

Abstract
Group A streptococcus (GAS) infection may cause severe life-threatening diseases, including necrotizing fasciitis and streptococcal toxic shock syndrome. Despite the availability of effective antimicrobial agents, there has been a worldwide increase in the incidence of invasive GAS infection. Kallistatin (KS), originally found to be a tissue kallikrein-binding protein, has recently been shown to possess anti-inflammatory properties. However, its efficacy in microbial infection has not been explored. In this study, we transiently expressed the human KS gene by hydrodynamic injection and investigated its anti-inflammatory and protective effects in mice via air pouch inoculation of GAS. The results showed that KS significantly increased the survival rate of GAS-infected mice. KS treatment reduced local skin damage and bacterial counts compared with those in mice infected with GAS and treated with a control plasmid or saline. While there was a decrease in immune cell infiltration of the local infection site, cell viability and antimicrobial factors such as reactive oxygen species actually increased after KS treatment. The efficiency of intracellular bacterial killing in neutrophils was directly enhanced by KS administration. Several inflammatory cytokines, including tumor necrosis factor alpha, interleukin 1β, and interleukin 6, in local infection sites were reduced by KS. In addition, KS treatment reduced vessel leakage, bacteremia, and liver damage after local infection. Therefore, our study demonstrates that KS provides protection in GAS-infected mice by enhancing bacterial clearance, as well as reducing inflammatory responses and organ damage.
INTRODUCTION
Group A streptococcus (GAS) is an important human pathogen that causes considerable morbidity and mortality worldwide. GAS infection causes a wide spectrum of diseases, ranging from mild pharyngitis, impetigo, and scarlet fever to life-threatening diseases such as necrotizing fasciitis and streptococcal toxic shock syndrome (1–3). Various virulence factors have been studied for their association with the severity of GAS infection (4–7). Although there are major advances in genome-wide system biology analysis of host and GAS interactions, limited therapeutic progress has been made, and no vaccine is available (8–11).
Kallistatin (KS), a human tissue kallikrein-binding protein, was first identified and characterized by Chao et al. (12, 13). KS is a serine proteinase inhibitor (serpin) and plays a role in many physiological processes. Previous studies showed that KS may function independently of its interaction with kallikrein, such as lowering blood pressure and inhibiting angiogenesis and tumor growth (14–17). Recent reports showed a novel role of KS in protection against ischemia-reperfusion myocardial and salt-induced renal and cardiovascular injuries by preventing apoptosis, oxidative stress, and inflammation (18–20). Studies in inflammatory bowel disease and obesity indicated that KS was significantly decreased in patients with inflamed intestine and adiposity compared to noninflammatory controls (21, 22). Furthermore, adenovirus-mediated human KS gene therapy inhibited arthritis in a rat model (23). The processes of angiogenesis and inflammation were accompanied by reduced levels of tumor necrosis factor alpha (TNF-α) and interleukin 1β (IL-1β) in joint homogenates. The anti-inflammatory properties of KS were shown to be mediated through its heparin-binding domain to inhibit TNF-α response and increase NF-κB degradation in endothelial cells (24). Using rat KS-transgenic mice, KS was shown to provide protection against lipopolysaccharide (LPS) endotoxic shock (25). In addition, KS was associated with a decreased risk of developing acute kidney injury in patients with sepsis shock (26). Despite these interesting results, the mechanisms behind the anti-inflammatory effects of KS in infectious diseases remain unclear.
We previously established an air pouch mouse model of GAS infection which mimics clinical severe disease such as necrotizing fasciitis (27, 28). Using this animal model, we have shown the protective effects of immunomodulators against GAS infection (29, 30). In the present study, we used hydrodynamic injection to transiently express human KS in mice followed by GAS infection and to demonstrate the protective and anti-inflammatory effects of KS.
MATERIALS AND METHODS
Bacterial strain.
Streptococcus pyogenes NZ131 (type M49, T14) was a gift from D. R. Martin, New Zealand Communicable Disease Center, Porirua, New Zealand. A fresh colony was inoculated into tryptic soy broth containing 0.5% yeast extract (TSBY) (Difco Laboratories, Detroit, MI) overnight and then diluted to 1:50 and cultured for 8 h at 37°C. The bacteria were harvested by centrifugation and resuspended in sterile phosphate-buffered saline (PBS), and the bacterial density was determined by measuring the absorbance at 600 nm. The bacterial suspension was then diluted with PBS to 109 CFU/ml, using a standard growth curve to relate the measured absorbance to the bacterial concentration.
Mice.
BALB/c mice were obtained from Jackson Laboratories, Bar Harbor, ME, and maintained on standard laboratory food and water in the Laboratory Animal Center of National Cheng Kung University Medical College. Their progeny, ranging from 8 to 10 weeks of age, were used for the experiments. Housing, breeding, and experimental use of the animals were performed in strict accordance with the Experimental Animal Committee of National Cheng Kung University.
Preparation of plasmid DNA and hydrodynamic injection techniques.
Human KS gene expression plasmid pcDNA3.1(+)-KS was prepared in L. Chao and J. Chao's lab, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, USA, and amplified by Escherichia coli DH5α. The plasmid was prepared using a Qiagen EndoFree Plasmid Giga kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. The plasmid DNA was administered to mice by hydrodynamic injection (31, 32). In brief, 10 μg of pcDNA3.1(+)-KS or control pcDNA3.1(+) plasmid DNA was diluted in 2 ml (approximately 1/10 of the body weight) of saline and injected into the mouse tail vein using a 26.5-gauge needle within an 8-s injection time.
Air pouch infection model and air pouch exudate collection.
The air pouch model of infection was previously described (27, 28). Mice were subcutaneously injected with 2 ml of air to form an air pouch and were inoculated with 0.3 ml of 2.4 × 108 to 3.5 × 108 CFU of bacterial suspension. The survival rate was monitored every day. The local tissue damage area was measured and photographed. At various time periods postinfection, air pouch exudates were collected by injecting 1 ml PBS into the air pouch and aspirating the exudates. Infiltrating cell number was measured with a hemocytometer, and cell viability was determined by trypan blue staining. Bacterial numbers were quantified by counting on TSBY plates.
Determination of KS and cytokines by ELISA.
KS levels in the air pouch exudates, sera, and organ homogenates were measured by enzyme-linked immunosorbent assay (ELISA) as described previously (13). The cytokines (TNF-α, IL-1β, and IL-6) were measured using ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions.
ROS and superoxide (O2−) detection.
Infiltrating cells in the air pouch exudates were collected from GAS-infected mice and directly stained with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) (2 μM) for reactive oxygen species (ROS) or dihydroethidium (DHE) (4 μM) for O2− detection (Sigma-Aldrich, Michigan). After 30 min of incubation on ice, the increased fluorescence of cells was detected by flow cytometry (BD FACSCalibur, New Jersey).
Histopathology.
At various times postinoculation, mice were sacrificed and tissues from the skin and liver were fixed in 4% neutral buffered formalin solution and dehydrated in graded alcohol. The fixed tissues were then embedded in paraffin and sectioned at a 4-μm thickness. The sections were mounted onto regular glass slides and stained with hematoxylin and eosin (H&E).
Statistics.
Statistical analysis was performed using the paired t test. The survival rate was displayed as a Kaplan-Meier survival curve and assessed by log-rank test. Significant differences were set at P values of <0.05 (GraphPad Prism software version 5; La Jolla, CA).
RESULTS
Expressed human KS increases survival of GAS-infected mice.
The in vivo clearance profile of human KS showed a nonlinear pattern with a short half-life of 65 min (33). We used hydrodynamic injection of pcDNA3.1-KS via the tail vein to transiently express KS in mice. After 12 h, mouse sera as well as liver, kidney, and spleen homogenates were prepared and determined for human KS levels by ELISA. Mice treated with pcDNA3.1-KS showed high levels of KS in the sera and various organs compared with those treated with saline or the pcDNA3.1(+) control plasmid (Fig. 1a). Time kinetics of KS expression in the sera after hydrodynamic injection showed a gradual decrease up to 5 days (Fig. 1b). KS protein was detectable in the plasma and air pouch after GAS infection but also showed a gradual decrease (see Fig. S1 in the supplemental material). At 12 h after hydrodynamic injection, mice were inoculated with a lethal dose of GAS, NZ131, via air pouch infection. Results showed that mice expressing KS had a higher survival rate than mice treated with saline or control plasmid after GAS infection (Fig. 1c). We conclude that KS offers significant protection in the early stage of GAS infection.
Fig 1.
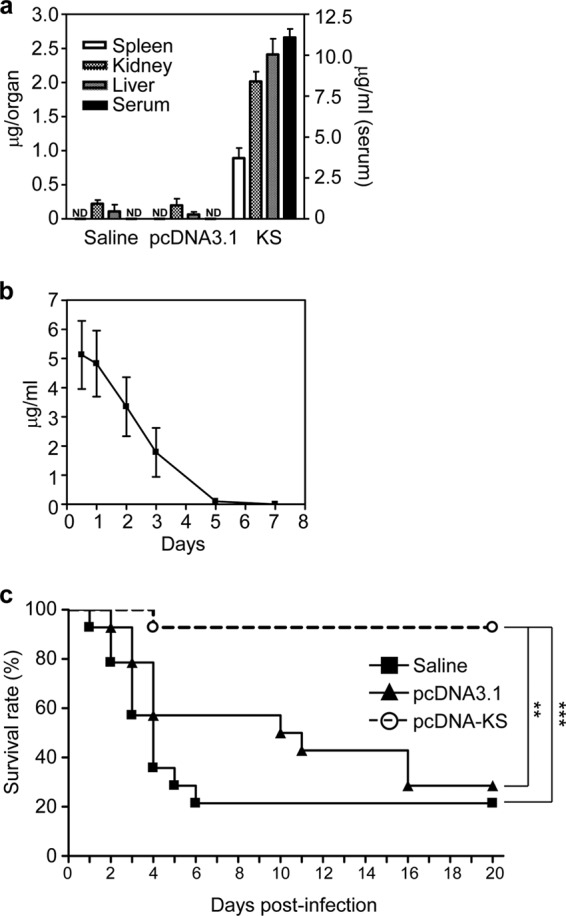
Hydrodynamic injection with pcDNA-KS increases the survival rate of GAS-infected mice. KS plasmid pcDNA3.1-KS or control plasmid pcDNA3.1 (10 μg in 2 ml saline) was transfected in BALB/c mice through hydrodynamic injection. The expression levels of KS in organs or serum after 12 h (a) and in serum at various time points (b) were determined by ELISA. Saline and pcDNA3.1 plasmid were used as negative controls. ND, not detectable. (c) After 12 h, GAS was inoculated in the air pouch and the survival rate was determined. (n = 14 mice for each group). Experiments were performed three times, and one representative experiment is shown. **, P < 0.01; ***, P < 0.001.
KS reduces skin damage in GAS-infected mice.
GAS infection through a skin wound may cause severe necrotic fasciitis and subsequent systemic infection. Hence, restrictions of local infection and tissue damage are important for disease control. In the air pouch infection model, the diapedetic areas of the air pouch showed more severe damage in control mice than in KS-treated mice (Fig. 2a and b). From skin section histological analysis, KS treatment showed a clear boundary of bacteria (Fig. 2c, top, arrows). There were no boundaries shown in the control groups. In addition, the enlarged image showed that the dermal structure was relatively intact in the KS treatment group, while the saline and control plasmid treatment groups showed tissue destruction after GAS infection (Fig. 2c, bottom).
Fig 2.
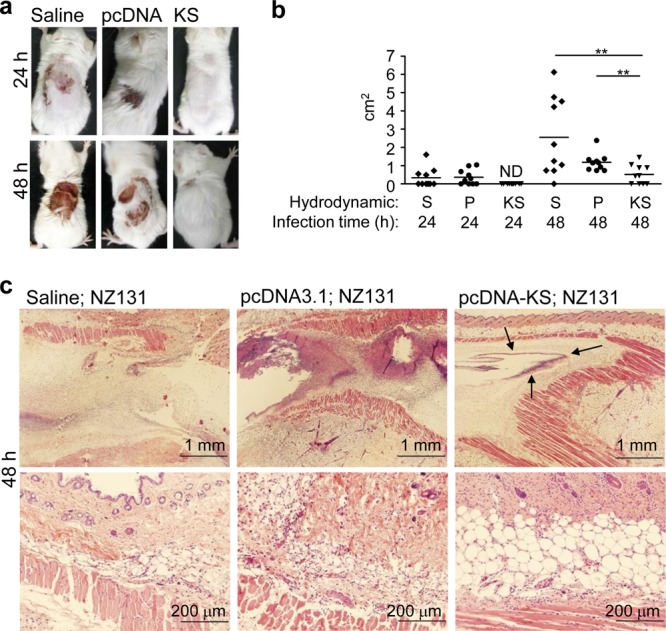
KS decreases skin damage of GAS-infected mice. At 12 h after administration of the KS plasmid, control plasmid, or saline via hydrodynamic injection, mice were infected with GAS using the air pouch infection model. At 24 and 48 h after GAS infection, the diapedetic areas were photographed (a) and measured (b), and the 48-h skin sections were prepared and stained with hematoxylin and eosin (H&E) (c). The arrows indicate the boundaries. Experiments were performed at least twice, and one representative experiment is shown. ND, not detectable; S, saline; P, control plasmid pcDNA3.1; KS, KS plasmid pcDNA3.1-KS. **, P < 0.01.
KS decreases bacterial counts in the local infection site.
Consistent with the occurrence of skin lesions, we found that KS-treated mice had lower bacterial counts in the air pouch exudates than in control mice both 24 and 48 h after GAS infection (Fig. 3a). To rule out the possibility that KS directly attacks and kills bacteria, we showed that the GAS growth curve was not affected by KS (see Fig. S2 in the supplemental material).
Fig 3.
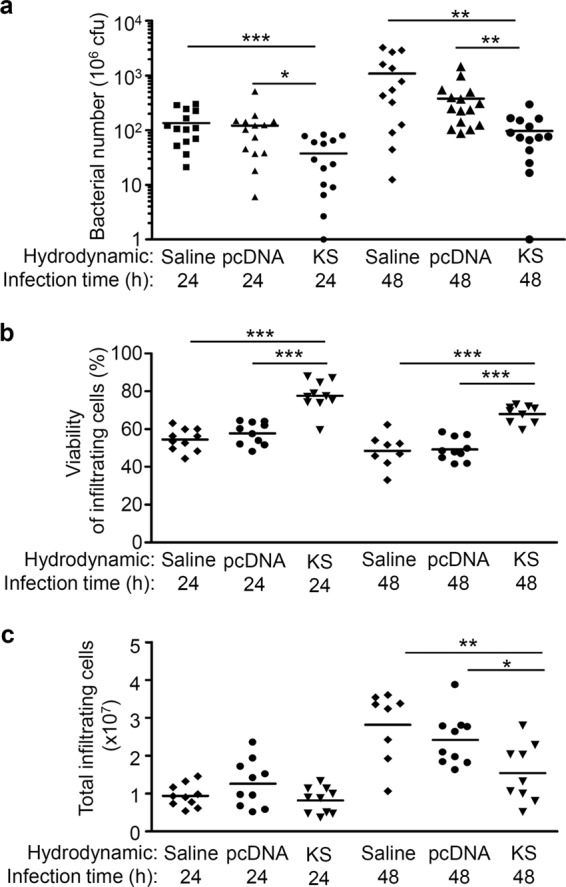
KS enhances immune cell survival and decreases bacterial number in the air pouch exudates of GAS-infected mice. At 12 h after hydrodynamic administration of the KS plasmid, control plasmid, or saline, GAS was inoculated in the air pouch, and air pouch exudates were collected 24 and 48 h after infection. (a) Bacterial counts were determined by plating, and the data were pooled from three independent experiments. The infiltrating cell viability (b) and number (c) were measured by trypan blue staining. Each point indicates one mouse sample. Experiments were performed at least twice, and one representative experiment is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
KS enhances infiltrating cell viability in the local infection site.
Immune cells are the first line of host defense against bacteria. However, immune cells can undergo apoptosis after interacting with pathogens, and GAS accelerates apoptosis in human neutrophils (34, 35). Consistent with our previous findings (27, 30), we found about 90% of infiltrating immune cells of GAS-infected mice were neutrophils (data not shown), and the viability of infiltrating immune cells in KS-treated mice was higher than in control mice both 24 and 48 h after GAS infection (Fig. 3b). KS has been reported to possess an antiapoptotic function in mammalian cells (18, 19). We also found that KS increased the viability of infiltrating cells and that this was correlated with reduced apoptosis in those cells (see Fig. S3a and b in the supplemental material). Immune cell infiltration into sites of infection assists defense against pathogens on the one hand but can cause local tissue damage on the other. We found that even though higher infiltrating cell viability was maintained, the number of infiltrating cells was lower in the presence of KS 48 h after GAS infection (Fig. 3c). Since GAS secretes a number of toxins that have been shown to affect viability of immune cells, including neutrophils and macrophages, the KS effect could be indirectly related to inactivation of these toxins. The increased viability of infiltrating cells by KS could be accounted for by the inhibition of GAS virulence factors. The interrelationships between KS and GAS virulence factors need to be verified.
KS enhances the bacterial clearance activity of immune cells.
Maintaining immune cell viability by KS might promote bacterial clearance. In addition, antimicrobial reagents such as ROS are critical in bacterial killing by neutrophils (36, 37). We found that the infiltrating immune cells (over 90% neutrophils) in mice treated with KS have higher levels of ROS, including O2−, after GAS infection (Fig. 4a and b). The beneficial effects of KS were further confirmed by showing that blood cells collected from KS-treated mice (Fig. 4c) and KS-pretreated human isolated neutrophils (Fig. 4d) both enhanced intracellular killing of GAS. Although the direct interaction between KS and immune cells requires further investigation, the results show that KS not only maintains the viability of immune cells but also is involved in superoxide production to promote immune cell antimicrobial activity.
Fig 4.
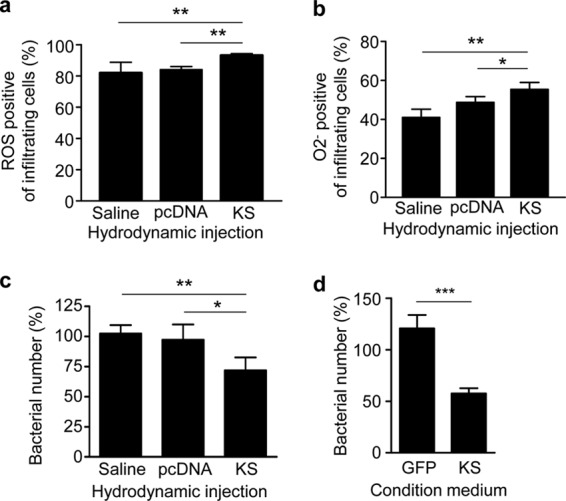
KS enhances reactive oxygen species (ROS) production and bacterial killing activity of immune cells. Reactive oxygen species (a) and peroxide radical (O2−) (b) were detected from the infiltrating immune cells in air pouch exudates after 12 h of infection. (c) Mouse whole blood (n = 3) was collected after hydrodynamic administration of KS plasmid, control plasmid, or saline and incubated with GAS (106 CFU/ml) for 1 h. (d) Neutrophils isolated from human peripheral blood were pretreated with conditioned medium which was prepared from culture supernatant of TE647 cells infected with lentiviruses encoding GFP (as a control) and KS. After 6 h of pretreatment, GAS at a multiplicity of infection (MOI) of 0.1 was added for 1 h. Both GAS-infected whole blood cells and neutrophils were followed by gentamicin (50 μg/ml) treatment for 1 h to kill extracellular bacteria and further lysed for intracellular bacterial detection. The percentage of bacterial number was determined by comparing with initially infected bacterial number. Experiments were performed twice, and one representative experiment is shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
KS decreases cytokine expression in GAS-infected mice.
The KS-mediated increase of antibacterial factors and viability of immune cells in the GAS infection may potentially result in hyperinflammation. However, it has been reported that KS decreases the secretion of proinflammatory cytokines through direct interaction with endothelial cells (18, 24). We collected the air pouch exudates after GAS infection and determined the cytokine and chemokine levels. Results showed that the proinflammatory cytokine TNF-α, IL-1β, and IL-6 levels were all significantly lower in KS-treated mice than in control mice 24 h after GAS infection (Fig. 5). Chemokines macrophage inflammatory protein 2 (MIP-2), monocyte chemoattractant protein 1 (MCP-1), and RANTES in the air pouch exudates did not show any significant difference between the three groups after GAS infection (data not shown). Taken together, we found that KS decreased the cytokine expression and reduced inflammation in local infected tissue.
Fig 5.
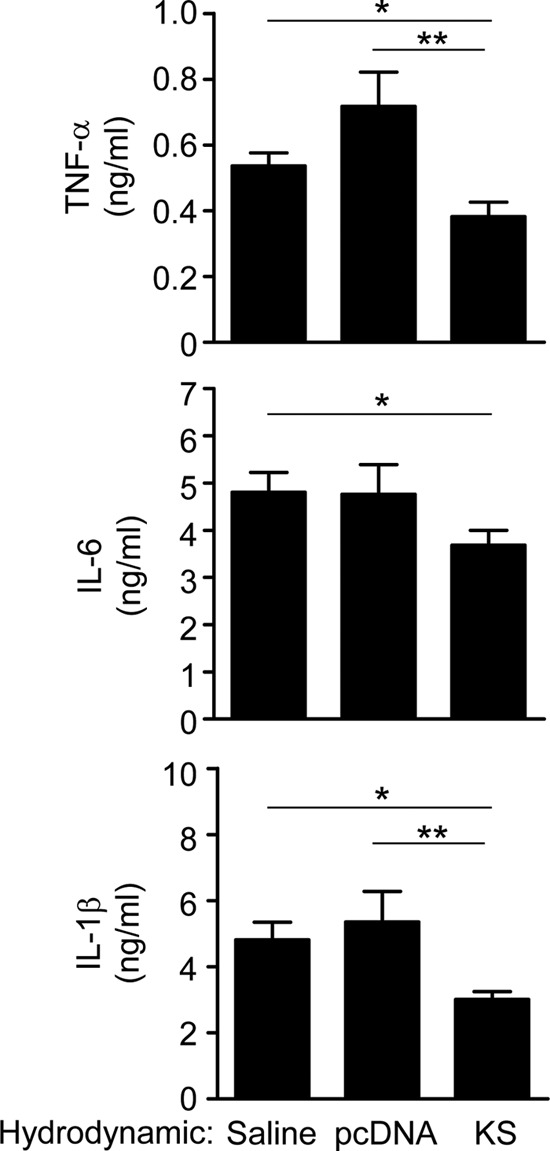
KS reduces cytokine levels in the air pouch exudates of GAS-infected mice. At 12 h after hydrodynamic administration of the KS plasmid, control plasmid, or saline, mice were infected with GAS through the air pouch, and air pouch exudates were collected after 24 h of infection (n = 10). The cytokine levels in air pouch exudates were determined using ELISA. Experiments were done twice, and one representative experiment is shown. *, P < 0.05; **, P < 0.01.
KS reduces vessel leakage, bacteremia, and liver pathology in GAS-infected mice.
A local GAS infection may further disseminate systemically, and multiple organ failure may occur either from bacteria or their virulence factors. The interaction between KS and endothelial cells has been shown not only to decrease inflammatory cytokine production but also to decrease membrane permeability and vascular leakage (24, 38). Here, we also found that KS could reduce streptococcal pyrogenic exotoxin B (SPE B)-induced membrane permeability and vascular leakage in vitro and in vivo (Fig. 6). When GAS was infected in the local tissue for 48 h, KS also reduced the number of mice exhibiting bacteremia to 6.7% (1/15 mice), while 40% (6/15) of saline-treated mice and 26.7% (4/15) of plasmid control mice had bacteria in the blood. In addition, our previous studies showed that systemic GAS infection caused histopathologic changes in mouse liver due to the disseminated bacteria or virulence factor(s) (28). Using hydrodynamic injection to express KS, the liver is a major source of KS expression (Fig. 1a). We found that, after GAS infection for 24 and 48 h, mice treated with saline or control plasmid showed hepatic necrosis (Fig. 7a). Although some necrotic hepatocytes were observed, KS-treated mice showed much less liver damage. Using biochemical analysis, we also showed that KS treatment reduced aspartate aminotransferase (AST) levels after GAS infection (Fig. 7b). This indicates that the systemic distribution of KS may reduce vessel leakage and provide organ protection.
Fig 6.
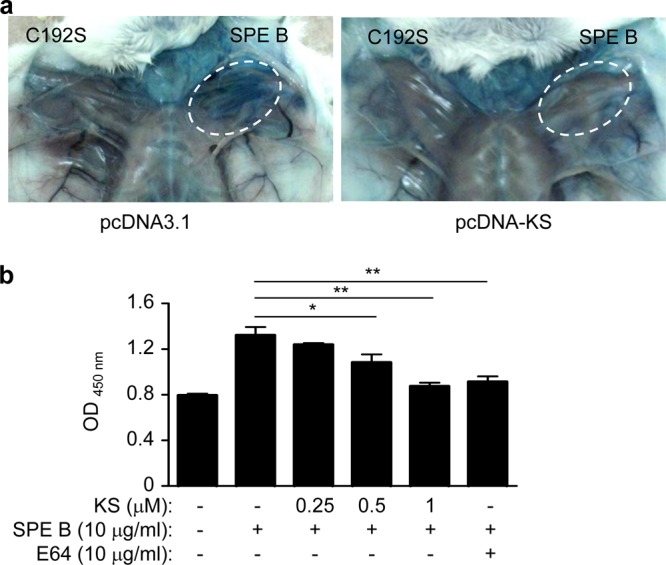
KS reduces vessel permeability induced by GAS virulence factor SPE B. (a) At 12 h after hydrodynamic administration of the KS plasmid or control plasmid, mice were subcutaneously injected with SPE B wild-type or C192S mutant protein without protease activity (25 μg in 50 μl PBS) for 1 h and then injected with Evans blue (10 mg/ml, 200 μl/mouse) through the tail vein. Mice were sacrificed after 30 min, and the local tissue was observed. (b) A monolayer of human microvascular endothelial cells (HMEC-1) in a Transwell was pretreated with KS for 12 h, and the supernatant was placed in fresh medium with SPE B for 1 h. E64 (10 μg/ml), a cysteine protease inhibitor, was used to inhibit SPE B activity as a control. Horseradish peroxidase (HRP) was added in the upper well for 30 min, and the lower-well culture medium was collected to react with tetramethylbenzidine (TMB) substrate. The amount of penetrating HRP activity was measured with absorbance at 450 nm. Experiments were performed three times, and one representative experiment is shown. *, P < 0.05; **, P < 0.01.
Fig 7.
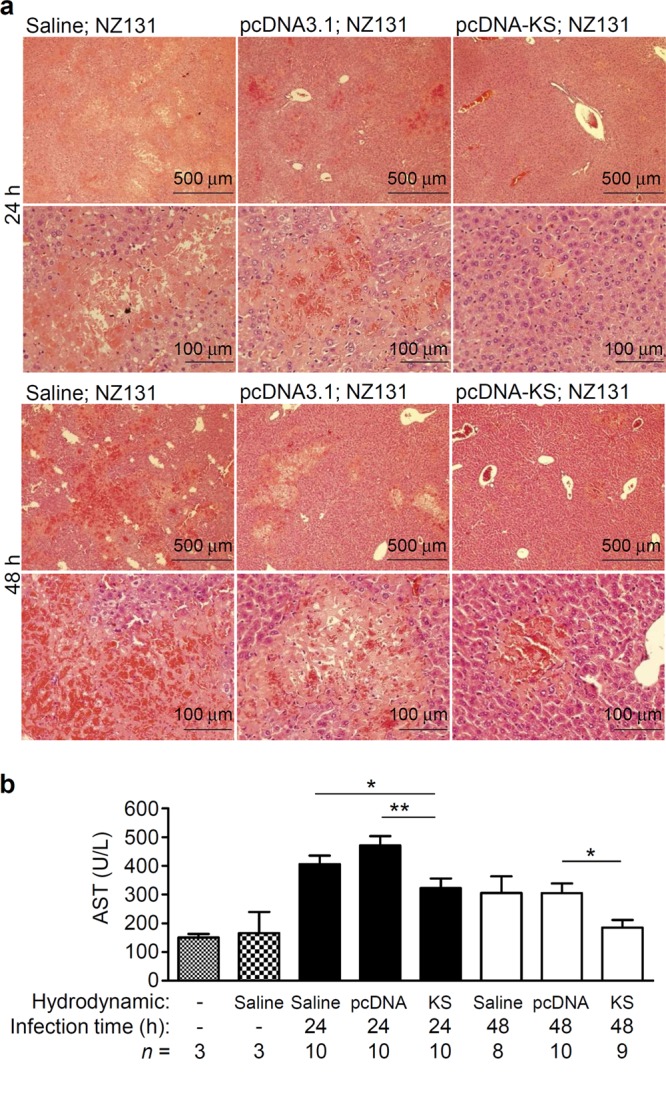
KS reduces liver damage of GAS-infected mice. Mouse blood and liver were collected 24 and 48 h after GAS infection. Liver tissue sections were stained with H&E (a), and plasma was assayed for aspartate aminotransferase (AST) levels (b). “n =” indicates mouse number in each group. Experiments were done twice, and one representative experiment is shown. *, P < 0.05; **, P < 0.01.
DISCUSSION
In the present study, we demonstrate the potential efficacy of KS not only as an anti-inflammatory agent but also as an immune cell modulator for protection against GAS infection. We used an air pouch model of GAS infection to mimic clinical disease, such as necrotizing fasciitis, which might lead to toxic shock syndrome (39). Overproduction of inflammatory cytokines might cause shock or even death of the host in severe systemic diseases. The present study showed that expression of human KS enhanced host immune cell defense, reduced inflammatory cytokine production, and protected mice from GAS infection, suggesting that an increase in KS levels may protect patients from bacteremia and severe toxic shock syndrome.
In an infectious environment, KS may play a dual role in modifying the immune response to GAS infection. We first excluded the possibility of direct action of KS on GAS growth (see Fig. S2 in the supplemental material). Next, concerning KS as a serine proteinase inhibitor, host tissue damage and inflammation might be reduced through inhibition of GAS virulence factors and surface binding of specific host plasma proteins. SpyCEP is a well-known serine proteinase which cleaves human IL-8 (a neutrophil attractant) as well as other chemokines (40). We detected the levels of mouse MIP-2, which is a homolog of human IL-8, in the air pouch exudates 24 and 48 h after GAS infection. The results showed that there was no significant difference between the KS-treated and control groups (data not shown). This finding suggests that KS has no effect on SpyCEP activity, at least for MIP-2, in the air pouch animal model.
Another virulence factor, SPE B, a cysteine proteinase, was shown to facilitate bacterial dissemination, colonization, and invasion and inhibit wound healing (7, 27, 28). Because the KS gene sequence is highly conserved with α-antitrypsin (33), and α-antitrypsin inhibits not only serine proteinase but also cysteine proteinase, we speculated that, even though SPE B is a cysteine proteinase, KS might have an inhibitory effect on SPE B. Surprisingly, instead of an effect of KS on SPE B activity, KS was actually cleaved by SPE B (see Fig. S4 in the supplemental material). In addition, our preliminary results showed that KS still increased mouse survival and decreased skin lesions in mice infected with SPE B-deficient GAS (see Fig. S5 in the supplemental material). This indicates that KS treatment resulted in a decreased bacterial count and reduced skin and organ injury but not through blockage of SPE B. Nevertheless, other virulence factors still need to be tested; our results do not rule out the possibility that the effects of KS could be bidirectional on both the host and pathogen.
As for the mechanisms of protection conferred by KS, we showed that KS did not influence GAS growth directly (see Fig. S2 in the supplemental material). When GAS was cocultured with the plasma collected from KS-treated mice, there was, nevertheless, higher bacterial clearance (data not shown). The detailed mechanism for KS-containing mouse plasma to clear bacteria remains to be further explored. In addition, the protective effects of KS may be mediated through interactions with host cells and promoting host defense mechanisms. Our study showed that blood cells collected from KS-treated mice and KS-pretreated neutrophils both enhanced intracellular bacterial killing (Fig. 4c and d). Previous reports showed a novel role of KS in binding to the TNF-α receptor on endothelial cell surface and preventing inflammation, including inhibiting adhesion molecule expression and cytokine production (18, 24). KS binding to the TNF-α receptor may also block excess cytokine-stimulated apoptotic cell death. We hypothesize that KS may bind to the TNF-α receptor on infiltrating neutrophils, resulting in higher survival of neutrophils, and may further contribute to more efficient bacterial clearance (Fig. 3a and b; see also Fig. S3 in the supplemental material). Figure 3c shows lower cell infiltration after KS treatment than that in control mice at 48 h, which may be due to several possibilities, including KS-mediated inhibition of adhesion molecule expression on endothelial cells and reduction of inflammatory cytokines at the local site. Furthermore, the anti-inflammatory effect of KS may occur through inhibition of the NF-κB signaling pathway (24). Based on previous findings (30, 41, 42), GAS can induce NF-κB activation in various cells. The inhibitory effect of KS on GAS-induced NF-κB activation requires further clarification.
Previous studies showed that the administration of carboxyfullerene (43) and dextromethorphan (DM) (30) were able to protect mice from GAS-induced death. Further investigation revealed that the survival of infiltrating neutrophils was prolonged, resulting in elimination of bacteria. Here, we also found prolonged survival of neutrophils and enhanced bacterial clearance in KS-treated mice. Although the detailed mechanisms need to be further explored, KS-induced prolonged survival of neutrophils is correlated with bacterial clearance. Neutrophils destroy pathogens through a number of mechanisms, including ROS production. However, previous studies showed that KS decreased ROS production in endothelial cells (18, 24). In the present study, we show that the ROS production was increased by KS in air pouch-infiltrating immune cells (Fig. 4a and b). It remains to be further investigated whether the enhanced bacterial clearance is, at least in part, due to the increased ROS production.
Elevated inflammatory cytokine production may be associated with severe invasive GAS infection. The increased levels of proinflammatory cytokines in GAS-infected mice most likely reflect the sustained systemic infection leading to sepsis. In this study, the anti-inflammatory activity of KS was demonstrated by the lowering of proinflammatory cytokines, including TNF-α, IL-1β, and IL-6. KS was previously shown to provide protection against endotoxic shock induced by Gram-negative bacterial LPS (25). The present study further demonstrates the efficacy and novel mechanisms of KS in protecting against Gram-positive bacterium-elicited skin and organ injury and septic shock. Although infecting bacteria can be treated by antibiotics, components released from dead bacteria may be responsible for systemic inflammation and cause organ failure and sepsis. Our results show that KS reduces the inflammatory response to protect mice against systemic GAS infection, which suggests that KS can be considered an adjunct agent for antimicrobial treatment. KS protein is routinely expressed and can be detected in human plasma under normal conditions. Our recent report showed that lower levels of KS have a trend toward predicting worse clinical outcomes in community-acquired pneumonia (44). When KS plasmid DNA was delivered 12 h following the initiation of GAS infection, the survival rate results showed protection by KS compared to the control groups, with a significant difference between the KS-treated group and saline control on days 7 and 8 after GAS infection (see Fig. S6 in the supplemental material). In conclusion, using a GAS infection animal model, the therapeutic and pharmacological effects of the anti-inflammatory drug KS has been demonstrated. This may provide valuable information for new therapeutic strategies in bacterial infections.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ai-Li Shiau for providing green fluorescent protein (GFP) and kallistatin-conditioned medium. We also thank Robert Anderson for critical reading of the manuscript.
This work was supported by grant NHRI-EX95-9429SP from National Health Research Institutes, Taiwan, grant NSC101-2320-B006-021-MY3 from National Science Council, Taiwan, and grants NCKUH-9903004, NCKUH-9902001, and NCKUH-10004003 from National Cheng Kung University Hospital.
Footnotes
Published ahead of print 19 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00322-13.
REFERENCES
- 1.Cole JN, Barnett TC, Nizet V, Walker MJ. 2011. Molecular insight into invasive group A streptococcal disease. Nat. Rev. Microbiol. 9:724–736 [DOI] [PubMed] [Google Scholar]
- 2.O'Connell D. 2006. Bacterial pathogenesis: triggering GAS invasive disease. Nat. Rev. Microbiol. 4:644–645 [Google Scholar]
- 3.Steer AC, Lamagni T, Curtis N, Carapetis JR. 2012. Invasive group A streptococcal disease: epidemiology, pathogenesis and management. Drugs 72:1213–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olsen RJ, Musser JM. 2010. Molecular pathogenesis of necrotizing fasciitis. Annu. Rev. Pathol. 5:1–31 [DOI] [PubMed] [Google Scholar]
- 5.Bisno AL, Brito MO, Collins CM. 2003. Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3:191–200 [DOI] [PubMed] [Google Scholar]
- 6.Chiappini N, Seubert A, Telford JL, Grandi G, Serruto D, Margarit I, Janulczyk R. 2012. Streptococcus pyogenes SpyCEP influences host-pathogen interactions during infection in a murine air pouch model. PLoS One 7:e40411. 10.1371/journal.pone.0040411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung CH, Tsao N, Zeng YF, Lu SL, Chuan CN, Lin YS, Wu JJ, Kuo CF. 2012. Synergistic effects of streptolysin S and streptococcal pyrogenic exotoxin B on the mouse model of group A streptococcal infection. Med. Microbiol. Immunol. 201:357–369 [DOI] [PubMed] [Google Scholar]
- 8.Thomas CL, Lee SW. 2012. Knowing is half the battle: targeting virulence factors of group A streptococcus for vaccine and therapeutics. Curr. Drug Targets 13:308–322 [DOI] [PubMed] [Google Scholar]
- 9.Musser JM, DeLeo FR. 2005. Toward a genome-wide systems biology analysis of host-pathogen interactions in group A streptococcus. Am. J. Pathol. 167:1461–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Currie BJ. 2006. Group A streptococcal infections of the skin: molecular advances but limited therapeutic progress. Curr. Opin. Infect. Dis. 19:132–138 [DOI] [PubMed] [Google Scholar]
- 11.Steer AC, Batzloff MR, Mulholland K, Carapetis JR. 2009. Group A streptococcal vaccines: facts versus fantasy. Curr. Opin. Infect. Dis. 22:544–552 [DOI] [PubMed] [Google Scholar]
- 12.Chao J, Tillman DM, Wang MY, Margolius HS, Chao L. 1986. Identification of a new tissue-kallikrein-binding protein. Biochem. J. 239:325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao J, Schmaier A, Chen LM, Yang Z, Chao L. 1996. Kallistatin, a novel human tissue kallikrein inhibitor: levels in body fluids, blood cells, and tissues in health and disease. J. Lab. Clin. Med. 127:612–620 [DOI] [PubMed] [Google Scholar]
- 14.Chao J, Stallone JN, Liang YM, Chen LM, Chao L. 1997. Kallistatin is a potent new vasodilator. J. Clin. Invest. 100:11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao RQ, Murakami H, Song Q, Chao L, Chao J. 2000. Kallistatin stimulates vascular smooth muscle cell proliferation and migration in vitro and neointima formation in balloon-injured rat artery. Circ. Res. 86:418–424 [DOI] [PubMed] [Google Scholar]
- 16.Miao RQ, Agata J, Chao L, Chao J. 2002. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood 100:3245–3252 [DOI] [PubMed] [Google Scholar]
- 17.Shiau AL, Teo ML, Chen SY, Wang CR, Hsieh JL, Chang MY, Chang CJ, Chao J, Chao L, Wu CL, Lee CH. 2010. Inhibition of experimental lung metastasis by systemic lentiviral delivery of kallistatin. BMC Cancer 10:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chao J, Yin H, Yao YY, Shen B, Smith RS, Chao L. 2006. Novel role of kallistatin in protection against myocardial ischemia-reperfusion injury by preventing apoptosis and inflammation. Hum. Gene Ther. 17:1201–1213 [DOI] [PubMed] [Google Scholar]
- 19.Shen B, Gao L, Hsu YT, Bledsoe G, Hagiwara M, Chao L, Chao J. 2010. Kallistatin attenuates endothelial apoptosis through inhibition of oxidative stress and activation of Akt-eNOS signaling. Am. J. Physiol. Heart Circ. Physiol. 299:1419–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Bledsoe G, Hagiwara M, Shen B, Chao L, Chao J. 2012. Depletion of endogenous kallistatin exacerbates renal and cardiovascular oxidative stress, inflammation, and organ remodeling. Am. J. Physiol. Renal Physiol. 303:1230–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stadnicki A, Mazurek U, Plewka D, Wilczok T. 2003. Intestinal tissue kallikrein-kallistatin profile in inflammatory bowel disease. Int. Immunopharmacol. 3:939–944 [DOI] [PubMed] [Google Scholar]
- 22.Zhu H, Chao J, Kotak I, Guo D, Parikh SJ, Bhagatwala J, Dong Y, Patel SY, Houk C, Chao L, Dong Y. 2013. Plasma kallistatin is associated with adiposity and cardiometabolic risk in apparently healthy African American adolescents. Metabolism 62:642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang CR, Chen SY, Wu CL, Liu MF, Jin YT, Chao L, Chao J. 2005. Prophylactic adenovirus-mediated human kallistatin gene therapy suppresses rat arthritis by inhibiting angiogenesis and inflammation. Arthritis Rheum. 52:1319–1324 [DOI] [PubMed] [Google Scholar]
- 24.Yin H, Gao L, Shen B, Chao L, Chao J. 2010. Kallistatin inhibits vascular inflammation by antagonizing tumor necrosis factor-α-induced nuclear factor κB activation. Hypertension 56:260–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen LM, Chao L, Chao J. 1997. Beneficial effects of kallikrein-binding protein in transgenic mice during endotoxic shock. Life Sci. 60:1431–1435 [DOI] [PubMed] [Google Scholar]
- 26.Frank AJ, Sheu CC, Zhao Y, Chen F, Su L, Gong MN, Bajwa E, Thompson BT, Christiani DC. 2012. BCL2 genetic variants are associated with acute kidney injury in septic shock. Crit. Care Med. 40:2116–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo CF, Wu JJ, Lin KY, Tsai PJ, Lee SC, Jin YT, Lei HY, Lin YS. 1998. Role of streptococcal pyrogenic exotoxin B in the mouse model of group A streptococcal infection. Infect. Immun. 66:3931–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo CF, Luo YH, Lin HY, Huang KJ, Wu JJ, Lei HY, Lin MT, Chuang WJ, Liu CC, Jin YT, Lin YS. 2004. Histopathologic changes in kidney and liver correlate with streptococcal pyrogenic exotoxin B production in the mouse model of group A streptococcal infection. Microb. Pathog. 36:273–285 [DOI] [PubMed] [Google Scholar]
- 29.Kuo CF, Chen CC, Luo YH, Huang RY, Chuang WJ, Sheu CC, Lin YS. 2005. Cordyceps sinensis mycelium protects mice from group A streptococcal infection. J. Med. Microbiol. 54:795–802 [DOI] [PubMed] [Google Scholar]
- 30.Li MH, Luo YH, Lin CF, Chang YT, Lu SL, Kuo CF, Hong JS, Lin YS. 2011. Dextromethorphan efficiently increases bactericidal activity, attenuates inflammatory responses and prevents group A streptococcal sepsis. Antimicrob. Agents Chemother. 55:967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu F, Song YK, Liu D. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6:1258–1266 [DOI] [PubMed] [Google Scholar]
- 32.Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, Dean DA, Liu D. 2004. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 11:675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong W, Tang CQ, Zhou GX, Chao L, Chao J. 1992. In vivo catabolism of human kallikrein-binding protein and its complex with tissue kallikrein. J. Lab. Clin. Med. 119:514–521 [PubMed] [Google Scholar]
- 34.Kobayashi SD, Braughton KR, Whitney AR, Voyich JM, Schwan TG, Musser JM, DeLeo FR. 2003. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U. S. A. 100:10948–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voyich JM, Musser JM, DeLeo FR. 2004. Streptococcus pyogenes and human neutrophils: a paradigm for evasion of innate host defense by bacterial pathogens. Microbes Infect. 6:1117–1123 [DOI] [PubMed] [Google Scholar]
- 36.Miller RA, Britigan BE. 1997. Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukaszewicz AC, Gontier G, Faivre V, Ouanounou I, Payen D. 2012. Elevated production of radical oxygen species by polymorphonuclear neutrophils in cerebrospinal fluid infection. Ann. Intensive Care 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao G, Shao C, Zhang SX, Dudley A, Fant J, Ma JX. 2003. Kallikrein-binding protein inhibits retinal neovascularization and decreases vascular leakage. Diabetologia 46:689–698 [DOI] [PubMed] [Google Scholar]
- 39.Stevens DL. 2000. Streptococcal toxin shock syndrome associated with necrotizing fasciitis. Annu. Rev. Med. 51:271–288 [DOI] [PubMed] [Google Scholar]
- 40.Hidalgo-Grass C, Mishalian I, Dan-Goor M, Belotserkovsky I, Eran Y, Nizet V, Peled A, Hanski E. 2006. A streptococcal protease that degrades CXC chemokines and impairs bacterial clearance from infected tissues. EMBO J. 25:4628–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medina E, Anders D, Chhatwal GS. 2002. Induction of NF-κB nuclear translocation in human respiratory epithelial cells by group A streptococci. Microb. Pathog. 33:307–313 [DOI] [PubMed] [Google Scholar]
- 42.Tsai PJ, Chen YH, Hsueh CH, Hsieh CH, Liu YH, Wu JJ, Tsou CC. 2006. Streptococcus pyogenes induces epithelial inflammatory responses through NF-κB/MAPK signaling pathways. Microbes Infect. 8:1440–1449 [DOI] [PubMed] [Google Scholar]
- 43.Tsao N, Lin TY, Chou CK, Wu JJ, Lin YS, Lei HY. 2001. Inhibition of group A streptococcus infection by carboxyfullerene. Antimicrob. Agents Chemother. 45:1788–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin WC, Lu SL, Lin CF, Chen CW, Chao L, Chao J, Lin YS. 8 February 2013. Plasma kallistatin levels in patients with severe community-acquired pneumonia. Crit. Care 17:R27. [Epub ahead of print.] 10.1186/cc12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.