

Abstract
In vitro and in vivo activities against Trypanosoma cruzi were evaluated for two sesquiterpene lactones: psilostachyin A and cynaropicrin. Cynaropicrin had previously been shown to potently inhibit African trypanosomes in vivo, and psilostachyin A had been reported to show in vivo effects against T. cruzi, albeit in another test design. In vitro data showed that cynaropicrin was more effective than psilostachyin A. Ultrastructural alterations induced by cynaropicrin included shedding events, detachment of large portions of the plasma membrane, and vesicular bodies and large vacuoles containing membranous structures, suggestive of parasite autophagy. Acute toxicity studies showed that one of two mice died at a cynaropicrin dose of 400 mg/kg of body weight given intraperitoneally (i.p.). Although no major plasma biochemical alterations could be detected, histopathology demonstrated that the liver was the most affected organ in cynaropicrin-treated animals. Although cynaropicrin was as effective as benznidazole against trypomastigotes in vitro, the treatment (once or twice a day) of T. cruzi-infected mice (up to 50 mg/kg/day cynaropicrin) did not suppress parasitemia or protect against mortality induced by the Y and Colombiana strains. Psilostachyin A (0.5 to 50 mg/kg/day given once a day) was not effective in the acute model of T. cruzi infection (Y strain), reaching 100% animal mortality. Our data demonstrate that although it is very promising against African trypanosomes, cynaropicrin does not show efficacy compared to benznidazole in acute mouse models of T. cruzi infection.
INTRODUCTION
American trypanosomiasis, also known as Chagas disease (CD), is a neglected disease caused by the intracellular protozoan Trypanosoma cruzi. Despite a large reduction of new acute cases in recent years due to vectorial transmission control policies, other routes of infection, including vertical transmission (mother to fetus) and ingestion of contaminated food and drinks (1), are still a concern. T. cruzi is restricted to Latin America, where about 10 million people are infected and 25 to 90 million are at risk of acquiring the infection (2). According to the Pan American Health Organization (PAHO) and the World Health Organization (WHO), CD causes between 10,000 and 14,000 deaths per year (3). Currently, only two drugs, benznidazole (Bz) and nifurtimox, are available for chagasic patients (4). However, the poor tolerance to and limited efficacy of these drugs, especially in the later stage of the chronic phase, make the search for novel compounds and therapeutic strategies ever more important as a means to offer alternative therapies to patients refractory to both these nitro derivatives (5).
Natural products are an excellent source of active compounds. In fact, despite them comprising <1 percent of known chemical compounds (DNP 2013 [http://dnp.chemnetbase.com/tour/]; Reaxys 2013 [https://www.reaxys.com/documentation/about_query.htm]), they have supplied or inspired more than half of all new drugs introduced to the market in the last 3 decades (6). The largest known group of natural products are the sesquiterpenes, of which about 10,000 have been described (7, 8). About 6,000 of them are sesquiterpene lactones (STLs), meaning that they possess at least one lactone group (7). STLs are a chemotaxonomic marker of the Asteraceae, the largest family of flowering plants, with about 24,000 species (http://tolweb.org/Asteraceae/20780). STLs have shown pharmacological effects, including anti-inflammatory, antitumor, and antimicrobial effects, in a large number of biological test systems (7–12). Due to notable activities against different protozoan human pathogens, such as Leishmania spp. (13–15), Plasmodium falciparum (16, 17), Trypanosoma brucei rhodesiense (12–18), and even T. cruzi (19, 20), further exploration of the trypanocidal effects of selected STLs by use of in vivo assays appeared worthwhile.
Zimmermann et al. (12) recently described that cynaropicrin, an STL found in artichokes (Cynara scolymus L.) and some species of cornflowers (Centaurea spp.), potently inhibits T. b. rhodesiense in vivo in an acute mouse model. A subsequent study demonstrated that the potent antitrypanosomal effects were due to the formation of cynaropicrin Michael adducts with trypanothione, which the trypanosomes depend on for redox regulation, as well as due to inhibition of ornithine decarboxylase (S. Zimmermann, M. Oufir, A. Leroux, L. Krauth-Siegel, K. Becker, R. Brun, M. Hamburger, and M. Adams, submitted for publication). Another STL, psilostachyin A from Ambrosia tenuifolia, was recently reported to have in vivo effects against T. cruzi (21). The aim of this study was to investigate cynaropicrin and psilostachyin A in highly stringent in vitro and in vivo experimental models of T. cruzi infection, with the goal of identifying novel candidates for possible future alternative therapies for CD.
MATERIALS AND METHODS
Compounds.
Cynaropicrin was isolated from Cynara scolymus L. (Asteraceae) as described by Zimmermann et al. (12). Psilostachyin A was isolated from Ambrosia tenuifolia Spreng. (Asteraceae). The structures of the compounds are shown in Fig. 1. The purity of both was determined by proton nuclear magnetic resonance (NMR) spectroscopy and was higher than 95%. Benznidazole (Bz; Laboratório Farmacêutico do Estado de Pernambuco [LAFEPE], Brazil) was used as a reference drug (22). Stock solutions of the compounds (1 mg/ml) were prepared in dimethyl sulfoxide (DMSO), and fresh final solutions were prepared with the DMSO concentration never exceeding 0.6%, which is not toxic for both parasites and mammalian cells. For in vivo studies, a stock solution of each compound was first prepared in DMSO and then diluted using sterile distilled water. The final concentration of DMSO never exceeded 10%, a concentration which is well tolerated by mice (22).
Fig 1.
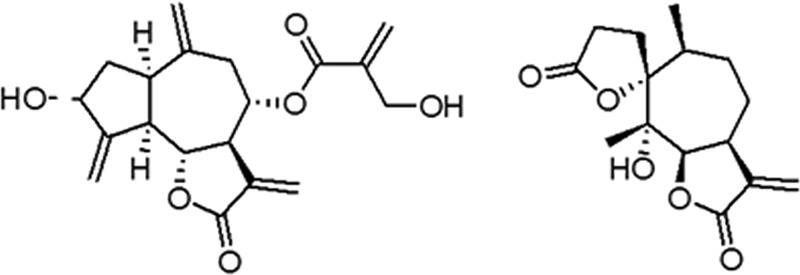
Structures of cynaropicrin and psilostachyin A.
Structure elucidation NMR.
NMR data were measured at room temperature on a Bruker Avance III 500-MHz spectrometer (Bruker, Fällanden, Switzerland). 1H and 13C experiments were performed as previously described (23). Topspin 2.0 was used as software for data processing and evaluation. The chemical shifts of cynaropicrin were 1H-NMR (500 MHz, CDCl3) δ 1.73 (1H, ddd, J = 10.9, 10.8, 9.8 Hz, H-2b), 2.2 (1H, ddd, J = 13.9, 6.9, 6.9 Hz, H-2a), 2.4 (1H, dd, J = 14.5, 3.8 Hz, H-9a), 2.73 (1H, dd, J = 14.5, 5.0 Hz, H-9b), 2.8 (1H, dd, J = 10.2, 9.0 Hz, H-5), 2.94 (1H, dd, J = 8.1, 8.0, 8.0, H-1), 3.17 (1H, dddd, J = 9.3, 9.3, 3.0, 3.0, H-7), 4.25 (1H, dd, J = 10.5, 9.1 Hz, H-pre6), 4.36 (2H, s, H-4′), 4.54 (1H, ttt, J = 7.2, 1.9, 1.9 Hz, H-3), 4.95 (1H, d, J = 1.0 Hz, H-14b), 5.15 (2H, m, H-8 and H-14a), 5.35 (1H, t, J = 1.7, H-15b), 5.47 (1H, t, J = 1.8 Hz, H-15a), 5.59 (1H, d, J = 3.1 Hz, H-13b), 5.92 (1H, d, J = 0.9 Hz, H-3b′), 6.18 (1H, d, J = 3.4 Hz, H-13a), 6.3 (1H, s, H-3a′), as reported previously (24).
The chemical shifts of psilostachyin C were as follows: 1H NMR (500 MHz, CDCl3), δ 6.23 (d, J = 3.6 Hz, 1H), 5.52 (d, J = 3.2 Hz, 1H), 4.92 (d, J = 9.6, Hz 1H), 3.37 (dddd, J = 12.7, 9.6, 6.7, and 3.3 Hz, 1H), 1.19 (s, 3H), 1.00 (d, J = 7.4 Hz, 3H) (25); and 13C NMR (125 MHz, CDCl3), δ 177.40, 169.72, 138.92, 121.87, 93.6, 83.33, 79.3, 41.62, 40.20, 30.43, 27.30, 26.91, 24.27, 15.03, 21.71, as reported previously (26).
Cell cultures.
Primary cultures of cardiac cells (CM) were obtained as reported previously (27). The cultures were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum (FBS), 2.5 mM CaCl2, 1 mM l-glutamine, and 2% chicken embryo extract. Cell cultures were maintained at 37°C in an atmosphere of 5% CO2 and air, and assays were run at least three times in duplicate.
Parasites.
The Y and Colombiana strains of Trypanosoma cruzi were used throughout the experiments. Bloodstream trypomastigotes (BT) were harvested from T. cruzi-infected Swiss mice by heart puncture on the day of parasitemia peak (27). Intracellular amastigotes lodged within cardiac cell cultures were employed as reported previously (28).
In vitro cytotoxicity assays.
Uninfected cardiac cultures were incubated for 24 and 48 h at 37°C in the presence or absence of each compound diluted in DMEM. The cardiac cells' morphology and spontaneous contractibility were evaluated by light microscopy. Cell death rates were measured by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; Sigma-Aldrich] colorimetric assay (29). The absorbance was measured on a spectrophotometer (VersaMax tunable microplate reader; Molecular Devices), which allowed for determination of the LC50 (compound concentration that reduces 50% of cellular viability).
Trypanocidal analysis.
BT were incubated at 37°C for 24 h in the presence of increasing nontoxic concentrations of the tested compounds diluted in RPMI 1640 medium (Sigma-Aldrich) supplemented with 5% fetal bovine serum. Death rates were determined by light microscopy through direct quantification of numbers of live parasites in a Neubauer chamber, and the EC50 (drug concentration that reduces 50% of the number of treated parasites) was calculated. For analysis of the effect against intracellular amastigotes, the infected cultures were washed to remove free parasites after 24 h of parasite-host cell interaction (10:1 parasite/CM ratio) and then were incubated for another 48 h with increasing but nontoxic doses of the test compounds. CM were maintained at 37°C under a 5% CO2 atmosphere, and the medium was replaced every 24 h. Untreated and treated infected CM were then fixed and stained with Giemsa solution, and the mean numbers of infected host cells and parasites per infected cell were scored as reported previously (30). As light microscopy of untreated T. cruzi-infected cell cultures allows the identification of parasites through their typical nuclei and KDNA structures, only intracellular forms that displayed these characteristic cellular elements were considered live/viable parasites (28). Control samples included parasites and infected cell cultures incubated only with vehicle solution (DMSO; final concentration not exceeding 0.6%). The drug activity was estimated by calculating the infection index (percentage of infected cells times average number of intracellular amastigotes per infected host cell). All in vitro assays were run at least twice, in duplicate.
TEM analysis.
BT that were treated or not for 2 h at 37°C with drug at the corresponding EC50 were fixed for 60 min at 4°C with 2.5% glutaraldehyde and 2.5 mM CaCl2 in 0.1 M cacodylate buffer, pH 7.2, and postfixed for 1 h at 4°C with 1% OsO4, 0.8% potassium ferricyanide, and 2.5 mM CaCl2, using the same buffer. Control samples were created using parasites incubated only with vehicle solution (DMSO; final concentration not exceeding 0.6%). Samples were routinely processed for transmission electron microscopy (TEM) and examined in a Zeiss EM10C electron microscope (Zeiss, Oberkochen, Germany) (31).
Acute toxicity for mice.
The maximum tolerated dose (MTD) was evaluated using female Swiss-Webster mice (20 to 23 g). On day 1, one female mouse and one male mouse were treated with each compound intraperitoneally (i.p.) or orally (p.o.) at doses ranging from 25 to 400 mg/kg of body weight (23). Mice were then inspected for toxic and subtoxic symptoms according to Organisation for Economic Co-operation and Development (OECD) guidelines. Forty-eight hours after compound injection, the MTD values were determined based on animal survival rates and behavior alterations (32).
Mouse infection and treatment schemes.
Male Swiss mice were obtained from the Fundação Oswaldo Cruz (Fiocruz) animal facilities (Rio de Janeiro, Brazil). Mice were housed at a maximum of 8 per cage and kept in a conventional room at 20 to 24°C under a 12-h–12-h light-dark cycle. The animals were provided with sterilized water and chow ad libitum. Infection was performed by i.p. injection of 104 (Y strain) or 5 × 103 (Colombiana strain) bloodstream trypomastigotes (22). The animals (18 to 21 g) were divided into the following groups (at least five mice per group): uninfected (noninfected and untreated), untreated (infected with T. cruzi but treated only with vehicle), and treated (infected and treated i.p. with 0.5 to 50 mg/kg/day compound or 100 mg/kg/day benznidazole). Mice received 0.1 ml (i.p.) at 5 and 8 days postinfection (dpi), or at 11, 12, and 13 dpi for the dose of 25 mg/kg, twice a day (b.i.d.). For Bz treatment, infected mice received a 0.2-ml oral dose (gavage) following the same therapeutic scheme as that described above.
Parasitemia, mortality rates, and ponderal curve analysis.
Parasitemia was checked individually by direct microscopic counting of parasites in 5 μl of blood, as described before (33). At 7, 14, 21, 28, and 38 dpi, body weight was recorded and the different mouse groups compared. Mortality was checked daily until 30 days posttreatment and is expressed as the percent cumulative mortality (22).
Biochemical analysis.
Forty-eight hours after compound administration, biochemical analysis of the plasmatic levels of calcium, creatine kinase, phosphorus, blood urea nitrogen, and alanine aminotransferase (ALT) was performed with a CECAL/Fiocruz platform using Vitros 250 (Ortho Clinical-Johnson & Johnson), as reported previously (32).
Histopathological analysis.
For acute toxicity analysis, mouse tissues (heart, spleen, liver, and kidneys) were removed after 48 h of cynaropicrin administration (25 to 400 mg/kg), cut longitudinally, rinsed in ice-cold phosphate-buffered saline (PBS), and fixed in Millonig-Rosman solution (10% formaldehyde in PBS). The tissues were dehydrated and embedded in paraffin. Sections (3 μm) were stained by routine hematoxylin-eosin staining and analyzed by light microscopy as reported previously (22).
Ethics.
All procedures were carried out in accordance with the guidelines established by the Fiocruz Committee of Ethics for the Use of Animals (CEUA 0028/09).
Statistical analysis.
Statistical analysis was performed by analysis of variance (ANOVA) as reported previously (28).
RESULTS
The activities of both STLs were evaluated in vitro against BT and intracellular amastigotes (Y strain), which are the relevant parasite forms for mammalian infection. Psilostachyin A showed only low trypanocidal activity, exhibiting an EC50 at 24 h (EC50/24 h) of 33 ± 1 μg/ml against bloodstream trypomastigotes (Table 1). The bloodstream forms of T. cruzi were more susceptible to cynaropicrin, with an EC50/24 h value of 1 ± 0.2 μg/ml, an activity quite similar to that of the positive control, Bz. Against intracellular proliferative forms, cynaropicrin was moderately effective, exhibiting an EC50/48 h of >0.75 μg/ml (Table 1). Cytotoxicity analysis of uninfected cardiac cultures showed that cynaropicrin was also the more toxic compound against mammalian cells, showing LC50s of >6 μg/ml (data not presented), and thus a low selectivity index (SI) of 6 to 8 (Table 1).
Table 1.
Trypanocidal effects of sesquiterpene lactones and benznidazole against T. cruzi (Y strain)a
| Compound | BT (24 h) |
Intracellular parasites (48 h) |
||
|---|---|---|---|---|
| EC50 (μg/ml) | SI | II (μg/ml) | SI | |
| Cynaropicrin | 1 ± 0.2b | 5.8 | >0.75 | 8 |
| Psilostachyin A | 33 ± 1c | 3 | ND | ND |
| Bz | 3.3 ± 0.5b,c | >78 | 0.9 ± 0.4 | >274 |
The activities of the compounds against BT and intracellular parasites were evaluated during their incubation at 37°C for 24 h and 48 h, respectively. All assays were run at least two times, in duplicate. EC50, compound concentration that reduces the number of parasites by 50%; II, infection index (percentage of infected cells times average number of intracellular amastigotes per infected host cell); SI, selectivity index (LC50/EC50 ratio for BT and intracellular parasites, calculated on LC50s from 24 and 48 h of incubation at 37°C, respectively); ND, not determined.
Statistical evaluation by ANOVA showed statistical significance (P = 0.0003).
Statistical evaluation by ANOVA showed statistical significance (P = 0.009).
Ultrastructural analysis of cynaropicrin-treated BT with a test concentration equaling the EC50/24 h, but for only 2 h of incubation in order to evaluate initial alterations induced by cynaropicrin, demonstrated that while untreated parasites exhibited normal morphology for the mitochondrion, nucleus, endoplasmic reticulum, and kinetoplast (Fig. 2A and B), cynaropicrin-treated BT showed intense intracellular vacuolization, with a great number of membrane blebs and with shedding of the intracellular contents, alongside the occurrence of large multivesicular bodies (Fig. 2C to F).
Fig 2.

Transmission electron microscopy analysis of cynaropicrin's effects on bloodstream trypomastigotes. BT were left untreated (A and B) or exposed to this STL (EC50/24 h) for 2 h (C to F). Untreated parasites displayed typical morphology, while cynaropicrin-treated parasites showed vacuolization (*), swelling of the mitochondrion and endoplasmic reticulum (white arrows), and plasma membrane shedding (black arrows). M, mitochondrion; G, Golgi complex; N, nucleus.
In the next step, in vivo assays were conducted to explore some aspects of acute toxicity, aiming to determine the MTD values. Escalating dose schemes were used in Swiss female mice (one per dose, from 25 mg/kg up to 400 mg/kg) treated by different routes (i.p. and p.o.). Forty-eight hours after application of cynaropicrin by different routes (i.p. and p.o.), no major difference was observed after p.o. application (Table 2). On the other hand, a higher tested dose of cynaropicrin (400 mg/kg) resulted in the death of one of two animals after i.p. application (Table 2). Acute toxicity assays using psilostachyin A did not show adverse effects until 48 h of treatment (Table 2). However, due to the compound solubility limitation, only doses of ≤100 mg/kg could be evaluated.
Table 2.
Acute toxicity analysis of Swiss male mice (20 to 23 g) with escalating doses of cynaropicrin and psilostachyin A by the i.p. and p.o. routesa
| Compound | Route of administration | Toxicity of dose (mg/kg) |
MTD (mg/kg) | ||||
|---|---|---|---|---|---|---|---|
| 25 | 50 | 100 | 200 | 400 | |||
| Cynaropicrin | i.p. | NDE | NDE | NDE | NDE | Deathb | 200 |
| p.o. | NDE | NDE | NDE | NDE | NDE | 400 | |
| Psilostachyin A | i.p. | NDE | NDE | NDE | ND | ND | 100 |
NDE, no detectable effect; MTD, maximum tolerated dose; ND, not done. All assays were run at least two times.
Death of one mouse inoculated with dose of 400 mg/kg.
Biochemical analysis of plasmatic proteins showed only a slight, dose-dependent but non-statistically significant (P ≥ 0.05) increase in alanine aminotransferase levels in mice exposed to cynaropicrin treatment by the i.p. route (Table 3), suggestive of mild hepatic damage. Histopathological analyses of liver, kidney, heart, and spleen samples collected after 48 h of cynaropicrin administration showed that the most frequent alterations were in the liver and spleen samples from treated mice, with the former being the most reactive organ. Cynaropicrin caused dose-dependent liver alterations (especially at doses of ≥100 mg/kg), including (i) Kupffer cell hyperplasia, (ii) vascular congestion, (iii) hepatocyte microvacuolization, (iv) inflammatory infiltrates, (v) hepatocyte regeneration, and (vi) mild focal hepatic necrosis (Fig. 3B and C). Mice treated with vehicle (DMSO) only presented little reactivity, displaying low-intensity hyperplasic activity of Kupffer cells without increased degeneration and/or regeneration aspects (Fig. 3A). Also, although no major histopathological alterations were found in the spleens of control (vehicle) group mice, a mild splenic reactivity with white pulp activation and an increased number of megakaryocytes were observed in cynaropicrin-treated mice (data not shown). No significant changes in heart and kidney morphology were noticed in any studied animals (data not shown).
Table 3.
Plasma biochemical analysis of female mice after 48 h of treatment with cynaropicrina
| Parameter | Route of cynaropicrin administration | Value for cynaropicrin dose (mg/kg) |
|||||
|---|---|---|---|---|---|---|---|
| 0 | 25 | 50 | 100 | 200 | 400 | ||
| Ca | i.p. | 10 ± 0.3 | 11 ± 0.07 | 11 ± 0.2 | 11 ± 0.07 | 11 ± 0.7 | 11b |
| p.o. | 11 ± 0.9 | 11 ± 1.3 | 11 ± 0.7 | 11 ± 0 | 11 ± 0.4 | 11 ± 0.3 | |
| CK | i.p. | 81 ± 23 | 100 ± 44 | 64 ± 36 | 72 ± 22 | 116 ± 30 | 82b |
| p.o. | 110 ± 17 | 95 ± 32 | 55 ± 30 | 89 ± 3 | 54 ± 40 | 128 ± 36 | |
| P | i.p. | 9 ± 0.7 | 10 ± 0.9 | 9 ± 1 | 8 ± 0.4 | 9 ± 2 | 11b |
| p.o. | 9 ± 0.3 | 9 ± 0.6 | 8 ± 0.3 | 9 ± 1 | 8 ± 0.07 | 9 ± 0.7 | |
| BUN | i.p. | 31 ± 13 | 26 ± 5 | 30 ± 3 | 26 ± 17 | 16 ± 7 | 20b |
| p.o. | 34 ± 4 | 29 ± 0.7 | 39 ± 10 | 28 ± 8 | 39 ± 9 | 36 ± 8 | |
| ALT | i.p. | 40 ± 5 | 37 ± 5 | 35 ± 6 | 40 ± 6 | 63 ± 35 | 86b |
| p.o. | 66 ± 24 | 51 ± 13 | 37 ± 1 | 38 ± 0 | 43 ± 5 | 40 ± 6 | |
Data are means ± standard deviations for two independent assays. P, phosphorus; CK, creatine kinase; Ca, calcium; BUN, blood urea nitrogen; ALT, alanine aminotransferase. The following reference values (mg/kg) (CECAL/Fiocruz) were used: P, 6.1 to 10.1; CK, 68 to 107; Ca, 5.9 to 9.4; BUN, 18 to 29; and ALT, up to 132.
Data from one mouse.
Fig 3.
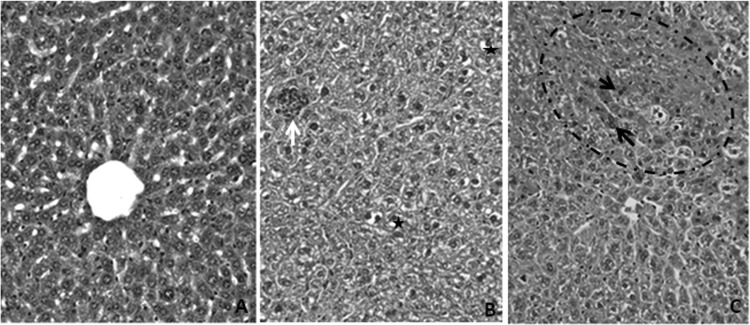
Histopathologic analysis of untreated female mice (A) and female mice treated with 200 (B)- and 400 (C)-mg/kg doses of cynaropicrin. (A) Liver section from an untreated animal, stained with hematoxylin-eosin and showing characteristic morphology. (B) Liver section from a treated mouse, exhibiting an inflammatory infiltrate (white arrow) in addition to the occurrence of hepatocyte vacuolization (black stars). (C) Liver regeneration area (circle) with binucleated cells (black arrows). Original magnification, ×400.
Next, the efficacy of the two sesquiterpene lactones was analyzed in vivo by employing different T. cruzi acute infection models (Y and Colombiana strains). Psilostachyin A was administered at 5 (onset) and 8 (peak of parasitemia) dpi, using 0.5, 5, and 50 mg/kg/day (i.p.), and Bz was administered at 50 mg/kg/day (p.o.). While this STL displayed reductions in parasitemia levels of about 40 to 70% for doses of 0.5 to 50 mg/kg, Bz administration resulted in a 75% parasitemia decrease (Fig. 4A). However, none of the doses of psilostachyin A protected against animal mortality, while Bz resulted in 80% survival (Fig. 4B).
Fig 4.

In vivo effects of psilostachyin A (A and B) and cynaropicrin (C to F) in acute mouse models of infection using different T. cruzi strains: Y (A to D) and Colombiana (E and F). Parasitemia (A, C, and E) and cumulative mortality (B, D, and F) data are shown. (A and B) The effects of psilostachyin A (Psilo) (i.p.) and Bz (p.o.) were followed using doses of up to 50 mg/kg/day, administered at 5 and 8 dpi. (C and D) The effects of cynaropicrin (cyna) and Bz were followed using doses of up to 50 mg/kg/day for cynaropicrin (i.p.) and 100 mg/kg/day for Bz (p.o.), administered at 5 and 8 dpi. (E and F) The effects of cynaropicrin and Bz were followed using doses of 25 mg/kg/day for cynaropicrin (i.p., b.i.d.) and 100 mg/kg/day for Bz (p.o.), administered for 2 consecutive days starting at the onset of parasitemia (11 dpi).
Next, the in vivo activity of cynaropicrin was evaluated in the same animal model, using nontoxic doses of 25 and 50 mg/kg/day (i.p.), with Bz administered at 100 mg/kg/day (p.o.). We found no parasitemia reduction with both doses of cynaropicrin, achieving 100% mortality in all groups except the uninfected and Bz mouse groups, with the latter also suppressing parasitemia (Fig. 4C and D).
In their experiments using cynaropicrin in an acute T. b. rhodesiense mouse model, Zimmermann et al. (12) applied a dose of 10 mg/kg twice daily. Therefore, we also evaluated the efficacy of cynaropicrin given twice a day (every 12 h), starting the therapy at the onset of parasitemia. Swiss male mice were inoculated with 5 × 103 parasites (Colombiana strain) and then treated with 25 mg/kg/day of cynaropicrin. However, on the third day of cynaropicrin administration, 4 of 6 mice died, displaying an earlier mortality than that of the untreated mouse group (Fig. 4E and F). The two surviving mice of the cynaropicrin-treated group were then euthanized due to toxic effects such as tremors and ataxia. Bz-treated mice suppressed parasitemia and showed 100% survival, while the untreated group showed a 70% mortality rate (Fig. 4E and F).
DISCUSSION
In the present study, the antiprotozoal activity of two sesquiterpene lactones (cynaropicrin and psilostachyin A) was investigated. Our data showed that although they presented quite considerable trypanocidal effects in vitro, with cynaropicrin being more effective than psilostachyin A, both STLs failed to control in vivo infection. Our data do not confirm a previous study performed with psilostachyin A on T. cruzi (21). The authors of that study found that psilostachyin A had an EC50 of 0.76 μg/ml on trypomastigotes, which is about 43 times more effective than what we found here (33 μg/ml against BT). However, we cannot rule out the possibility that this discrepancy may be due to the different parasite strains used (RA and Y strains, respectively). Our in vivo study also gave different results, since both cynaropicrin and psilostachyin A were not able to protect against mortality induced by acute T. cruzi infection of Swiss male mice, in contrast to the reference drug (Bz), which suppressed parasitemia levels. Thus, although our present data demonstrated that psilostachyin A could reduce parasitemia by 40 to 70%, it was not able to increase mouse survival. A previous study also reported a 60% parasitemia reduction after 5 days of psilostachyin A (isolated from Ambrosia tenuifolia) administration by the i.p. route, but the authors of that study found a protection against mortality that we did not observe (21). It is possible that the contrasting results are due to the different models that were employed, since the previous study used a longer treatment period (5 consecutive days), a different dose (1 mg/kg/day), and a different parasite strain (RA strain). In the present study, besides applying a more stringent protocol (two doses, at the onset and the peak of parasitemia), a moderately resistant parasite strain (Y strain) was also used, as recommended for the identification of novel promising anti-T. cruzi candidates (34). Additionally, in a previous study (21), Bz was used at a low and nontrypanocidal dose (1 mg/kg/day) and given by an unusual route (i.p.), while in our present analysis, this reference drug was used at higher doses (≥50 mg/kg/dose), following the protocol for acute experimental T. cruzi infection (34).
Thus, using our standardized and stringent protocols (22, 32), we found that both STLs are not promising candidates for CD, since both were nonefficacious in our murine models of T. cruzi acute infection at the doses and regimens presently investigated. In fact, in a recent study, the in vitro activity of cynaropicrin against T. cruzi was evaluated, and the data showed a similar 50% inhibitory concentration (IC50) (4.4 μM) to that reported by Zimmermann et al. (12). The authors also reported cynaropicrin to be about 15 times less active against T. cruzi than against African trypanosomes, corroborating the limited activity of this STL, as presently noted. Studies with this STL in a rodent model of acute T. brucei infection demonstrated a high in vivo efficacy, with 92% parasitemia reduction (12). These findings are in contrast to our current studies using two strains of T. cruzi (Y and Colombiana) which are considered moderately to highly resistant to nitro derivatives such as Bz (34).
Another important issue to be discussed is the toxicity of STLs toward mammalian cells, as well as their therapeutic indexes. Both studied STLs showed low selectivity indexes in vitro (≤8), and acute toxicity analysis (followed for up to 48 h post-STL administration) showed that the higher studied dose of cynaropicrin (400 mg/kg) resulted in animal mortality (1 of 2 mice), while no major adverse effect could be noticed with lower doses or when psilostachyin A was administered (≤100 mg/kg due to its low solubility). Also, histopathology assays revealed that the liver was the most reactive organ, corroborating the biochemical plasma analysis, which demonstrated a slight increase in ALT levels at doses of ≥200 mg/kg cynaropicrin administered via the i.p. route. The most frequent adverse events included a dose-dependent mononuclear cell infiltration, necrosis, and hepatocyte regeneration, especially in those animals that received the higher doses (≥200 mg/kg) by the i.p. route. Cynaropicrin also caused a mild dose-dependent alteration in the spleens of treated mice, with white pulp activation and an increased number of megakaryocytes. While assaying its efficacy in parasite infection, we noticed that administration of this STL twice a day at 25 mg/kg resulted in earlier death (4 of 6 mice) than that of untreated and infected mice (Colombiana strain), while the remaining 2 mice showed considerable adverse effects, such as tremors, that made it necessary to euthanize the animals.
Thus, the low efficacy and overt toxicity of cynaropicrin do not indicate the need for further studies with this natural compound as an agent for treatment of CD. On the other hand, our ultrastructural analysis corroborates previous findings obtained using STLs such as psilostachyin A against T. cruzi (21). As previously reported, the incubation of bloodstream trypomastigotes with cynaropicrin also induced an intense cytoplasmic vacuolization, with the frequent appearance of multivesicular bodies suggestive of autophagy, a type II programmed cell death (PCD) mechanism. In fact, this type of PCD has already been reported by others during the treatment of T. cruzi with different natural and synthetic trypanocidal compounds, including posaconazole, amiodarone (35), and triazolic naphthofuranquinone (36), among others (37). Interestingly, other types of PCD were also reported when STLs were used against different pathogens. The STL xanthatin, which had good activity against Trypanosoma brucei brucei (IC50 of 2.6 μg/ml), triggered apoptosis in treated trypanosomes (38). Leishmania mexicana treated with 3 different STLs (helenalin, mexicanin, and dehydroleucodine) presented fragmented DNA, a hallmark of apoptosis-like death (14). Additionally, cynaropicrin induced the appearance of blebs and apoptotic bodies, indicating apoptotic cell death, in leukocyte cancer cell lines (9). In summary, our data revealed that neither cynaropicrin, which was very promising against African trypanosomes, nor psilostachyin A, which was shown by another group to have in vivo trypanocidal effects, showed efficacy in mouse models of acute T. cruzi infection compared to the reference drug benznidazole. However, although both STLs are not promising candidates for CD therapy, additional literature data showing the effects of sesquiterpenes against other microbial agents justify further studies (including modifications of their chemical structures) aiming to improve their activity and selectivity toward parasitic infections.
ACKNOWLEDGMENTS
This study was funded by PDTIS/Fiocruz, PROEP/CNPq, FAPERJ (Estudo de Doenças Negligenciadas Reemergentes 2012, APQ1, e Apoio a Instituições de Ensino e Pesquisa Sediadas no Estado do Rio de Janeiro 2011), CNPq, and Fiocruz. S.Z. gratefully acknowledges a grant from the Swiss South African Joint Research Programme (grant JRP 03).
We thank the Program for Technological Development in Tools for Health (PDTIS), Fiocruz, for use of its facilities.
The present work was part of activities conducted within the research network Natural Products against Neglected Diseases (ResNetNPND).
Footnotes
Published ahead of print 12 August 2013
REFERENCES
- 1.Salomon CJ. 2011. First century of Chagas' disease: an overview on novel approaches to nifurtimox and benzonidazole delivery systems. J. Pharm. Sci. 101:888–894 [DOI] [PubMed] [Google Scholar]
- 2.Díaz-Chiguer DL, Márquez-Navarro A, Nogueda-Torres B, de la Luz León-Ávila G, Pérez-Villanueva J, Hernández-Campos A, Castillo R, Ambrosio JR, Nieto-Meneses R, Yépez-Mulia L, Hernández-Luis F. 2012. In vitro and in vivo trypanocidal activity of some benzimidazole derivatives against two strains of Trypanosoma cruzi. Acta Trop. 122:108–112 [DOI] [PubMed] [Google Scholar]
- 3.Rassi A, Jr, Rassi A, Marcondes de Rezende J. 2012. American trypanosomiasis (Chagas disease). Infect. Dis. Clin. N. Am. 26:275–291 [DOI] [PubMed] [Google Scholar]
- 4.Soeiro MN, de Castro SL. 2009. Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin. Ther. Targets 13:105–121 [DOI] [PubMed] [Google Scholar]
- 5.Le Loup G, Pialoux G, Lescure FX. 2011. Update in treatment of Chagas disease. Curr. Opin. Infect. Dis. 24:428–434 [DOI] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 75:311–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt TJ, Khalid SA, Romanha AJ, Alves TM, Biavatti MW, Brun R, Da Costa FB, de Castro SL, Ferreira VF, de Lacerda MV, Lago JH, Leon LL, Lopes NP, das Neves Amorim RC, Niehues M, Ogungbe IV, Pohlit AM, Scotti MT, Setzer WN, de Soeiro NCM, Steindel M, Tempone AG. 2012. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases. Part I. Curr. Med. Chem. 19:2128–2175 [DOI] [PubMed] [Google Scholar]
- 8.Schmidt TJ, Khalid SA, Romanha AJ, Alves TM, Biavatti MW, Brun R, Da Costa FB, de Castro SL, Ferreira VF, de Lacerda MV, Lago JH, Leon LL, Lopes NP, das Neves Amorim RC, Niehues M, Ogungbe IV, Pohlit AM, Scotti MT, Setzer WN, de Soeiro NCM, Steindel M, Tempone AG. 2012. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases. Part II. Curr. Med. Chem. 19:2176–2228 [PubMed] [Google Scholar]
- 9.Cho JY, Kim AR, Jung JH, Chun T, Rhee MH, Yoo ES. 2004. Cytotoxic and pro-apoptotic activities of cynaropicrin, a sesquiterpene lactone, on the viability of leukocyte cancer cell lines. Eur. J. Pharmacol. 492:85–94 [DOI] [PubMed] [Google Scholar]
- 10.Otoguro K, Iwatsuki M, Ishiyama A, Namatame M, Nishihara-Tukashima A, Kiyohara H, Hashimoto T, Asakawa Y, Omura S, Yamada H. 2011. In vitro antitrypanosomal activity of plant terpenes against Trypanosoma brucei. Phytochemistry 72:2024–2030 [DOI] [PubMed] [Google Scholar]
- 11.Julianti T, Hata Y, Zimmermann S, Kaiser M, Hamburger M, Adams M. 2011. Antitrypanosomal sesquiterpene lactones from Saussurea costus. Fitoterapia 82:955–959 [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann S, Kaiser M, Brun R, Hamburger M, Adams M. 2012. Cynaropicrin: the first plant natural product with in vivo activity against Trypanosoma brucei. Planta Med. 78:553–556 [DOI] [PubMed] [Google Scholar]
- 13.Berger I, Passreiter CM, Cáceres A, Kubelka W. 2001. Antiprotozoal activity of Neurolaena lobata. Phytother. Res. 15:327–330 [DOI] [PubMed] [Google Scholar]
- 14.Barrera PA, Jimenez-Ortiz V, Tonn C, Giordano O, Galanti N, Sosa MA. 2008. Natural sesquiterpene lactones are active against Leishmania mexicana. J. Parasitol. 94:1143–1149 [DOI] [PubMed] [Google Scholar]
- 15.Tiuman TS, Ueda-Nakamura T, Garcia Cortez DA, Dias Filho BP, Morgado-Díaz JA, de Souza W, Nakamura CV. 2005. Antileishmanial activity of parthenolide, a sesquiterpene lactone isolated from Tanacetum parthenium. Antimicrob. Agents Chemother. 49:176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nour AM, Khalid SA, Kaiser M, Brun R, Abdallah WE, Schmidt TJ. 2009. The antiprotozoal activity of sixteen Asteraceae species native to Sudan and bioactivity-guided isolation of xanthanolides from Xanthium brasilicum. Planta Med. 75:1363–1368 [DOI] [PubMed] [Google Scholar]
- 17.Ganfon H, Bero J, Tchinda AT, Gbaguidi F, Gbenou J, Moudachirou M, Frédérich M, Quetin-Leclercq J. 2012. Antiparasitic activities of two sesquiterpenic lactones isolated from Acanthospermum hispidum D.C. J. Ethnopharmacol. 141:411–417 [DOI] [PubMed] [Google Scholar]
- 18.Karioti A, Skaltsa H, Kaiser M, Tasdemir D. 2009. Trypanocidal, leishmanicidal and cytotoxic effects of anthecotulide-type linear sesquiterpene lactones from Anthemis auriculata. Phytomedicine 16:783–787 [DOI] [PubMed] [Google Scholar]
- 19.Brengio SD, Belmonte SA, Guerreiro E, Giordano OS, Pietrobon EO, Sosa MA. 2000. The sesquiterpene lactone dehydroleucodine (DhL) affects the growth of cultured epimastigotes of Trypanosoma cruzi. J. Parasitol. 86:407–412 [DOI] [PubMed] [Google Scholar]
- 20.Jimenez-Ortiz V, Brengio SD, Giordano O, Tonn C, Sánchez M, Burgos MH, Sosa MA. 2005. The trypanocidal effect of sesquiterpene lactones helenalin and mexicanin on cultured epimastigotes. J. Parasitol. 91:170–174 [DOI] [PubMed] [Google Scholar]
- 21.Sülsen VP, Frank FM, Cazorla SI, Anesini CA, Malchiodi EL, Freixa B, Vila R, Muschietti LV, Martino VS. 2008. Trypanocidal and leishmanicidal activities of sesquiterpene lactones from Ambrosia tenuifolia Sprengel (Asteraceae). Antimicrob. Agents Chemother. 52:2415–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Batista DG, Batista MM, de Oliveira GM, do Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Boykin DW, Soeiro MN. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob. Agents Chemother. 54:2940–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmermann S, Thomi S, Kaiser M, Hamburger M, Adams M. 2012. Antiprotozoal screening and HPLC based activity profiling for leads from European plants. Sci. Pharm. 80:205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ha TJ, Jang DS, Lee JK, Lee KD, Lee J, Hwang SW. 2003. Cytotoxic effects of sesquiterpene lactones from flowers of Hemisteptia lyrata B. Arch. Pharm. Res. 26:925–928 [DOI] [PubMed] [Google Scholar]
- 25.Werner H, Raulais D, Anderson GD. 1973. Sesquiterpene lactones from Ambrosia cordifolia. Phytochemistry 12:1415–1420 [Google Scholar]
- 26.Borges-del-Castillo J, Manresa-Ferrero MT, Rodríguez-Luis F, Vázquez-Bueno P, Joseph-Nathan P. 1981. 13C NMR study of psilostachyinolides from some Ambrosia species. Org. Magn. Res. 17:232–234 [Google Scholar]
- 27.Meirelles MN, Araujo-Jorge TC, Miranda CF, De Souza W, Barbosa HS. 1986. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur. J. Cell Biol. 41:198–206 [PubMed] [Google Scholar]
- 28.Da Silva CF, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Som P, Boykin DW, Soeiro MN. 2011. In vitro trypanocidal activity of DB745B and other novel arylimidamides against Trypanosoma cruzi. J. Antimicrob. Chemother. 66:1295–1297 [DOI] [PubMed] [Google Scholar]
- 29.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55–63 [DOI] [PubMed] [Google Scholar]
- 30.Silva CF, Batista MM, Mota RA, de Souza EM, Stephens CE, Som P, Boykin DW, Soeiro MN. 2007. Activity of “reversed” diamidines against Trypanosoma cruzi in vitro. Biochem. Pharmacol. 73:1939–1946 [DOI] [PubMed] [Google Scholar]
- 31.da Silva CF, Batista MM, Batista Dda de Souza GEM, da Silva PB, de Oliveira GM, Meuser AS, Shareef AR, Boykin DW, Soeiro MN. 2008. In vitro and in vivo studies of the trypanocidal activity of a diarylthiophene diamidine against Trypanosoma cruzi. Antimicrob. Agents Chemother. 52:3307–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.da Silva CF, Batista DG, Oliveira GM, de Souza EM, Hammer ER, da Silva PB, Daliry A, Araujo JS, Britto C, Rodrigues AC, Liu Z, Farahat AA, Kumar A, Boykin DW, Soeiro MN. 2012. In vitro and in vivo investigation of the efficacy of arylimidamide DB1831 and its mesylated salt form—DB1965—against Trypanosoma cruzi infection. PLoS One 7:e30356. 10.1371/journal.pone.0030356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Souza EM, Oliveira GM, Boykin DW, Kumar A, Hu Q, De Nazaré Soeiro CM. 2006. Trypanocidal activity of the phenyl-substituted analogue of furamidine DB569 against Trypanosoma cruzi infection in vivo. J. Antimicrob. Chemother. 58:610–614 [DOI] [PubMed] [Google Scholar]
- 34.Romanha AJ, Castro SL, Soeiro MN, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W, Andrade ZA. 2010. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem. Inst. Oswaldo Cruz 105:233–238 [DOI] [PubMed] [Google Scholar]
- 35.Veiga-Santos P, Desoti VC, Miranda N, Ueda-Nakamura T, Dias-Filho BP, Silva SO, Cortez DA, de Mello JC, Nakamura CV. 2013. The natural compounds piperovatine and piperlonguminine induce autophagic cell death on Trypanosoma cruzi. Acta Trop. 125:349–356 [DOI] [PubMed] [Google Scholar]
- 36.Fernandes MC, Da Silva EN, Pinto AV, De Castro SL, Menna-Barreto RF. 2012. A novel triazolic naphthofuranquinone induces autophagy in reservosomes and impairment of mitosis in Trypanosoma cruzi. Parasitology 139:26–36 [DOI] [PubMed] [Google Scholar]
- 37.Soeiro MNC, Daliry A, Silva CF, de Souza M, Oliveira E GG, Salomão K, Menna Barreto R, Castro SL. 2010. Electron microscopy approaches for the investigation of the cellular targets of trypanocidal agents in Trypanosoma cruzi, p 191–203 In Méndez-Vilas A, Díaz J. (ed), Microscopy: science, technology, applications and education, vol 1 Formatex Research Center, Badajoz, Spain [Google Scholar]
- 38.Nibret E, Youns M, Krauth-Siegel RL, Wink M. 2011. Biological activities of xanthatin from Xanthium strumarium leaves. Phytother. Res. 25:1883–1890 [DOI] [PubMed] [Google Scholar]