

Abstract
The spread of drug-resistant bacterial pathogens is a growing global concern and has prompted an effort to explore potential adjuvant and alternative therapies derived from nature's repertoire of bactericidal proteins and peptides. In humans, the airway surface liquid layer is a rich source of antibiotics, and lysozyme represents one of the most abundant and effective antimicrobial components of airway secretions. Human lysozyme is active against both Gram-positive and Gram-negative bacteria, acting through several mechanisms, including catalytic degradation of cell wall peptidoglycan and subsequent bacterial lysis. In the infected lung, however, lysozyme's dense cationic character can result in sequestration and inhibition by polyanions associated with airway inflammation. As a result, the efficacy of the native enzyme may be compromised in the infected and inflamed lung. To address this limitation, we previously constructed a charge-engineered variant of human lysozyme that was less prone to electrostatic-mediated inhibition in vitro. Here, we employ a murine model to show that this engineered enzyme is superior to wild-type human lysozyme as a treatment for mucoid Pseudomonas aeruginosa lung infections. The engineered enzyme effectively decreases the bacterial burden and reduces markers of inflammation and lung injury. Importantly, we found no evidence of acute toxicity or allergic hypersensitivity upon repeated administration of the engineered biotherapeutic. Thus, the charge-engineered lysozyme represents an interesting therapeutic candidate for P. aeruginosa lung infections.
INTRODUCTION
The spread of antibiotic resistance among bacterial pathogens represents a looming public health crisis (1). With a rise in multidrug-resistant bacteria and few new antimicrobials in the pipeline, there is a need to explore potential alternative and adjuvant therapies (1). Human lysozyme (hLYS) is a naturally occurring antimicrobial peptide found in a variety of tissues, cells, and secretions involved in the pathophysiology of lung infection, e.g., the airway surface liquid and cytoplasmic granules of neutrophils (2). It plays a key role in the innate immune response to infection, with levels rising in response to microbial invaders (3, 4). hLYS exerts its antimicrobial effect through catalytic hydrolysis of cell wall peptidoglycan (5) and muramidase-independent processes that have yet to be fully elucidated (6, 7). It has been shown to be effective against both Gram-positive and Gram-negative organisms, including Pseudomonas aeruginosa (8–10).
Several studies have examined lysozyme's potential as an exogenously administered biotherapeutic. Recently, Bhavsar et al. administered aerosolized recombinant hLYS as a treatment for P. aeruginosa lung infection in hamsters (11). They found that 2 h of treatment for 3 consecutive days decreased the bacterial burden in both bronchoalveolar lavage fluid (BALF) and lung homogenate. The enzyme treatment also decreased lung tissue inflammation, reduced BALF leukocytes and neutrophils, and decreased alveolar septal apoptosis (11). A follow-up study found that a single nebulized dose of coadministered hLYS and tobramycin decreased the lung and BALF bacterial burden and reduced markers of inflammation (12). They concluded that hLYS is an interesting therapeutic candidate for treatment of lung infections in humans.
While there is precedent for using inhaled lysozyme as an exogenously administered antibacterial, experimental evidence suggests that cationic antimicrobials such as lysozyme are sequestered by anionic biopolymers associated with inflammation. Moreover, it is thought that this electrostatic sequestration compromises antibacterial efficacy in the infected and inflamed lung (4, 13). We have previously shown that wild-type hLYS is inhibited in vitro by anionic biopolymers, and we have employed biomolecular engineering to remodel the enzyme's electrostatic potential field to mitigate this limitation. In brief, a library of charge-altered lysozyme variants was constructed by combinatorial mutagenesis of eight basic residues that possessed low-level evolutionary conservation. The library was screened, under inhibitory conditions, for lytic activity against Micrococcus luteus. The enzyme variant 2-3-7 retained wild-type levels of bactericidal activity toward M. luteus and P. aeruginosa strain PAO1, but it was found to exhibit far superior lytic activity in the presence of the inhibitory polyanions alginate, DNA, mucin, and F-actin. The details of the construction and in vitro characterization of 2-3-7 are reported elsewhere (14, 15).
In the current study, we employed a murine model of mucoid P. aeruginosa lung infection to assess the therapeutic potential of variant 2-3-7. The presence of alginate and extracellular DNA is a hallmark of chronic P. aeruginosa infection of the human airway (16, 17), and we have previously shown that these biopolymers do in fact accumulate in our mouse model of lung infection (14). Here, we describe a series of systematic studies that evaluate the in vivo toxicity and allergenicity, the antibacterial efficacy, and the anti-inflammatory properties of our engineered lysozyme. We conclude that exogenous administration of variant 2-3-7, which effectively evades inhibitory polyanions, provides a therapeutic advantage relative to wild-type hLYS.
MATERIALS AND METHODS
The protocol for animal infection and lysozyme administration was approved by the Institutional Animal Care and Use Committee of the University of Vermont (Burlington, VT), in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines. All surgeries were performed under pentobarbital anesthesia, and all efforts were made to minimize animal suffering. Wild-type hLYS and tobramycin were purchased from Sigma-Aldrich (St. Louis, MO), and the genetically engineered enzyme 2-3-7 was produced and purified as previously described (14). We employed P. aeruginosa strain FRD1, a mucoid clinical isolate (18) that exhibits antibiotic resistance under in vitro conditions relevant to lung infections (19–21).
In vitro quantitative culture.
For antipseudomonal assays, 25,000 CFU/ml of mid-log-phase P. aeruginosa strain FRD1 was mixed with 7.5 μg of purified enzyme in activity buffer (10% [vol/vol] Luria-Bertani broth [LB] in 10 mM potassium phosphate, pH 7.0) in a total reaction volume of 115 μl. Dilutions were plated after a 60-min incubation at 37°C, colonies were enumerated following overnight outgrowth, and results were compared to those for dilution plates sampled at time zero. Assays were performed in triplicate on each of two different days to yield biological replicates.
Pulmonary infection model and treatment regimen.
Overnight LB cultures of P. aeruginosa were pelleted, washed twice with phosphate-buffered saline (PBS; 137 mM NaCl, 2.6 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, pH 7.4), and resuspended to give 5 × 107 viable P. aeruginosa bacteria in 40 μl of PBS. The actual inoculum was determined by serial dilution of the input bacterial suspension on Pseudomonas isolation agar (Difco), followed by incubation at 37°C for 24 h. Adult male C57BL/6J mice (age, 8 to 12 weeks; Jackson Laboratories, Detroit, MI) were anesthetized briefly with isoflurane and inoculated with 40 μl (5 × 107 CFU) of P. aeruginosa via oropharyngeal aspiration. At 1 h postinfection, a second inoculation with either hLYS or 2-3-7 in 40 μl of PBS was administered in the same fashion. Enzyme doses were typically 100 μg, with the exception of the doses in the 2-3-7 dose-response study (1 μg, 10 μg, and 100 μg). Tobramycin, when applicable, was administered at the time of the second inoculation as an intraperitoneal injection of 75 μg in 200 μl PBS per mouse. Intraperitoneal injection is an accepted route for systemic drug delivery in rodent models.
BALF collection, cell count, and cell differential.
At 24 h postinfection, mice were anesthetized with intraperitoneal sodium pentobarbital, tracheas were cannulated, and BALF was collected using an instillation of 1 ml of cold PBS. The BALF was centrifuged, and mouse immune cells were enumerated using an Advia automated cell counter (Siemens, Berlin, Germany). The cell-free protein content of BALF was determined by the Bradford assay with bovine serum albumin as a standard. Cytokine levels in BALF were determined using Bio-Plex Pro assays according to the manufacturer's instructions (Bio-Rad, Hercules, CA).
Quantification of P. aeruginosa bacterial burden.
Once the BALF had been obtained as described above, lungs were excised and placed into 1 ml of cold PBS, followed by homogenization. Viable bacterial counts in the lung homogenate were determined by plating serial dilutions (100 μl) onto Pseudomonas isolation agar, followed by incubation at 37°C for 24 h.
Liver histology.
Livers were fixed in buffered formalin for 24 h, embedded in paraffin, sectioned, and stained with hematoxylin-eosin. Histological sections were reviewed by two independent observers blinded to treatment group. Representative images were obtained using an Olympus BX50 light microscope with an Optronics MagnaFire digital camera.
Toxicology model.
Adult male C57BL/6J mice (age, 8 to 12 weeks) were given 100 μg of enzyme 2-3-7 by oropharyngeal aspiration on one, two, or three consecutive days (for a total of one, two, or three doses). Control animals were treated with PBS. Replicate groups were sacrificed on day 4 and day 10. BALF was obtained as described above for determination of the white blood cell content. In addition, serum from anesthetized mice was collected by right heart puncture. On day 10, total IgG1 and IgE levels were determined by enzyme-linked immunosorbent assay using capture and detection antibodies, according to the manufacturer's instructions (BD Pharmingen, San Diego, CA).
Statistical analysis.
Results were analyzed using one-way analysis of variance (ANOVA) with Dunnett's post hoc comparison (22) to the PBS control. In all cases, statistical significance was assessed at an α level of 0.05.
RESULTS
Lysozyme in vitro activity toward P. aeruginosa strain FRD1.
In prior reports, we detailed the in vitro activity of engineered lysozyme 2-3-7, specifically highlighting its capacity to evade electrostatic inhibition by anionic biopolymers (14, 15). As a component of that work, we showed that 2-3-7 was equivalent to wild-type hLYS in quantitative culture experiments assessing bactericidal activity toward the nonmucoid P. aeruginosa strain PAO1. For the purpose of evaluating in vivo efficacy, we were primarily interested in P. aeruginosa strain FRD1, as this bacterium's mucoid phenotype has greater clinical relevance to chronic lung infections (23). Surprisingly, quantitative culture experiments with FRD1 showed the engineered enzyme to have reduced bactericidal activity compared to that of wild-type hLYS (Fig. 1). Because, however, the charge-engineered enzyme had been designed specifically for enhanced performance in the infected and inflamed lung environment, we continued to pursue in vivo studies, despite this unexpected preliminary result.
Fig 1.
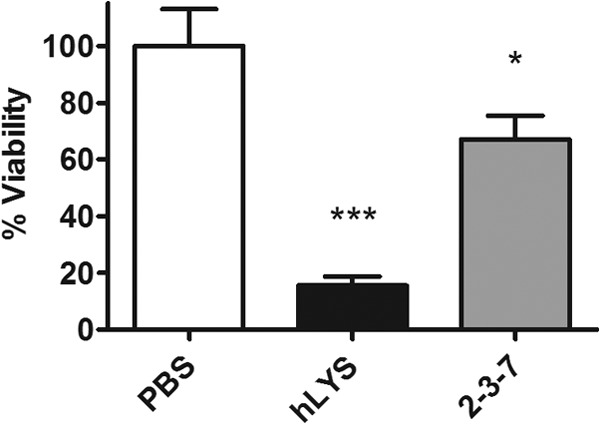
Lysozyme in vitro activity toward P. aeruginosa strain FRD1. Quantitative culture was performed on bacterial suspensions incubated for 1 h with 65 μg/ml enzyme or a PBS sham treatment. Viability was normalized to that for the PBS control. Mean values and SEMs are shown for triplicate measurements from two independent experiments (ANOVA, P < 0.0001). *, P < 0.05 compared with the PBS control group; ***, P < 0.001 compared with the PBS control group.
Repeated dosing of 2-3-7 is nontoxic and nonallergenic.
To assess the acute toxicity of the engineered enzyme 2-3-7, we examined lung and liver inflammation after repeated dosing (100 μg once per day for one, two, or three consecutive days). Compared to a PBS control, none of the dosing regimens caused significant increases in BALF immune cells, as measured on day 4 or day 10 (Fig. 2A and B). Similarly, there was no evidence of liver toxicity, as determined by histological sampling on day 4 or day 10 (Fig. 2C). To determine if 2-3-7 induced an allergic response, we quantified serum immunoglobulins on day 10 of the repeat dosing study. Again, none of the dosing regimens caused a significant increase in serum IgE or IgG1 levels (Fig. 3). Thus, there is no evidence that the engineered enzyme causes acute toxicity or allergic hypersensitivity during short-term repeated dosing.
Fig 2.

Charge-engineered lysozyme is nontoxic during repeat dosing. Groups of eight mice received 100 μg of enzyme daily. Experimental groups received a total of one, two, or three doses. Following treatment, four mice from each group were sacrificed for analysis on day 4 and four mice were sacrificed for analysis on day 10. (A) BALF immune cell concentration on day 4 (ANOVA, P = 0.377); (B) BALF immune cell concentration on day 10 (ANOVA, P = 0.192). The means ± SEMs are indicated, and there were no significant differences compared to the results for the PBS control. (C) Liver damage assessed by hematoxylin-eosin staining. Representative images are shown, and no differential pathology was observed between experimental groups and the PBS control. Magnifications, ×100.
Fig 3.
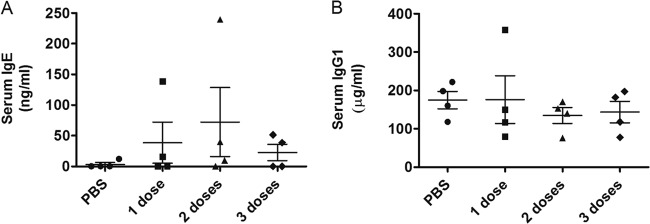
Charge-engineered lysozyme causes no allergic hypersensitivity during repeated dosing. Groups of four mice received 100 μg of enzyme daily for a total of one, two, or three doses. Serum antibody levels were quantified on day 10. (A) Total serum IgE (ANOVA, P = 0.535); (B) total serum IgG1 (ANOVA P = 0.810). The means ± SEMs are indicated, and no statistically significant differences were observed.
2-3-7 decreases P. aeruginosa burden and airway inflammation.
A dose-response study of enzyme 2-3-7 was conducted to assess the enzyme's therapeutic performance. The lungs of mice were infected with 5 × 107 P. aeruginosa FRD1 cells by oropharyngeal aspiration, and at 1 h postinfection escalating doses of 2-3-7 were administered by the same route. The numbers of bacterial CFU were quantified at 23 h posttreatment, and a dose-response effect showed that peak efficacy was approached between 10 and 100 μg (Fig. 4A). It bears noting that preliminary studies with nonmucoid P. aeruginosa strain PAO1 found a similar dose-response trend at lower enzyme concentrations (100 ng to 1 μg), although the results were not statistically significant at these low doses (one-way ANOVA, P = 0.188; data not shown).
Fig 4.
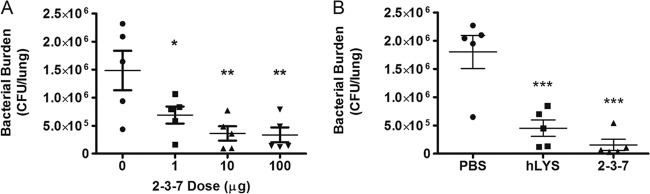
Efficacy of lysozyme treatments (n = 5 mice per group). (A) Reduction in the numbers of P. aeruginosa CFU with escalating doses of charge-engineered lysozyme 2-3-7 (ANOVA, P = 0.005); (B) numbers of lung P. aeruginosa CFU following treatment with PBS, 100 μg of wild-type hLYS, or 100 μg of variant 2-3-7 (ANOVA, P = 0.0002). The means ± SEMs are indicated. *, P < 0.05 compared with the PBS control group; **, P < 0.01 compared with the PBS control group; ***, P < 0.001 compared with the PBS control group.
Following the same protocol, the efficacy of variant 2-3-7 was compared head-to-head with that of wild-type hLYS at a 100-μg dosage. Mice treated with either enzyme showed a statistically significant reduction in P. aeruginosa lung burden compared to that achieved with a PBS sham treatment (Fig. 4B). While not significant at α equal to 0.05, mice treated with 2-3-7 trended toward lower bacterial burdens than mice treated with wild-type hLYS (P = 0.132, two-tailed t test). We also sought to determine if enzyme therapy reduced infection-associated lung inflammation and injury. Compared to the results for a PBS control group, treatment with 2-3-7 showed a strong trend toward fewer airway immune cells (Fig. 5A) and significantly less leakage of protein into the airway (Fig. 5B), whereas treatment with hLYS yielded no significant difference. Additionally, we found that treatment with 2-3-7 resulted in significantly reduced BALF concentrations of tumor necrosis factor alpha (TNF-α) (Fig. 5C) and keratinocyte-derived cytokine (KC) (Fig. 5D). In contrast, wild-type hLYS did not reduce lung cytokine levels to a statistically significant degree. In aggregate, these results provide evidence that engineered variant 2-3-7 outperforms wild-type hLYS in combating airway infections by mucoid P. aeruginosa.
Fig 5.
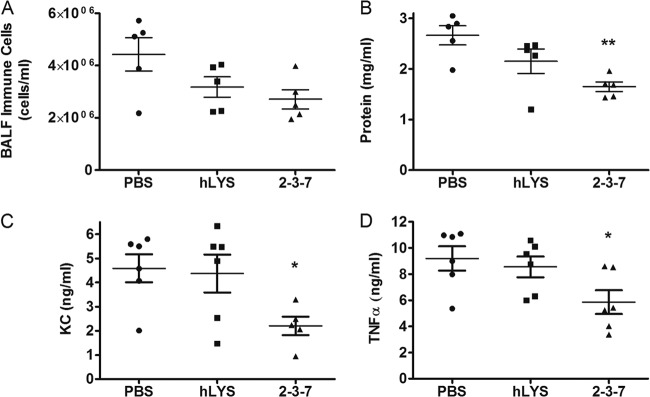
Assessment of lung inflammation and injury following P. aeruginosa infection and treatment with 100 μg wild-type or engineered lysozyme. (A) BALF immune cell concentration (n = 5 mice per group; ANOVA, P = 0.067); (B) BALF protein concentration as a surrogate of lung injury (n = 5 mice per group; ANOVA, P = 0.008); (C) BALF cytokine KC concentration as a marker of inflammation (n = 6 mice per group; ANOVA, P = 0.040); (D) BALF TNF-α concentration as a marker of inflammation (n = 6 mice per group; ANOVA, P = 0.039). The means ± SEMs are indicated. *, P < 0.05 compared with the PBS control group; **, P < 0.01 compared with the PBS control group.
2-3-7 is as effective as tobramycin in treating mucoid P. aeruginosa lung infection.
To determine if there was an added benefit to combining lysozyme treatment with standard antibacterial therapies, mice were infected with P. aeruginosa as described above and treated with either tobramycin, tobramycin combined with hLYS, or tobramycin combined with 2-3-7. A 75-μg intraperitoneal dose of tobramycin resulted in a significantly reduced P. aeruginosa burden compared to that achieved with a PBS sham treatment (ANOVA, P = 0.005). Surprisingly, when tobramycin was coadministered with hLYS, the combination therapy resulted in slightly higher mean bacterial counts than the PBS sham treatment (Fig. 6A). In contrast, the combination of variant 2-3-7 and tobramycin yielded a slightly lower bacterial burden than that achieved with tobramycin alone (average numbers of lung CFU = 90,636 versus 116,615, respectively). A similar pattern emerged in quantitative measures of airway inflammatory cells (Fig. 6B). We conclude that while 2-3-7 combined with tobramycin does not completely eradicate the model infection, neither does the engineered enzyme severely antagonize the aminoglycoside, as observed with wild-type hLYS.
Fig 6.
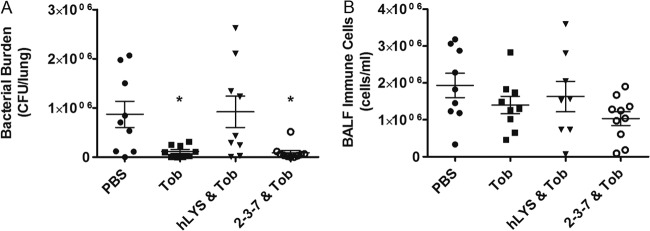
Efficacy of lysozymes combined with 75 μg intraperitoneal tobramycin (Tob) (n = 10 mice per group). One mouse each from the groups treated with PBS, tobramycin only, and hLYS and tobramycin exhibited acute morbidity and was removed prior to study completion. (A) Numbers of lung P. aeruginosa CFU following treatment with tobramycin alone, tobramycin combined with 100 μg wild-type hLYS, or tobramycin combined with 100 μg engineered variant 2-3-7 (ANOVA, P = 0.005); (B) analogous BALF immune cell concentrations (ANOVA, P = 0.170). The means ± SEMs are indicated. *, P < 0.05 compared with the PBS control group.
DISCUSSION
Colonization of the lower respiratory tract by various bacterial pathogens leads to a wide spectrum of pulmonary diseases, and as a whole, lung infections cause a greater global burden of disease than any other category, including HIV/AIDS, cancer, heart attacks, or malaria (24). Even in the United States, the evidence suggests that mortality rates from lung infections have failed to decline appreciably since the 1950s (25). One key contributor to this lack of progress is the emergence and rapid spread of antibiotic resistance among pathogenic bacteria (26, 27). Indeed, antibiotic resistance is a widespread health concern that often complicates ventilator-associated pneumonia (28, 29) and contributes to patient morbidity and mortality in cases of underlying chronic pulmonary disease, such as chronic obstructive pulmonary disease and cystic fibrosis (30–32). These observations are driving a medical imperative to develop new antimicrobial agents for bacterial infections of the lung.
Fueled in part by advances in recombinant protein production technologies (33, 34), endogenous antimicrobial proteins, such as lysozyme, have emerged as prospective therapeutic candidates. These agents are naturally occurring within humans, play important roles in innate immunity, exert broad-spectrum antimicrobial activity, and function via mechanisms distinct from those of traditional antibiotics. Additionally, relative to conventional chemotherapeutics, they are thought to have a lower propensity toward rapid induction of resistance phenotypes (35). While natural lysozymes advantageously possess bactericidal activity against both Gram-positive and Gram-negative bacteria, there exists considerable evidence that their efficacy within the infected and inflamed lung may be compromised by electrostatic interactions with disease-associated, anionic biopolymers (4, 13).
We have successfully redesigned the electrostatic potential field of hLYS, creating a charge-engineered variant that is less prone to electrostatic inhibition (15). A previous in vitro analysis had shown that variant 2-3-7 exerted wild-type or better bactericidal activity toward both Micrococcus luteus and P. aeruginosa strain PAO1 (14), but here, we found it to exhibit 2.6-fold lower in vitro activity toward the P. aeruginosa clinical isolate FRD1. We emphasize, however, that variant 2-3-7 was designed specifically for the infected and inflamed lung environment, and consistent with this objective, it outperformed wild-type hLYS in our murine model of FRD1 lung infection. Compared to a PBS sham treatment, mice treated with the engineered enzyme showed a significant reduction in measures of lung damage and inflammation, whereas treatment with the wild-type enzyme did not yield statistically significant results. In head-to-head comparisons of the engineered and wild-type enzymes, the engineered variant showed a strong trend toward a greater reduction in bacterial burden. Toxicology studies with the engineered enzyme showed no evidence of acute toxicity or allergic hypersensitivity. These observations suggest that the enhanced in vivo performance of the variant stems from a direct effect of the enzyme itself rather than some secondary effect of an inflammatory response elicited by the exogenous protein.
We also analyzed combination treatments of lysozyme and tobramycin, a frontline chemotherapeutic for cystic fibrosis patients. During acute respiratory exacerbations, patients are commonly given systemic tobramycin via intravenous administration (36), and as a single agent, tobramycin exhibits a 1-μg/ml in vitro MIC toward P. aeruginosa strain FRD1 (37). In our in vivo model, we found that a single 75-μg dose per mouse reduced the bacterial burden by 87%. We were surprised to find that the combination of wild-type hLYS and tobramycin failed to yield any reduction in the mean bacterial burden compared to that achieved with the PBS sham treatment. This suggests an antagonistic interaction in vivo, since each agent individually produced a 4- to 7-fold reduction in the numbers of lung CFU. In contrast, the combination of tobramycin with variant 2-3-7 yielded slightly greater efficacy than tobramycin alone, although the differences were not statistically significant. Thus, while the engineered enzyme fails to completely clear infections when combined with tobramycin, it also lacks the strong antagonistic effect observed with wild-type hLYS. This could prove to be an important point should clinical trials be pursued in the future. It bears noting that our results differ from those of Bhavsar et al., who showed that wild-type hLYS enhanced the efficacy of tobramycin in a hamster model of P. aeruginosa lung infection (12). We believe, however, that the discrepancies likely stem from differences in the two experimental systems: mouse versus hamster model, mucoid versus nonmucoid P. aeruginosa strain, and intraperitoneal versus inhalation routes of tobramycin administration.
In aggregate, we show here that our charge-engineered lysozyme variant decreases the infection-derived bacterial burden and lung inflammation without associated toxicity or hypersensitivity. These results suggest that the engineered enzyme is an interesting therapeutic candidate for treating P. aeruginosa infections of the airway, although studies with additional clinical isolates will be needed to rigorously assess the protein's broader therapeutic utility.
ACKNOWLEDGMENTS
This work was supported by an early career award (to K.E.G.) and a research grant (to K.E.G. and L.W.L.), both from the Wallace H. Coulter Foundation. C.C.T. and T.C.S. were supported in part by postdoctoral research awards from the Cystic Fibrosis Foundation.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Taubes G. 2008. The bacteria fight back. Science 321:356–361 [DOI] [PubMed] [Google Scholar]
- 2.Callewaert L, Michiels CW. 2010. Lysozymes in the animal kingdom. J. Biosci. 35:127–160 [DOI] [PubMed] [Google Scholar]
- 3.Sagel SD, Sontag MK, Accurso FJ. 2009. Relationship between antimicrobial proteins and airway inflammation and infection in cystic fibrosis. Pediatr. Pulmonol. 44:402–409 [DOI] [PubMed] [Google Scholar]
- 4.Sanders LK, Xian W, Guaqueta C, Strohman MJ, Vrasich CR, Luijten E, Wong GC. 2007. Control of electrostatic interactions between F-actin and genetically modified lysozyme in aqueous media. Proc. Natl. Acad. Sci. U. S. A. 104:15994–15999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koch AL. 2003. Bacterial wall as target for attack: past, present, and future research. Clin. Microbiol. Rev. 16:673–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim HR, Matsuzaki T, Aoki T. 2001. Genetic evidence that antibacterial activity of lysozyme is independent of its catalytic function. FEBS Lett. 506:27–32 [DOI] [PubMed] [Google Scholar]
- 7.Nash JA, Ballard TN, Weaver TE, Akinbi HT. 2006. The peptidoglycan-degrading property of lysozyme is not required for bactericidal activity in vivo. J. Immunol. 177:519–526 [DOI] [PubMed] [Google Scholar]
- 8.Akinbi HT, Epaud R, Bhatt H, Weaver TE. 2000. Bacterial killing is enhanced by expression of lysozyme in the lungs of transgenic mice. J. Immunol. 165:5760–5766 [DOI] [PubMed] [Google Scholar]
- 9.Markart P, Korfhagen TR, Weaver TE, Akinbi HT. 2004. Mouse lysozyme M is important in pulmonary host defense against Klebsiella pneumoniae infection. Am. J. Respir. Crit. Care Med. 169:454–458 [DOI] [PubMed] [Google Scholar]
- 10.Martinez JG, Waldon M, Huang Q, Alvarez S, Oren A, Sandoval N, Du M, Zhou F, Zenz A, Lohner K, Desharnais R, Porter E. 2009. Membrane-targeted synergistic activity of docosahexaenoic acid and lysozyme against Pseudomonas aeruginosa. Biochem. J. 419:193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhavsar T, Liu M, Hardej D, Liu X, Cantor J. 2010. Aerosolized recombinant human lysozyme ameliorates Pseudomonas aeruginosa-induced pneumonia in hamsters. Exp. Lung Res. 36:94–100 [DOI] [PubMed] [Google Scholar]
- 12.Bhavsar T, Liu M, Liu X, Cantor J. 2011. Aerosolized recombinant human lysozyme enhances the bactericidal effect of tobramycin in a hamster model of Pseudomonas aeruginosa-induced pneumonia. Exp. Lung Res. 37:536–541 [DOI] [PubMed] [Google Scholar]
- 13.Weiner DJ, Bucki R, Janmey PA. 2003. The antimicrobial activity of the cathelicidin LL37 is inhibited by F-actin bundles and restored by gelsolin. Am. J. Respir. Cell Mol. Biol. 28:738–745 [DOI] [PubMed] [Google Scholar]
- 14.Scanlon TC, Teneback CC, Gill A, Bement JL, Weiner JA, Lamppa JW, Leclair LW, Griswold KE. 2010. Enhanced antimicrobial activity of engineered human lysozyme. ACS Chem. Biol. 5:809–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill A, Scanlon TC, Osipovitch DC, Madden DR, Griswold KE. 2011. Crystal structure of a charge engineered human lysozyme having enhanced bactericidal activity. PLoS One 6:e16788. 10.1371/journal.pone.0016788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen SS, Kharazmi A, Espersen F, Hoiby N. 1990. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect. Immun. 58:3363–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulmer JS, Herzka A, Toy KJ, Baker DL, Dodge AH, Sinicropi D, Shak S, Lazarus RA. 1996. Engineering actin-resistant human DNase I for treatment of cystic fibrosis. Proc. Natl. Acad. Sci. U. S. A. 93:8225–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohman DE, Chakrabarty AM. 1981. Genetic mapping of chromosomal determinants for the production of the exopolysaccharide alginate in a Pseudomonas aeruginosa cystic fibrosis isolate. Infect. Immun. 33:142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alkawash MA, Soothill JS, Schiller NL. 2006. Alginate lyase enhances antibiotic killing of mucoid Pseudomonas aeruginosa in biofilms. APMIS 114:131–138 [DOI] [PubMed] [Google Scholar]
- 20.Walters MC, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. 2003. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 47:317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamppa JW, Griswold KE. 2013. Alginate lyase exhibits catalysis-independent biofilm dispersion and antibiotic synergy. Antimicrob. Agents Chemother. 57:137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunnett CW. 1964. New tables for multiple comparisons with a control. Biometrics 20:482–491 [Google Scholar]
- 23.Elkin S, Geddes D. 2003. Pseudomonal infection in cystic fibrosis: the battle continues. Expert Rev. Anti Infect. Ther. 1:609–618 [DOI] [PubMed] [Google Scholar]
- 24.Mizgerd JP. 2006. Lung infection—a public health priority. PLoS Med. 3:e76. 10.1371/journal.pmed.0030076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armstrong GL, Conn LA, Pinner RW. 1999. Trends in infectious disease mortality in the United States during the 20th century. JAMA 281:61–66 [DOI] [PubMed] [Google Scholar]
- 26.Furuya EY, Lowy FD. 2006. Antimicrobial-resistant bacteria in the community setting. Nat. Rev. Microbiol. 4:36–45 [DOI] [PubMed] [Google Scholar]
- 27.Mandell LA. 2005. Antimicrobial resistance and treatment of community-acquired pneumonia. Clin. Chest Med. 26:57–64 [DOI] [PubMed] [Google Scholar]
- 28.Craven DE. 2006. Preventing ventilator-associated pneumonia in adults: sowing seeds of change. Chest 130:251–260 [DOI] [PubMed] [Google Scholar]
- 29.Diaz E, Munoz E, Agbaht K, Rello J. 2007. Management of ventilator-associated pneumonia caused by multiresistant bacteria. Curr. Opin. Crit. Care 13:45–50 [DOI] [PubMed] [Google Scholar]
- 30.Macia MD, Blanquer D, Togores B, Sauleda J, Perez JL, Oliver A. 2005. Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother. 49:3382–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chernish RN, Aaron SD. 2003. Approach to resistant gram-negative bacterial pulmonary infections in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 9:509–515 [DOI] [PubMed] [Google Scholar]
- 32.Salunkhe P, Smart CH, Morgan JA, Panagea S, Walshaw MJ, Hart CA, Geffers R, Tummler B, Winstanley C. 2005. A cystic fibrosis epidemic strain of Pseudomonas aeruginosa displays enhanced virulence and antimicrobial resistance. J. Bacteriol. 187:4908–4920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hennegan K, Yang D, Nguyen D, Wu L, Goding J, Huang J, Guo F, Huang N, Watkins SC. 2005. Improvement of human lysozyme expression in transgenic rice grain by combining wheat (Triticum aestivum) puroindoline b and rice (Oryza sativa) Gt1 promoters and signal peptides. Transgenic Res. 14:583–592 [DOI] [PubMed] [Google Scholar]
- 34.Lamppa JW, Tanyos SA, Griswold KE. 2013. Engineering Escherichia coli for soluble expression and single step purification of active human lysozyme. J. Biotechnol. 164:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeaman MR, Yount NY. 2007. Unifying themes in host defence effector polypeptides. Nat. Rev. Microbiol. 5:727–740 [DOI] [PubMed] [Google Scholar]
- 36.Blumer JL, Saiman L, Konstan MW, Melnick D. 2005. The efficacy and safety of meropenem and tobramycin vs ceftazidime and tobramycin in the treatment of acute pulmonary exacerbations in patients with cystic fibrosis. Chest 128:2336–2346 [DOI] [PubMed] [Google Scholar]
- 37.Yu Q, Griffin EF, Moreau-Marquis S, Schwartzman JD, Stanton BA, O'Toole GA. 2012. In vitro evaluation of tobramycin and aztreonam versus Pseudomonas aeruginosa biofilms on cystic fibrosis-derived human airway epithelial cells. J. Antimicrob. Chemother. 67:2673–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]