

Abstract
Antibiotics excreted into the intestinal tract, such as broad-spectrum cephalosporins, disrupt the indigenous microflora, affect colonization resistance (CR), and promote intestinal colonization by resistant bacteria. We tested whether oral DAV131, a charcoal-based adsorbent, would prevent colonization by a cefotaxime (CTX)-resistant Klebsiella pneumoniae strain (PUG-2) in CTX-treated mice. Mice received CTX, saline, CTX and DAV131, or saline and DAV131 for 3 days before oral challenge with 106 CFU of PUG-2. The fecal CTX concentrations and counts of PUG-2 were assayed. Fecal CTX disappeared when DAV131 was given concomitantly with CTX (P < 0.05), and the area under the curve of PUG-2 fecal density was significantly reduced (P < 0.01). In conclusion, reducing intestinal antibiotic exposure with DAV131 may reduce colonization by resistant strains during treatment compared to treatment with CTX only. This might open new possibilities for decreasing the impact of antibiotics on the intestinal microbiota during treatments.
INTRODUCTION
Resistance to broad-spectrum cephalosporins is rising dramatically worldwide in Enterobacteriaceae. It is now a major public health concern, and new measures are urgently required (1).
Resistance may result from the selection of resistant mutants or from the horizontal transfer of resistance genes encoding products that destroy or inactivate antibiotics. The link between antibiotic exposure and the emergence of resistance is well demonstrated (2). Resistance in pathogenic bacteria can result from two different sequences. The first, known since the early use of antibiotics, is the selection of resistant mutants at a site of infection during treatment (3). More often, however, antibiotics favor the emergence of resistance by exerting a selective pressure on the intestinal commensal microbiota. Colonization of the intestinal microbiota by resistant bacteria plays a central role in the dissemination of resistance. The colonization and proliferation of resistant microorganisms are increased in all treated individuals, irrespective of the reason why the antibiotic is given, because part of the dose administered is excreted via the feces and impacts the intestinal microbiota (4).
In the absence of antibiotic exposure, the intestinal microbiota is stable and resists colonization by exogenous microorganisms; this so-called “colonization resistance” (CR) is due to the action of anaerobic intestinal bacteria (5–7). When anaerobes are affected during antibiotic treatments, colonization resistance is altered, resulting in the proliferation and excretion of resistant microorganisms, with further risks of infection for the patient himself and for dissemination (4, 8, 9).
In this context, it has been hypothesized that inactivation of residual antibiotics present in the colon during treatment could reduce the impact of such treatments on the intestinal microbiota. A clear proof of concept has been obtained using orally administered β-lactamases given together with parenteral β-lactams, both in animal models and in humans (10, 11). However, β-lactamases have a limited spectrum of activity and are not active on other classes of antibiotics that are often coadministered with β-lactams. An alternate means of inactivating antibiotics, the use of adsorbents targeted to the colon, could overcome this disadvantage. Indeed, it has been shown in vitro and in vivo in animal models that activated charcoal targeted to the colon was able to reduce exposure of the microbiota to fluoroquinolones during treatments (12–14). However, whether this approach could also work with broad-spectrum cephalosporins, which are currently a major driver of bacterial resistance to antibiotics (15), has not yet been tested.
As a first demonstration step, we tested in a mouse model whether oral DAV131, a formulated activated charcoal-based adsorbent, would prevent CR disruption and intestinal colonization by a cefotaxime (CTX)-resistant Klebsiella pneumoniae strain after CTX administration.
(This work has been presented as a poster at the 52nd ICAAC, 9 to 12 September 2012, San Francisco, CA [16].)
MATERIALS AND METHODS
Bacterial strain.
Klebsiella pneumoniae PUG-2 was a rectal strain isolated from a sample from an intensive care unit (ICU) patient in our laboratory. Its MICs, determined by Etest susceptibility assay, were >32 mg/liter for cefotaxime, 16 mg/liter for imipenem, and >32 mg/liter for ertapenem. Resistance was due to a plasmidic cephalosporinase, DHA-1, and to the lack of the OmpK36 outer membrane protein.
Characterization of DAV131, a formulated activated-charcoal-based adsorbent.
DAV131 was provided by Da Volterra (Paris, France). It was made of a charcoal-based adsorbent, chosen among several mineral and vegetal adsorbing materials for its capacity to efficiently and rapidly adsorb small molecules, such as antibiotics. The selected charcoal was then formulated for in vivo oral administration to obtain DAV131.
Experimental model.
Eight-week-old female Swiss mice weighing 30 ± 2 g (Janvier, Le Genest Saint Isle, France) were used. Upon arrival, the mice were allowed a 5-day period of rest before experiments began and were fed ad libitum.
Preliminary experiments (not shown) showed that a dose of 300 mg/kg of body weight of subcutaneous (SC) CTX once per day was the minimal dose that was followed by significant and reproducible fecal colonization of mice by the K. pneumoniae PUG-2 strain administered intragastrically.
The protocol was approved by the scientific board of our research unit in accordance with local regulations.
Mice, housed in one cage per group, were randomly assigned to receive parenteral CTX only (group 1, n = 8), parenteral saline only (group 2, n = 6), parenteral CTX and oral DAV131 (group 3, n = 8), or parenteral saline and oral DAV131 (group 4, n = 8). Parenteral CTX was given subcutaneously once a day for 3 days (D−2 to D0) at a dose of 300 mg/kg in 0.3 ml of saline. Oral DAV131 was given by gastric gavage (25 mg in 0.15 ml) twice a day (20 min before and 3 h after each CTX injection). Bacterial challenge was done in all mice by gastric gavage of 106 CFU of PUG-2 in 0.2 ml of saline on the last treatment day (D0). Fecal pellets were collected (i) before treatment and on every treatment day 4 h after CTX injection for the determination of CTX concentration in feces and (ii) before bacterial challenge and 1, 2, 5, 7, and 9 days thereafter for PUG-2 fecal counts.
Fecal counts of PUG-2.
Fecal samples (∼100 mg) were serially diluted 10-fold in physiological saline and plated onto ChromID ESBL agar (bioMérieux, Marcy l'Etoile, France). The plates were incubated aerobically at 37°C for 48 h, and the numbers of PUG-2 CFU per gram of feces were determined. The lower limit of detection was ∼ 3.00 log10 CFU/g of feces. PUG-2 colonies appeared blue on ChromID ESBL agar. Randomly selected clones were confirmed as PUG-2 by antibiotic susceptibility testing and matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry identification. No other broad-spectrum-cephalosporin-resistant strain was isolated.
Determination of fecal antibiotic concentrations.
The fecal concentrations of CTX were determined by high-performance liquid chromatography (HPLC) with UV detection. Weighed samples of feces from experimental mice were homogenized with 500 μl of water and 100 μl of 20% trichloroacetic acid solution. Samples were mixed for 2 min and centrifuged for 10 min at 10,500 × g. A 150-μl amount of the supernatant was added to 50 μl of 0.1 M acetoacetate buffer (pH 4) and mixed, and 40 μl was injected into the chromatographic system. Chromatographic separation was performed on a C18 Supelcosil column (5 μm, 250 mm by 4.6 mm; Supelco, St. Quentin Fallavier, France). The mobile phase consisted of 95% 0.1 M acetoacetate buffer, pH 4, 5% acetonitrile. The flow rate was set at 1 ml/min, and detection was achieved at 254 nm. The lower limit of quantification was 1 μg CTX/100 mg of feces.
Statistical analysis.
The mean fecal CTX concentrations from group 1 mice on D−1 and D0 were compared to those of group 3 mice using nonparametric Wilcoxon tests. Individual areas under the curve (AUC) above the detection limits of log counts of fecal PUG-2 were computed by trapezoidal approach from D−1 to D9 (AUC_PUG-2). The means for AUC_PUG-2 were also compared between group 1 and group 3 by Wilcoxon tests. All statistical analyses were performed using SAS software version 9.2 (SAS, Cary, NC).
RESULTS
Oral DAV131 reduced CTX concentrations in mouse feces.
CTX was undetectable in the feces of mice treated with CTX only (group 1) on the first day of treatment (D−2) but was detected in 4/8 mice (50%) on the two following days (D−1 and D0) 4 h after CTX injections at concentrations of 3.25 μg/100 mg of feces (95% confidence interval [CI], 0.004 to 6.50) and 5.75 μg/100 mg of feces (95% CI, −2.28 to 13.78) (Fig. 1A). No CTX was ever detected in the feces of mice that received oral DAV131 in addition to the antibiotic (group 3) or in the feces of mice treated with saline and oral DAV131 (group 4). Statistical analysis showed that fecal CTX concentrations were significantly reduced in mice receiving DAV131 in addition to CTX (group 3) in comparison to the concentrations in those receiving CTX only (group 1) on the third day of treatment (D0) (P < 0.05).
Fig 1.
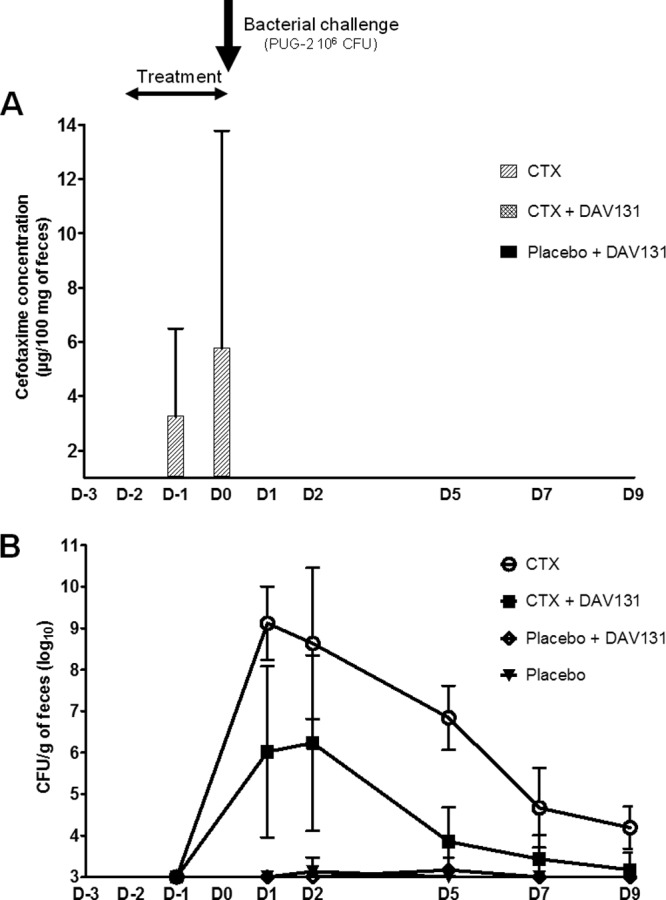
(A) Efficacy of DAV131 in adsorbing CTX residues in the colon. The CTX concentrations in feces (μg/100 mg of feces) during treatment (D−2 to D0) are shown. The vertical bars represent 95% CI. (B) Efficacy of DAV131 in reducing CTX-induced colonization by K. pneumoniae strain PUG-2. The densities (log10 CFU/g of feces) of PUG-2 after orogastric challenge with 106 CFU of PUG-2 on day 0 are shown. Prior to inoculation, none of the mice had detectable levels of PUG-2 (level of detection, ∼3 log10 CFU/g of feces). The whiskers represent 95% CI.
Oral DAV131 decreased intestinal colonization by K. pneumoniae PUG-2.
Mice that received SC CTX only (group 1) developed a high-density fecal colonization with PUG-2 as soon as 1 day after inoculation of the strain (peak value on D1, 9.12 log10 CFU/g of feces [95% CI, 8.24 to 10.01]). In contrast, no significant colonization was observed in mice treated with SC saline with (group 4) or without (group 2) oral DAV131 (Fig. 1B). In mice that received oral DAV131 in addition to SC CTX (group 3), the level of colonization was severely reduced, by close to 3 log10 on average at D1, D2, and D5 (peak value on D2, 6.23 log10 CFU/g of feces [95% CI, 4.12 to 8.35]). Statistical analysis showed that the AUC of PUG-2 colonization was highly significantly reduced by DAV131 (14.17 ± 10.74 log10 CFU/g × number of days for group 3 versus 34.59 ± 8.61 log10 CFU/g × number of days for group 1, P < 0.01).
DISCUSSION
Our most important result is that DAV131, an orally administered formulated activated-charcoal-based adsorbent, partially restored CR to a CTX-resistant strain in mice treated with SC CTX. Indeed, mice treated with CTX developed high-density fecal colonization after oral challenge with a CTX-resistant K. pneumoniae strain, whereas those treated with CTX in combination with oral DAV131 had a very significantly lower level of colonization. Analyses of CTX fecal concentrations suggest that, while CTX disturbed CR by disrupting the intestinal microbiota, DAV131 restored it, at least partially, thanks to the adsorption of the antibiotic residues in the colon. Compared to data from the literature, DAV131 appeared less effective than an orally administered β-lactamase preparation that was shown to fully restore CR when given to mice treated with piperacillin or piperacillin-tazobactam (11). However, this comparison suffers from the flaws that the models, strains, and antibiotics were different and, thus, cannot be considered fully valid.
Altogether, our findings provide further evidence that the inactivation of residual antibiotics in the colon can reduce the impact of antibiotics on the intestinal microbiota during treatment. Adsorbents might have the advantage over enzymatic preparations of exhibiting a larger spectrum of activity. This is of particular importance because of the key role of the intestinal microbiota in the emergence and spread of antibiotic resistance. It has recently been unequivocally shown that previous treatment with broad-spectrum cephalosporins is an independent risk factor for intestinal colonization by broad-spectrum-cephalosporin-resistant bacteria in ICU patients and that carriage of such bacteria is associated with episodes of ICU-associated infections (15).
Additional studies are needed to determine whether the inactivation of antibiotics in the colon during treatment is an effective strategy to limit the emergence and spread of resistant pathogens in humans.
ACKNOWLEDGMENTS
This work was supported in part by a grant from Da Volterra, in part by Oseo (Nosobio program), and in part by the FP7 program Evo-Tar.
A. Andremont gives scientific advice to Da Volterra within the frame of the French law for Innovation and Research. All other authors declare that they have no conflicts of interest.
Footnotes
Published ahead of print 19 August 2013
REFERENCES
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 2.Goossens H, Ferech M, Vander Stichele R, Elseviers M. 2005. Outpatient antibiotic use in Europe and association with resistance: a cross-national database study. Lancet 365:579–587 [DOI] [PubMed] [Google Scholar]
- 3.Anonymous 1948. Streptomycin treatment of pulmonary tuberculosis: a Medical Research Council investigation. Br. Med. J. 2:769–782 [PMC free article] [PubMed] [Google Scholar]
- 4.Tosh PK, McDonald LC. 2012. Infection control in the multidrug-resistant era: tending the human microbiome. Clin. Infect. Dis. 54:707–713 [DOI] [PubMed] [Google Scholar]
- 5.Servin AL. 2004. Antagonistic activities of lactobacilli and bifidobacteria against microbial pathogens. FEMS Microbiol. Rev. 28:405–440 [DOI] [PubMed] [Google Scholar]
- 6.O'Hara AM, Shanahan F. 2006. The gut flora as a forgotten organ. EMBO Rep. 7:688–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vollaard EJ, Clasener HA. 1994. Colonization resistance. Antimicrob. Agents Chemother. 38:409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lautenbach E, Patel JB, Bilker WB, Edelstein PH, Fishman NO. 2001. Extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae: risk factors for infection and impact of resistance on outcomes. Clin. Infect. Dis. 32:1162–1171 [DOI] [PubMed] [Google Scholar]
- 9.Donskey CJ. 2004. The role of the intestinal tract as a reservoir and source for transmission of nosocomial pathogens. Clin. Infect. Dis. 39:219–226 [DOI] [PubMed] [Google Scholar]
- 10.Welling GW, Holtrop A, Slootmaker-van der Meulen C, Meijer-Severs GJ, van Santen E, Tonk RH, de Vries-Hospers HG, van der Waaij D. 1992. Inactivation of ceftriaxone by faecal enzyme preparations during ceftriaxone treatment. J. Antimicrob. Chemother. 30:234–236 [DOI] [PubMed] [Google Scholar]
- 11.Pitout JD. 2009. IPSAT P1A, a class A beta-lactamase therapy for the prevention of penicillin-induced disruption to the intestinal microflora. Curr. Opin. Investig. Drugs 10:838–844 [PubMed] [Google Scholar]
- 12.Khoder M, Tsapis N, Huguet H, Besnard M, Gueutin C, Fattal E. 2009. Removal of ciprofloxacin in simulated digestive media by activated charcoal entrapped within zinc-pectinate beads. Int. J. Pharm. 379:251–259 [DOI] [PubMed] [Google Scholar]
- 13.Khoder M, Tsapis N, Domergue-Dupont V, Gueutin C, Fattal E. 2010. Removal of residual colonic ciprofloxacin in the rat by activated charcoal entrapped within zinc-pectinate beads. Eur. J. Pharm. Sci. 41:281–288 [DOI] [PubMed] [Google Scholar]
- 14.Nguyen TT, Chachaty E, Huy C, Cambier C, de Gunzburg J, Mentré F, Andremont A. 2012. Correlation between fecal concentrations of ciprofloxacin and fecal counts of resistant Enterobacteriaceae in piglets treated with ciprofloxacin: towards new means to control the spread of resistance? Antimicrob. Agents Chemother. 56:4973–4975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razazi K, Derde LP, Verachten M, Legrand P, Lesprit P, Brun-Buisson C. 2012. Clinical impact and risk factors for colonization with extended-spectrum β-lactamase-producing bacteria in the intensive care unit. Intensive Care Med. 38:1769–1778 [DOI] [PubMed] [Google Scholar]
- 16.Grall N, Ducrot N, Massias L, Sayah-Jeanne S, Chachaty E, Nguyen T, De Gunzburg J, Andremont A. 2012. Orally administered DAV131, a formulated activated charcoal, inhibits establishment of intestinal colonization by beta-lactamine resistant Klebsiella pneumoniae in mice treated by cefotaxime, poster F-2003. Abstr. 52nd Intersci. Conf. Antimicrob. Agents Chemother. American Society for Microbiology, Washington, DC [Google Scholar]