further consistent with our finding that 5FC treatment of C. glabrata induced petite mutants with dysfunctional mitochondria at high frequency. These petite mutants, similar to those induced by ethidium bromide (6, 17, 18), exhibit Pdr1-dependent CDR1 upregulation and, hence, reduced FLC susceptibility, revealing the likely mechanism behind 5FC-azole antagonism.
MATERIALS AND METHODS
Media, strains, and drugs.
The media used were RPMI (RPMI 1640 [Sigma-Aldrich], 0.165 M MOPS [morpholinepropanesulfonic acid] [pH 7.0], 2% dextrose) and YPD (1% yeast extract, 2% peptone, 2% dextrose). C. glabrata strains 66032 and 38326 were obtained from the American Type Culture Collection (Manassus, VA); all other strains were clinical isolates from diverse sources. Disruptants with pdr1Δ, cdr1Δ, and ade2Δ mutations in strain backgrounds 66032, BG2, and 200989 were previously described (5, 19). 5FC (Sigma-Aldrich), FLC (Pfizer), voriconazole (Pfizer), itraconazole (Jannsen), posaconazole (Merck), amphotericin B (Sigma-Aldrich), caspofungin (Merck), and terbinafine (Novartis) were dissolved in 50 or 100% dimethyl sulfoxide (diluted to ≤0.5% in all experiments) and stored at −20°C until use.
Broth microdilution assays.
Log-phase cultures in the media indicated below were diluted to 3 × 103 cells/ml and aliquoted to 6 tubes. 5FC was added to final concentrations of 0, 0.002, 0.004, 0.008, 0.016, and 0.032 μg/ml. Aliquots (200 μl in row A and 100 μl in row B through H) were distributed to wells of a 96-well plate. Antifungal (e.g., FLC) was added to well A at various concentrations and serially 2-fold diluted into wells B through G; well H served as control. Plates were incubated at 35°C, and the absorbance at 630 nm was read at 24 h. The MIC was defined as the concentration inhibiting growth by ≥80% relative to the growth of the FLC-free control.
RNA analysis.
Log-phase cultures in YPD (treated with 0.3 μg/ml 5FC for 0, 2, or 4 h) were aliquoted to microcentrifuge tubes, and the cells (5 × 107) pelleted and resuspended in 300 μl of 10 mM Tris, 10 mM EDTA, and 0.5% SDS. RNA was extracted by using hot SDS-phenol followed by ethanol precipitation as described previously (5), suspended in 50 μl nuclease-free water, and treated with RQ1 DNase (Promega) as recommended by the manufacturer. RNA (diluted to 50 ng/μl) was analyzed by quantitative reverse transcription (qRT)-PCR as described previously (20), using one-step qRT-PCR on the Stratagene Mx3000P QPCR system (Stratagene). Assays (25 μl) in triplicate contained 125 ng RNA, 0.6 μM each primer (CDR1F and CDR1R or ACT1F and ACT1R), 0.2 μM FAM-labeled probe, 12.5 μl Quanta one-step master mix (2×) (Quanta Biosciences), 6.5 μl water, and 0.5 μl Quanta Escript one-step reverse transcriptase. The PCR conditions consisted of an initial incubation at 50°C for 10 min and then 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 20 s and annealing/extension at 51°C for 1 min. Fluorescence acquisition was performed at the end of each cycle immediately following the annealing/extension step. Negative controls substituted water for RNA. CDR1 expression levels were normalized to ACT1 expression. The cycle threshold (CT) value of ACT1 was subtracted from that of CDR1 to obtain a ΔCT value, and expression relative to the wild-type expression was expressed as 2−ΔΔCT.
5FC induction and characterization of petite mutants.
YPD plates containing 1 or 8 μg/ml 5FC were spread with 1 × 107 cells of strains 66032 or BG2 and incubated for 3 days. Regions with no visible colonies were streaked for isolation on drug-free YPD, yielding a mixture of petite and normal-sized colonies. To assess mitochondrial function, ca. 1,000 cells of representative colonies in 3 μl were spotted in parallel on YPD and YP-glycerol (substituting 3% glycerol for the 2% dextrose) and the plates incubated for 2 to 3 days. Loss of mitochondrial DNA was assessed by PCR using COX1 primers 5′-AGCAACAATTTATGGAGGTTCT and 5′-GAATGAACAGCTGGTGGTGA (and ACT1F-ACT1R as a positive control), equivalent amounts of DNA (125 ng) prepared from log-phase cultures by bead beating and phenol-chloroform extraction, and 20 cycles of amplification with annealing at 56°C; products were analyzed by agarose gel electrophoresis and ethidium bromide staining.
To determine the frequency of petite induction, equivalent log-phase cultures (1 × 106 cells/ml) of strain 200989 ade2Δ (19) were incubated with 0, 1, or 5 μg/ml 5FC in YPD for 24 h at 35°C with aeration. Cultures were then equivalently diluted, spread on YPD plates, and incubated for an additional 3 days before red (wild-type mitochondria) and white (petite) colonies were counted.
RESULTS
5FC specifically antagonizes azole activity versus C. glabrata.
The interaction of FLC and 5FC was evaluated using checkerboard broth microdilution assays in RPMI medium with eight C. glabrata strains from diverse sources. The FLC MICs ranged from 2 to 32 μg/ml (median, 8 μg/ml). The 5FC MIC was 0.016 μg/ml for all strains. As shown in Fig. 1, in the presence of subinhibitory concentrations of 5FC, the FLC MIC increased in all eight strains. Specifically, in the presence of 0.008 μg/ml 5FC, the FLC MICs increased 4- to 16-fold (median, 8-fold). Conversely, subinhibitory concentrations of FLC had little or no effect (≤2-fold) on the 5FC MIC for all eight strains (data not shown).
Fig 1.
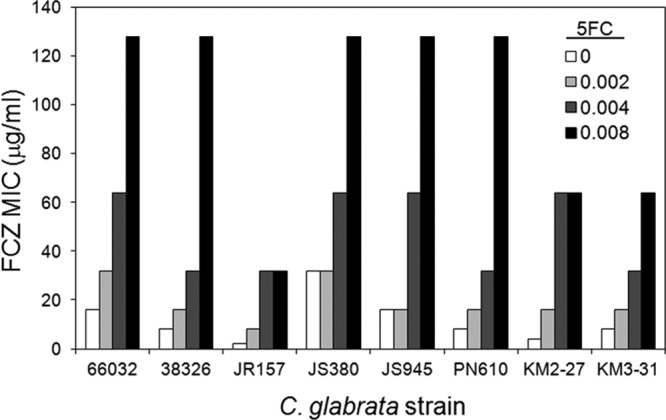
5FC antagonizes FLC activity versus 8 of 8 C. glabrata strains from diverse sources. Broth microdilution assays in RPMI medium were used to determine FLC MICs in the presence of the indicated concentrations of 5FC (μg/ml).
Further studies were conducted with representative strains. In YPD medium, 5FC exhibits substantially reduced activity compared to its activity in RPMI, presumably due to competition with endogenous cytosine. Nevertheless, FLC activity versus the two strains tested was similarly antagonized 4- to 16-fold at subinhibitory 5FC concentrations (0.5 to 2 μg/ml). Also, 5FC antagonism of azole activity versus the three strains tested extended to voriconazole (4- to 16-fold), itraconazole (2- to 8-fold), and posaconazole (2- to 4-fold). On the other hand, 5FC had additive or indifferent effects on the activity of the structurally distinct antifungals amphotericin B, caspofungin, and terbinafine (0.5-, 1-, and 1-fold changes in MIC, respectively).
5FC antagonism of FLC activity is mediated by Pdr1 and Cdr1.
To explore the basis for this 5FC-azole antagonism, we first tested mutants demonstrating resistance to one of these antifungals for cross-resistance to the other. We recently characterized 5FC-resistance-conferring mutations in C. glabrata (11). Representative mutants and their wild-type parent strain 66032 were tested for FLC susceptibility, and identical MICs were obtained (16 μg/ml). Similarly, 5FC activity was unaffected by CDR1 or PDR1 mutations known to modulate azole susceptibility (4, 5). Specifically, cdr1Δ and pdr1Δ disruptants are azole hypersusceptible (MICs of ≤8 μg/ml), while a pdr1-F15 gain-of-function mutant is azole resistant (MIC of >128 μg/ml); all three have unaltered 5FC susceptibility (0.016 μg/ml in RPMI) compared to that of the wild-type parent 66032. These results are consistent with a previous report that 5FC does not compete with FLC for transport (21).
Conversely, 5FC antagonism of FLC activity was Cdr1 and Pdr1 dependent. Specifically, the 8- to 16-fold increase in FLC MICs in the presence of subinhibitory 5FC observed for the parent strain was decreased to ≤2-fold in its pdr1Δ and cdr1Δ derivatives (Table 1). This effect was reproduced in both RPMI and YPD medium and in two different strain backgrounds.
Table 1.
5FC antagonism of FLC activity is PDR1 and CDR1 dependent
| Strain background | Genotype | Medium | MIC (μg/ml) of: |
|
|---|---|---|---|---|
| FLC | FLC + 5FCa | |||
| 66032 | Wild type | RPMI | 16 | 128 |
| pdr1Δ | 8 | 8 | ||
| cdr1Δ | 4 | 8 | ||
| 66032 | Wild type | YPD | 16 | 128 |
| pdr1Δ | 4 | 4 | ||
| cdr1Δ | 2 | 4 | ||
| BG2 | Wild type | YPD | 8 | 128 |
| pdr1Δ | 4 | 8 | ||
| cdr1Δ | 2 | 4 | ||
5FC concentrations were subinhibitory at 0.008 and 0.5 μg/ml in RPMI and YPD media, respectively.
5FC antagonism correlates with Pdr1-dependent induction of CDR1 expression.
Previous studies in our laboratory demonstrated that diverse compounds are capable of upregulating the expression of C. albicans multidrug transporter genes, resulting in antagonism of azole activity versus this yeast (22). We hypothesized that 5FC antagonism of azole activity versus C. glabrata is mediated by an analogous mechanism; specifically, activation of Pdr1 leading to CDR1 upregulation. To test this, CDR1 expression was compared in log-phase YPD cultures not treated or treated for 2 and 4 h with subinhibitory (0.3 μg/ml) 5FC. Indeed, 5FC treatment resulted in 6.4-fold CDR1 upregulation in wild-type strain 66032 (Table 2). In its pdr1Δ derivative, CDR1 expression was negligible both in the absence and presence of 5FC.
Table 2.
Pdr1-dependent CDR1 upregulation following 5FC treatment and in petite mutantsa
| Strain description | 5FC treatment time (h) | CDR1 expression (fold change ± SD) |
|---|---|---|
| Wild type | 0 | 1 |
| 2 | 4.0 ± 1.7 | |
| 4 | 6.4 ± 2.7 | |
| pdr1Δ | 0 | 0.11 ± 0.03 |
| 2 | 0.13 ± 0.04 | |
| 4 | 0.09 ± 0.08 | |
| Petite | 0 | 14 ± 5.3 |
| 2 | 16 ± 3.8 | |
| 4 | 8.4 ± 5.1 | |
| Petite pdr1Δ | 0 | 0.76 ± 0.85 |
| 2 | 0.36 ± 0.28 | |
| 4 | 0.53 ± 0.36 |
Log-phase cultures in YPD were treated with 0.3 μg/ml 5FC for 0, 2, or 4 h as indicated, and then RNA was extracted and analyzed for CDR1 expression (normalized to ACT1 and to results for untreated wild-type parent 66032).
5FC exposure induces petite mutants.
Since 5FC is not a substrate for Cdr1 transport, it was not obvious how 5FC treatment induced CDR1 expression, as shown above. During our studies of C. glabrata 5FC resistance (11), however, we noted that many of the cells representing background growth on 5FC-containing selection plates formed small colonies following transfer to drug-free plates. Their size suggested a petite (dysfunctional mitochondria) phenotype, which was confirmed by replica plating on medium containing fermentative (dextrose) or respiratory (glycerol) carbon sources (Fig. 2A). Furthermore, loss of mitochondrial DNA was demonstrated in a PCR assay targeting COX1 (CaglfMp04) (Fig. 2B). These observations are consistent with studies published in 1976 of 5FC-induced petite formation in Saccharomyces cerevisiae, a close relative of C. glabrata (23).
Fig 2.
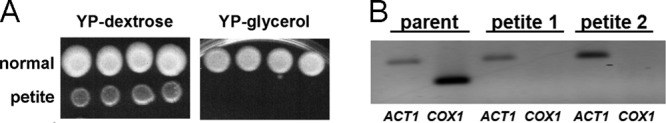
C. glabrata petite colonies induced by 5FC exposure have mitochondrial defects indicated by lack of growth on respiratory carbon source glycerol and absence of mitochondrially encoded COX1 DNA. YPD plates containing 1 or 8 μg/ml 5FC were spread with 1 × 107 cells of strains 66032 or BG2 and incubated for 3 days. Regions with no visible colonies were streaked for isolation on drug-free YPD, yielding an apparent mixture of petite and normal-sized colonies. (A) Approximately 1,000 cells of representative colonies in 3 μl were spotted in parallel on YP-dextrose (YPD) and YP-glycerol (substituting 3% glycerol for the 2% dextrose), and the plates incubated for 2 to 3 days before being photographed. (B) DNA was prepared from two representative petite colonies and their wild-type 66032 parent and used as the template in PCRs with ACT1 and COX1 primers (nuclear and mitochondrially encoded, respectively). Products were analyzed by agarose gel electrophoresis and ethidium bromide staining.
To confirm and quantitate this observation, we extended to C. glabrata a method developed in S. cerevisiae in which the red pigmentation associated with ade2 mutation is lost upon petite formation (24). A C. glabrata ade2Δ strain similarly forms red colonies (19), with a 0.4% (3 of 780) background frequency of readily distinguished white colonies following 24 h of incubation in drug-free YPD (see Fig. S1 in the supplemental material). This frequency increased to 8% (65 of 775) and 24% (185 of 765) (see Fig. S1) following incubation in 1 and 5 μg/ml 5FC, respectively, concentrations which had no detectable effect on growth under these conditions. Representative white colonies from these plates were confirmed as petites by using the replica plating assay.
5FC-induced petites exhibit CDR1 upregulation and reduced azole susceptibility.
It was previously shown that ethidium bromide-induced petite mutants of both C. glabrata and S. cerevisiae exhibit upregulated expression of the multidrug transporter gene CDR1 or, respectively, its ortholog PDR5 (6, 17, 18, 25). Furthermore, this upregulation is dependent on Pdr1 or, respectively, its paralog Pdr3. Similarly, a representative 5FC-induced petite mutant of C. glabrata exhibited pronounced CDR1 upregulation which was fully Pdr1 dependent (Table 2).
Consistent with CDR1 upregulation, broth microdilution assays in YPD medium of two representative 5FC-induced petite mutants of strain 66032 demonstrated 8- to 16-fold reduced susceptibilities to FLC, voriconazole, and posaconazole (MICs of 128, 4, and 4 μg/ml, respectively) but wild-type susceptibilities to amphotericin B, caspofungin, and 5FC itself (MICs of 0.5, 0.03, and 4 μg/ml, respectively). An equivalent susceptibility profile was obtained for the Pdr1 gain-of-function mutant F15, tested in parallel.
DISCUSSION
C. glabrata emerged in the azole era as a major cause of life-threatening fungal infections, potentially as a direct consequence of its intrinsically low susceptibility and high capacity for acquired resistance to this class of antifungals. C. glabrata is susceptible to echinocandins and amphotericin B; however, these agents require intravenous administration. Orally administered 5FC represents an attractive alternative to azoles, although its use has diminished in recent years due to concerns regarding resistance and the related concern of toxicity associated with the high doses often used to counter resistance. 5FC-azole combination therapy would in theory reduce the likelihood of resistance to either agent, but previous studies observed, albeit inconsistently, that 5FC-azole combinations are antagonistic in C. glabrata (12, 14, 15, 16). Here, we confirmed 5FC antagonism of azole activity and, furthermore, identified its likely mechanism.
5FC at a subinhibitory concentration antagonized FLC activity 4- to 16-fold versus all eight C. glabrata strains tested. It similarly antagonized the activity of other triazoles but had only indifferent effects on amphotericin B, caspofungin, and terbinafine activity. Since azole resistance in C. glabrata results from transcription factor Pdr1-dependent upregulation of multidrug transporter gene CDR1, we reasoned that 5FC antagonism might be similarly mediated. Indeed, 5FC-FLC antagonism was abrogated in pdr1Δ and cdr1Δ strains. In further support of this hypothesis, 5FC exposure induced CDR1 expression 6-fold, and this upregulation was Pdr1 dependent. In contrast to azoles, 5FC is not a Cdr1 substrate and so its activation of Pdr1 was unexpected. We observed, however, that 5FC exposure readily induced petite mutants, which exhibit Pdr1-dependent CDR1 upregulation. Thus, mitochondrial dysfunction resulting in Pdr1 activation is the likely basis for 5FC antagonism of azole activity versus C. glabrata.
Oliver and Williamson reported in 1976 that 5FC induced petite mutants of S. cerevisiae with high efficiency (23), and so its ability to do the same in the closely related yeast C. glabrata is, in retrospect, not unexpected. These authors further argued that the effect was mediated by 5-fluoro-modified RNA, rather than inhibition of dTMP synthesis. Regardless, these observations suggest that 5FC, as is the case with petite-phenotype-inducing ethidium bromide, preferentially inhibits mitochondrial versus nuclear replication. In support of this, PCR analysis of two 5FC-induced petite mutants showed loss of mitochondrially encoded COX1. It would be of interest to see whether specific inhibitors of mitochondrial function other than replication (e.g., respiration) similarly activate Pdr1 to lead to CDR1 upregulation. We anticipate that this will not be the case, since studies in S. cerevisiae have shown that a respiratory-deficient atp2Δ mutant did not exhibit PDR5 upregulation or multidrug resistance (25).
How mitochondrial dysfunction activates C. glabrata Pdr1 also remains to be clarified, although studies in S. cerevisiae suggest a potential mechanism. In S. cerevisiae petite mutants, PDR5 upregulation is mediated at least in part by the retrograde (mitochondrion-to-nucleus) signaling components Rtg1, a transcription factor, and Rtg2, a component of the SLIK histone acetyltransferase complex (25, 26). Disruption of the C. glabrata ortholog of RTG2 conferred modestly increased FLC susceptibility in a wild-type background (27); its effects in a petite background warrant examination.
In contrast to C. glabrata, for its distant relative C. albicans, several studies have reported that 5FC-azole combinations are additive or even synergistic (13, 14). This could reflect a reduced effect of 5FC on mitochondrial function in this yeast or, perhaps more likely, differences in retrograde signaling. Indeed, BLASTP analysis (unpublished data) revealed that C. albicans and related yeasts in the CTG clade (species in which CTG is translated as serine rather than leucine) lack an RTG2 homolog, in contrast to all other ascomycetes.
In conclusion, both microbiological and, now, molecular analyses argue against the clinical use of 5FC-azole combinations in the treatment of C. glabrata infection. On the other hand, these data provide further impetus for the development of Pdr1 inhibitors, since these would reverse not only azole resistance but also 5FC-azole antagonism in this challenging pathogen.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Cormack, K. Marr, P. Nyirjesy, J. Rex, and J. Sobel for strains.
This work was supported by NIH grant AI073794 to T.E.
Footnotes
Published ahead of print 26 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02394-12.
REFERENCES
- 1.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39:309–317 [DOI] [PubMed] [Google Scholar]
- 2.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20:133–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfaller MA, Diekema DJ, International Fungal Surveillance Participant Group 2004. Twelve years of fluconazole in clinical practice: global trends in species distribution and fluconazole susceptibility of bloodstream isolates of Candida. Clin. Microbiol. Infect. 10(Suppl 1):11–23 [DOI] [PubMed] [Google Scholar]
- 4.Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 48:3773–3781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61:704–722 [DOI] [PubMed] [Google Scholar]
- 6.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268. 10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pappas PG, Kauffman CA, Andes D, Benjamin DK, Calandra TF, Edwards JE, Filler SG, Fisher JF, Kullberg BJ, Ostrosky-Zeichner L, Reboli AC, Rex JH, Walsh TJ, Sobel JD, Infectious Diseases Society of America 2009. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 48:503–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfaller MA, Messer SA, Boyken L, Huynh H, Hollis RJ, Diekema DJ. 2002. In vitro activities of 5-fluorocytosine against 8,803 clinical isolates of Candida spp.: global assessment of primary resistance using National Committee for Clinical Laboratory Standards susceptibility testing methods. Antimicrob. Agents Chemother. 46:3518–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vermes A, Guchelaar HJ, Dankert J. 2000. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 46:171–179 [DOI] [PubMed] [Google Scholar]
- 11.Edlind TD, Katiyar SK. 2010. Mutational analysis of flucytosine resistance in Candida glabrata. Antimicrob. Agents Chemother. 54:4733–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alves IA, Bandeira LA, Mario DA, Denardi LB, Neves LV, Santurio JM, Alves SH. 2012. Effects of antifungal agents alone and in combination against Candida glabrata strains susceptible or resistant to fluconazole. Mycopathologia 174:215–221 [DOI] [PubMed] [Google Scholar]
- 13.Johnson MD, MacDougall C, Ostrosky-Zeichner L, Perfect JR, Rex JH. 2004. Combination antifungal therapy. Antimicrob. Agents Chemother. 48:693–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Te Dorsthorst DTA, Verweij PE, Meletiadis J, Bergervoet M, Punt NC, Meis JFGM, Mouton JW. 2002. In vitro interaction of flucytosine combined with amphotericin B or fluconazole against thirty-five yeast isolates determined by both the fractional inhibitory concentration index and the response surface approach. Antimicrob. Agents Chemother. 46:2982–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siau H, Kerridge D. 1998. The effect of antifungal drugs in combination on the growth of Candida glabrata in solid and liquid media. J. Antimicrob. Chemother. 41:357–366 [DOI] [PubMed] [Google Scholar]
- 16.Barchiesi F, Spreghini E, Maracci M, Fothergill AW, Baldassarri I, Rinaldi MG, Scalise G. 2004. In vitro activities of voriconazole in combination with three other antifungal agents against Candida glabrata. Antimicrob. Agents Chemother. 48:3317–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanglard D, Ischer F, Bille J. 2001. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45:1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brun S, Bergès T, Poupard P, Vauzelle-Moreau C, Renier G, Chabasse D, Bouchara JP. 2004. Mechanisms of azole resistance in petite mutants of Candida glabrata. Antimicrob. Agents Chemother. 48:1788–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edlind TD, Henry KW, Vermitsky JP, Edlind MP, Raj S, Katiyar SK. 2005. Promoter-dependent disruption of genes: simple, rapid, and specific PCR-based method with application to three different yeast. Curr. Genet. 48:117–125 [DOI] [PubMed] [Google Scholar]
- 20.Gygax SE, Vermitsky JP, Chadwick SG, Self MJ, Zimmerman JA, Mordechai E, Adelson ME, Trama JP. 2008. Antifungal resistance of Candida glabrata vaginal isolates and development of a quantitative reverse transcription-PCR-based azole susceptibility assay. Antimicrob. Agents Chemother. 52:3424–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mansfield BE, Oltean HN, Oliver BG, Hoot SJ, Leyde SE, Hedstrom L, White TC. 2010. Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 6:e1001126. 10.1371/journal.ppat.1001126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henry KW, Cruz MC, Katiyar SK, Edlind TD. 1999. Antagonism of azole activity against Candida albicans following induction of multidrug resistance genes by selected antimicrobial agents. Antimicrob. Agents Chemother. 43:1968–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliver SG, Williamson DH. 1976. The molecular events involved in the induction of petite yeast mutants by fluorinated pyrimidines. Mol. Gen. Genet. 146:253–259 [DOI] [PubMed] [Google Scholar]
- 24.Kim G, Sikder H, Singh KK. 2002. A colony color method identifies the vulnerability of mitochondria to oxidative damage. Mutagenesis 17:375–381 [DOI] [PubMed] [Google Scholar]
- 25.Hallstrom TC, Moye-Rowley WS. 2000. Multiple signals from dysfunctional mitochondria activate the pleiotropic drug resistance pathway in Saccharomyces cerevisiae. J. Biol. Chem. 275:37347–37356 [DOI] [PubMed] [Google Scholar]
- 26.Pray-Grant MG, Schieltz D, McMahon SJ, Wood JM, Kennedy EL, Cook RG, Workman JL, Yates JR, Grant PA. 2002. The novel SLIK histone acetyltransferase complex functions in the yeast retrograde response pathway. Mol. Cell. Biol. 22:8774–8786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaur R, Castaño I, Cormack BP. 2004. Functional genomic analysis of fluconazole susceptibility in the pathogenic yeast Candida glabrata: roles of calcium signaling and mitochondria. Antimicrob. Agents Chemother. 48:1600–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.