

Abstract
Dolutegravir (DTG) and GSK1265744 are HIV integrase inhibitors (INIs) in clinical development. The oral formulation of rilpivirine (RPV), a nonnucleoside reverse transcriptase inhibitor (NNRTI), has been approved for treatment-naive HIV infection. Long-acting depot injections of GSK1265744 and RPV are also being developed. This study evaluated the potential for drug interactions between RPV and these INIs. This phase 1, open-label, two-cohort, three-period, single-sequence crossover study evaluated oral coadministration of RPV with DTG or GSK1265744. Healthy subjects received DTG (50 mg every 24 h for 5 days) or GSK1265744 (30 mg every 24 h for 12 days) in period 1 followed by a washout, RPV (25 mg every 24 h for 11 or 12 days) in period 2, immediately followed by RPV (25 mg every 24 h) plus DTG (50 mg every 24 h) for 5 days or GSK1265744 (30 mg every 24 h) for 12 days in period 3. Steady-state pharmacokinetic (PK) parameters were estimated using noncompartmental analysis of data collected on the last day of each period. The combinations of RPV and DTG (n = 16) and of RPV and GSK1265744 (n = 11) were well tolerated; no grade 3 or 4 adverse events (AEs) or AE-related discontinuations were observed. The 90% confidence intervals for the area under the curve from time zero until the end of the dosage interval [AUC0–τ] and maximum concentration of drug in serum (Cmax) geometric mean ratios were within 0.8 to 1.25. Following administration of DTG + RPV, DTG and RPV Cτ increased by 22% and 21%, respectively. Following administration of GSK1265744 + RPV, RPV Cτ decreased 8%. DTG and GSK1265744 can be administered with RPV without dosage adjustment for either agent. These results support coadministration of RPV with DTG or GSK1265744 as either oral or long-acting depot injection regimens. (This study has been registered at ClinicalTrials.gov under registration no. NCT01467531.)
INTRODUCTION
There remains a need for simple, effective, and well-tolerated regimens for HIV infection. Oral, low-dose, once-daily regimens without pharmacokinetic (PK) boosters and long-acting injectable regimens are examples of antiretroviral treatments that have the potential to demonstrate significant advantages in tolerability and convenience. A nonstandard two-drug combination of an integrase inhibitor (INI) and a nonnucleoside reverse transcriptase inhibitor (NNRTI) may be an attractive option to achieve these goals.
Dolutegravir (DTG) is an HIV INI that has demonstrated efficacy in treatment-naive, treatment-experienced but INI-naive, and INI-resistant subjects (1–3). DTG is primarily metabolized via UDP-glucuronosyltransferase (UGT) 1A1, with cytochrome P450 (CYP) 3A4 being a notable pathway, and is not a significant inhibitor of phase I or II metabolism (4).
GSK1265744 is another INI currently in clinical development as oral and long-acting parenteral formulations. GSK1265744 has produced significant −2.2 to −2.5 log10 reductions in HIV RNA following short-term oral monotherapy at doses of 5 to 30 mg once daily in treatment-naive subjects infected with HIV (5). In addition, single intramuscular and subcutaneous injections have achieved prolonged plasma exposures to GSK1265744 for up to 52 weeks in healthy subjects (6). Preliminary findings suggest that GSK1265744 is primarily metabolized via UGT, with low potential for drug interactions as a perpetrator or victim. Coadministration with etravirine (a CYP3A inducer and an inhibitor of CYP2C9, CYP2C19, and Pgp) did not affect the concentrations of GSK1265744 in plasma (7).
Rilpivirine (RPV, TMC278), an NNRTI, is approved for once-daily oral administration in combination with other antiretroviral agents in treatment-naive patients and has a long-acting parenteral formulation currently in development that has shown sustained concentrations in plasma upon single and repeated monthly injections (8). RPV is a substrate of CYP3A. Standard clinical doses of RPV (25 mg once daily) do not affect CYP3A enzyme activity, and RPV has demonstrated no significant effect on the exposure of CYP3A4 substrates and does not induce or inhibit UGT (9). This study evaluated the potential for drug interactions between RPV and either DTG or GSK1265744. Confirmation of the lack of any drug interaction between these drugs would support future coadministration of oral or long-acting parenteral formulations.
MATERIALS AND METHODS
The bidirectional drug-drug interactions between RPV and DTG or GSK1265744 were evaluated in two separate cohorts of healthy subjects. Written informed consent was obtained from all subjects, and the institutional review board of the study site, IntegReview, Inc. (Austin, TX), approved the protocol. The study (ClinicalTrials.gov registration no. NCT01467531) was initiated on 7 November 2011 (first subject, first visit) and was completed on 20 February 2012 (last subject, last visit).
This was a phase 1, open-label, two-panel, single-sequence, crossover study in healthy male and female adults. A steady-state design was selected to address any potential induction effects and simulate clinically relevant exposures. Sixteen healthy subjects enrolled in cohort 1 received DTG (50 mg every 24 h) for 5 days alone in period 1 followed by a ≥7-day washout, RPV (25 mg every 24 h) for 11 days alone in period 2, immediately followed by DTG (50 mg) plus RPV (25 mg) every 24 h for 5 days in period 3. Differences in duration of each drug were based on half-lives and time to steady-state (RPV, DTG, and 744 half-lives of 50, 14, and 40 h, respectively). No washout was needed between periods 2 and 3 as steady-state data had been collected on the integrase inhibitor alone (period 1) and on RPV alone (period 2). The integrase inhibitor was then added back in with RPV (period 3) and dosed to steady state. Twelve healthy subjects enrolled in cohort 2 received GSK1265744 (30 mg every 24 h) for 12 days alone in period 1 followed by a ≥14-day washout and then RPV (25 mg every 24 h) for 12 days alone in period 2, immediately followed by GSK1265744 (30 mg) plus RPV (25 mg) every 24 h for 12 days in period 3. Sample sizes were selected for both cohorts based on known within-subject variability of DTG, GSK1265744, and RPV PK parameters such that the 90% confidence interval (CI) would be approximately 0.806 to 1.24 if the point estimate of the geometric mean treatment ratio were 1.
Subject eligibility was determined by medical history, physical examination, and laboratory screening tests. Healthy volunteers of either sex between the ages of 18 and 55 years (inclusive) at the time of signing the informed consent and with a body mass index (BMI) within the range of 18.5 to 31.0 kg/m2 (inclusive) were eligible to participate in the study. Male subjects with female partners of childbearing potential agreed to use specified contraception methods from the time of the first dose of the study medication until 14 days after the last dose of the study medication. Female subjects were required to be of non-childbearing potential, defined as premenopausal females with a documented tubal ligation or hysterectomy or postmenopausal, defined as 12 months of spontaneous amenorrhea with confirmatory follicle-stimulating hormone of >40 mIU/ml and estradiol of <40 pg/ml (<147 pmol/liter).
Subjects were ineligible if they had hypersensitivity to any of the study medications, a positive prestudy hepatitis B surface antigen, a positive hepatitis C antibody result within 3 months of screening, a positive test for HIV antibody, history of liver disease, or known hepatobiliary abnormalities. Pregnant or lactating females were not eligible. Subjects could not have participated in a clinical trial and received an investigational product within 30 days prior to the first dosing day in the current study or had a current history of regular use of tobacco- or nicotine-containing products within 6 months of screening. Use of prescription or nonprescription drugs, including vitamins or herbal and dietary supplements (e.g., St. John Wort), was precluded within 7 days (or 14 days if the drug was a potential enzyme inducer) prior to the first dose of study medication. Subjects had screening visits within 30 days prior to the first dose of the study drug and prior to the three treatment periods and a follow-up visit 7 to 14 days after the last dose of the study drug.
RPV was administered with food, given that exposures are increased ∼40% following a meal compared with a fasted state (9). Although GSK1265744 and DTG do not have clinically significant food effects, all doses in this study were given in the morning with a moderate-fat (30%) meal.
In cohort 1, blood samples (2 ml) were collected in K3EDTA tubes for determination of DTG concentrations in plasma on day 5 of periods 1 and 3 at predose (within 15 min prior to dosing) and 1, 2, 3, 4, 8, 12, and 24 h after the morning dose. In cohort 2, blood samples (2 ml) were collected in K3EDTA tubes for determination of plasma GSK1265744 concentrations during periods 1 and 3 at predose on days 10, 11, and 12 and at 1, 2, 3, 4, 8, 12, and 24 h after the morning dose on day 12. In both cohorts, blood samples (3 ml) were collected in Li-heparin tubes for determination of RPV concentrations in plasma on the final day of dosing during periods 2 and 3 at predose and 1, 2, 3, 4, 5, 6, 9, 12, 16, and 24 h after the morning dose. In addition, predose blood samples were collected on the 2 days prior to each intensive PK day for the assessment of steady-state conditions for all analytes. Safety evaluations, including adverse event (AE) assessments, vital signs, laboratory testing, and electrocardiographic studies, were performed at regular intervals throughout the trial.
Bioanalytical methods. (i) Dolutegravir.
DTG concentrations were determined by QPS, LLC (Newark, DE), under the management of Bioanalytical Sciences and Toxicokinetics, Drug Metabolism and Pharmacokinetics of GlaxoSmithKline (Research Triangle Park, NC) using validated, high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) methods following extraction from plasma by protein precipitation. Data were acquired and processed (integrated) using the proprietary software application Analyst (version 1.4 or higher; MDS Sciex, Concord, Ontario, Canada) and Watson LIMS (version 6.4.0.04; Thermo Scientific, Waltham, MA). ERC-349572-d7 15N1 was used as an internal standard. The validated linear concentration range was 20 to 20,000 ng/ml, and quality control (QC) samples were included in each run at 60, 1,600, 16,000, and 20,000 ng/ml. Based on the results of the QC sample analysis, the bias (% relative error) ranged from −5.7% to 3.6%, and precision (percent coefficient of variation [CV%]) was <5.0%.
(ii) GSK1265744.
GSK1265744 concentrations were determined by Bioanalytical Sciences and Toxicokinetics, Drug Metabolism and Pharmacokinetics of GlaxoSmithKline using a validated HPLC-MS/MS assay with a TurboIonSpray (AB Sciex, Framingham, MA) interface and positive-ion multiple-reaction monitoring following extraction from plasma by protein precipitation using acetonitrile. Data were acquired and processed using the proprietary software application Analyst (version 1.4 or higher; MDS Sciex, Canada) and the GlaxoSmithKline Study Management System, SMS2000 (version 2.3; GlaxoSmithKline). The internal standard was [13C2H215N]-GSK1265744. This method was validated over the range of 10 to 10,000 ng/ml. QC samples were included in each run at 30, 800, and 8,000 ng/ml. Based on the results of the QC sample analysis, the bias ranged from 4.3% to 9.4%, and precision ranged from 2.4% to 3.9%.
(iii) Rilpivirine.
RPV concentrations were determined by Frontage Laboratories (Shanghai) Co., Ltd. (Zhangjiang High-Tech Park, Shanghai, People's Republic of China), under the management of Janssen Research & Development, a division of Janssen Pharmaceutica N.V. (Beerse, Belgium), using a validated HPLC-MS/MS method following protein precipitation. Data were acquired and quantified using the proprietary software application Analyst (version 1.4.2; Applied Biosystems, Foster City, CA) and Watson 7.3 LIMS (Thermo Scientific). The internal standard was JNJ-ZBJOB358. This method was validated over the range of 1.0 to 2,000 ng/ml. QC samples were included in each run at 2.73, 54.6, and 1,560 ng/ml. Based on the results of the QC sample analysis, the bias ranged from −5.9% to 2.6%, and intra- and interrun precision ranged from 0.4% to 6.8% and 3.1% to 5.8%, respectively.
Pharmacokinetic analysis.
Individual plasma concentration-time data were analyzed with model 200 for extravascular administration of the WinNonlin Professional software (version 5.2; Pharsight Corp., Mountain View, CA). Actual recorded sampling times for each individual profile were used to determine plasma DTG, GSK1265744, and RPV PK parameters, which included the area under the curve from time zero until the end of the dosage interval (AUC0–τ) using the linear-up/log-down approach to the trapezoidal rule, the observed maximum plasma concentration (Cmax), the time to observed maximum concentration in plasma (Tmax), and the concentration in plasma at the end of the dosage interval (Cτ).
Statistical analysis.
PK parameter values for each analyte were summarized by treatment. Analysis of variance, considering the treatment as a fixed effect and the subject as a random effect, was performed using a mixed-linear models procedure (version X; SAS Institute, Cary, NC) to compare the plasma DTG, GSK1265744, and RPV parameters when drugs were administered in combination (test treatments) to those following administration of each drug alone (reference treatments). All of the PK parameters were log-transferred, except Tmax, for treatment comparisons. Point estimates and their associated 90% CIs were constructed for the differences in test and reference treatments.
RESULTS
Demographics.
Sixteen subjects were enrolled and completed all treatments in cohort 1. Twelve subjects were enrolled in cohort 2, and 11 completed all three treatments. One subject was withdrawn in period 3 by the investigator for noncompliance with study procedures. Baseline patient demographics are summarized in Table 1. Both cohorts had more than 80% male participants, and there were no relevant demographic differences between the cohorts.
Table 1.
Baseline demographics
| Demographic parameter | Cohort 1 (n = 16) | Cohort 2 (n = 12) | Overall (n = 28) |
|---|---|---|---|
| Age (yr), mean (SD) | 34.4 (11.33) | 27.4 (10.57) | 31.4 (11.37) |
| Sex, n (%) | |||
| Female | 2 (13) | 2 (17) | 4 (14) |
| Male | 14 (88) | 10 (83) | 24 (86) |
| BMI (kg/m2), mean (SD) | 26.51 (2.58) | 27.03 (3.24) | 26.73 (2.83) |
| Height (cm), mean (SD) | 178.50 (10) | 175.75 (12.71) | 177.32 (11.11) |
| Weight (kg), mean (SD) | 84.46 (10.56) | 83.74 (15.23) | 84.15 (12.51) |
| Race, n (%) | |||
| African American/African heritage | 4 (25) | 7 (58) | 11 (39) |
| American Indian or Alaska native | 1 (6) | 0 | 1 (4) |
| Native Hawaiian or other Pacific Islander | 1 (6) | 0 | 1 (4) |
| White/Caucasian/European heritage | 10 (63) | 5 (42) | 15 (54) |
Safety.
No serious AEs (SAEs) or grade 3 or 4 AEs were reported, and no subjects were discontinued due to AEs. In addition, no laboratory abnormalities were designated AEs during the study. The incidence of drug-related AEs was low. The only drug-related AE reported in more than one subject was headache, occurring in 1 of the 16 subjects in the RPV-alone arm of cohort 1 and in 3 of the 16 subjects in the DTG-plus-RPV arm. Other AEs occurring in one subject each included abnormal dreams and insomnia (RPV alone), dizziness and dyspepsia (RPV plus DTG), and decreased appetite (GSK1265744 alone and GSK1265744 plus RPV).
Pharmacokinetics.
PK parameters for all analytes are summarized in Table 2, and statistical analyses are presented in Table 3. Overall, PK was not significantly altered for any of the drugs when given in combination compared with administration alone. Mean (standard deviation [SD]) steady-state plasma DTG concentration-time profiles following administration of DTG with and without RPV are shown in Fig. 1. Coadministration of RPV with DTG had no effect on DTG AUC0–τ or Cmax and increased DTG Cτ by 22% (Table 3).
Table 2.
DTG, GSK1265744, and RPV PK parameter summary
| Plasma PK parameter | Geometric mean [95% CI] (CVb%)c |
|||||||
|---|---|---|---|---|---|---|---|---|
| DTG |
GSK1265744 |
RPV |
||||||
| DTG (n = 16) | DTG + RPV (n = 16) | GSK1265744 (n = 11) | GSK1265744 + RPV (n = 11) | RPV (n = 16) | DTG + RPV (n = 16) | RPV (n = 11) | GSK1265744 + RPV (n = 11) | |
| AUC0–τa | 48.8 [40.8, 58.4] (35) | 54.7 [46.1, 64.9] (33) | 142 [118, 171] (28) | 159 [138, 183] (21) | 2,227 [1,872, 2,649] (33) | 2,368 [1,985, 2,825] (34) | 2,473 [2,034, 3,008] (30) | 2,441 [1,916, 3,110] (37) |
| Cmaxb | 3.46 [2.96, 4.04] (30) | 3.90 [3.43, 4.44] (25) | 8.22 [6.83, 9.89] (28) | 8.65 [7.69, 9.72] (18) | 148 [128, 173] (29) | 164 [136, 197] (36) | 171 [137, 213] (34) | 165 [120, 226] (50) |
| Cτb | 1.07 [0.850, 1.34] (45) | 1.31 [1.04, 1.65] (46) | 4.65 [3.73, 5.78] (33) | 5.29 [4.49, 6.23] (25) | 74.5 [60.3, 92.2] (41) | 90.5 [70.9, 115] (48) | 87.4 [66.8, 114] (42) | 80.3 [58.6, 110] (50) |
Values are in μg × h/ml for DTG and GSK1265744 and in ng × h/ml for RPV.
Values are in μg/ml for DTG and GSK1265744 and in ng/ml for RPV.
CVb%, between-subject variability.
Table 3.
PK parameter treatment comparisons
| Plasma PK parameter | Geometric least-squares mean ratio [90% CI] |
|||
|---|---|---|---|---|
| DTG + RPV versus DTG | GSK1265744 + RPV versus GSK1265744 | DTG + RPV versus RPV | GSK1265744 + RPV versus RPV | |
| AUC0–τ | 1.12 [1.05, 1.19] | 1.12 [1.05, 1.19] | 1.06 [0.976, 1.16] | 0.987 [0.890, 1.09] |
| Cmax | 1.13 [1.06, 1.21] | 1.05 [0.963, 1.15] | 1.10 [0.992, 1.22] | 0.963 [0.849, 1.09] |
| Cτ | 1.22 [1.15, 1.30] | 1.14 [1.04, 1.24] | 1.21 [1.07, 1.38] | 0.919 [0.789, 1.07] |
Fig 1.
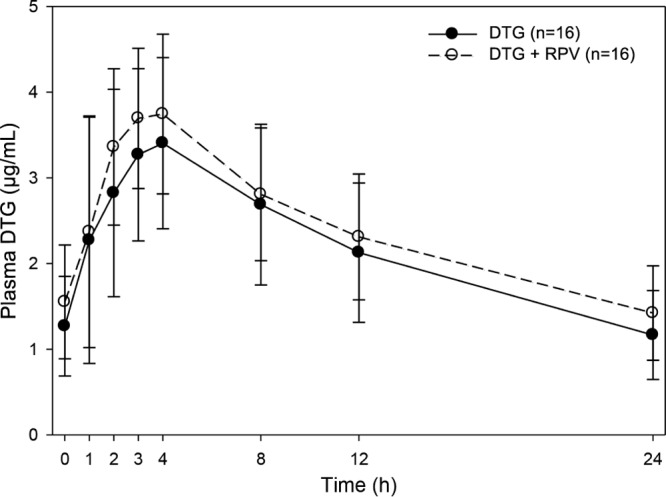
Mean (SD) steady-state DTG concentration-time profiles following administration of DTG with and without RPV.
Mean (SD) steady-state plasma GSK1265744 concentration-time profiles following administration of GSK1265744 with and without RPV are shown in Fig. 2. Coadministration of RPV with GSK1265744 had no effect on GSK1265744 PK parameters (Table 3).
Fig 2.
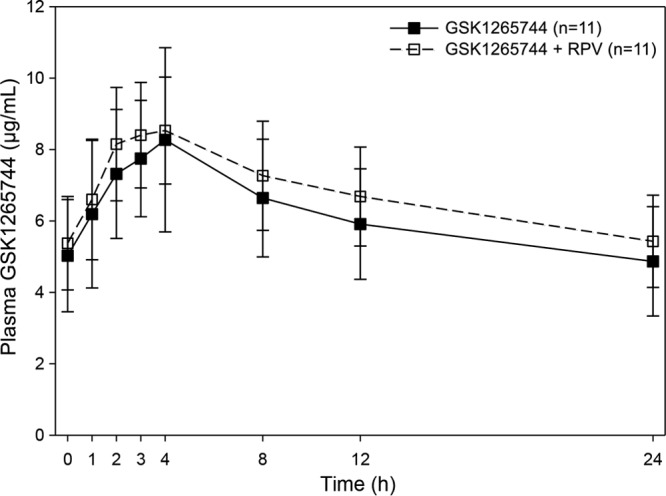
Mean (SD) steady-state GSK1265744 concentration-time profiles following administration of GSK1265744 with and without RPV.
Mean steady-state plasma RPV concentration-time profiles following administration of RPV alone in cohorts 1 and 2 and with DTG or GSK1265744 are shown in Fig. 3. Coadministration of DTG with RPV had no effect on RPV AUC0–τ or Cmax and increased RPV Cτ by 21%. Coadministration of GSK1265744 with RPV had no effect on RPV AUC0–τ or Cmax and decreased Cτ by 8% (Table 3).
Fig 3.

Mean (SD) steady-state plasma RPV concentration-time profiles following administration of RPV with and without DTG or GSK1265744.
DISCUSSION
RPV and GSK1265744 are being developed as long-acting injectable antiretroviral agents, capable of being administered on an infrequent basis (e.g., once a month). This type of novel regimen may extend opportunities for therapeutic intervention to underserved patient populations and could be an attractive option for certain subjects infected with HIV at risk of poor adherence to an oral regimen. The combination of DTG and RPV could make an attractive oral regimen since both are administered once daily at low doses. Therefore, a drug-interaction study of these new INIs with RPV using oral formulations was conducted to determine whether the PK of either drug was altered during concomitant dosing. This evaluation was warranted to guide future dosing and support clinical trials of these combinations in patients infected with HIV.
The likelihood of a significant drug interaction between RPV and DTG was considered to be low. Several clinical studies (10–13) in healthy subjects have shown that no dosage adjustments are necessary for DTG when combined with other antiretroviral agents. Coadministration with the NNRTI etravirine, a known UGT and CYP3A4 inducer, is not recommended due to the observed 88% reduction in DTG Cτ, although the interaction is mitigated by concomitant administration of lopinavir and RTV or of darunavir and RTV (14). Unlike etravirine, RPV does not induce UGTs and was not expected to interact with DTG, which is primarily metabolized by UGT 1A1.
No clinically significant changes in the PK of DTG or RPV were observed. Mean plasma AUC0–τ and Cmax of each drug were modestly increased during coadministration compared with dosing alone. The 90% confidence intervals for AUC and Cmax were contained within the interval of 0.8 to 1.25, which is generally considered not to be of clinical concern as per FDA guidelines on drug interaction studies (15). DTG and RPV Cτ were increased 22% and 21%, respectively. These modest increases are not clinically relevant or likely to be associated with safety concerns, as neither drug is considered to have a narrow therapeutic window for safety. It is unknown whether an ∼20% increase in Cτ would provide an advantage for efficacy.
Similarly, no interaction was expected between RPV and GSK1265744. In preliminary investigations, GSK1265744 has demonstrated no direct inhibition (half-maximal inhibitory concentration, >33 μM) for CYP1A2, 2D6, and 3A4 and no metabolism-dependent inhibition of CYP3A4 in vitro. In addition, there is a low potential for GSK1265744 to cause activation of the PXR target gene and cause CYP2B or CYP3A induction in vivo. RPV at the recommended dose does not inhibit or induce CYP enzyme activity and does not interfere with UGT. Following coadministration of GSK1265744 and RPV, PK parameters of each drug were similar to those when dosed alone. CIs associated with modest increases in geometric mean GSK1265744 AUC0–τ, Cmax, and Cτ met the criteria for bioequivalence. The modest 8% decrease in RPV Cτ following coadministration with GSK1265744 was not considered clinically relevant. It should be noted that, in general, the inter- and intraindividual variability for the Cτ of RPV is larger than that forAUC0–τ, and the AUC0–τ within-subject ratio is considered a more robust comparison within the setting of intensive PK evaluation for RPV. The clinical data in this study confirm a previous study of GSK1265744 and RPV in rats and rhesus macaques that demonstrated similar exposures of each drug in combination compared with dosing alone (unpublished data). In addition, the results were consistent with expectations given the lack of interaction between the known inducer (UGT and CYP3A4) etravirine and GSK1265744 (7).
Both combinations were well tolerated in this study, with a low incidence of drug-related AEs. The only drug-related AE in cohort 2 (GSK1265744 plus RPV) was a report of decreased appetite during period 3 of coadministration. The most common AE in the DTG plus RPV cohort was headache, which is a common AE in phase 1 studies given the caffeine restrictions. As such, the short-term safety and tolerability of RPV with GSK1265744 and DTG support longer-term studies. No new or clinically significant safety issues were identified for DTG, GSK1265744, or RPV compared with their known safety profiles (16).
Overall, the observed lack of PK interactions and the favorable short-term safety profile of RPV and DTG or GSK1265744 support coadministration of either INI with RPV in oral or long-acting depot injection regimens.
ACKNOWLEDGMENTS
Funding for this work was provided by ViiV Healthcare and Janssen Pharmaceuticals. The authors are employees of GlaxoSmithKline and Janssen, the manufacturers of the study drugs. All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors.
We acknowledge Sherri Damlo for editorial assistance during the development of the manuscript.
Footnotes
Published ahead of print 26 August 2013
REFERENCES
- 1.Raffi F, Rachlis A, Stellbrink H-J, Hardy WD, Torti C, Orkin C, Bloch M, Podzamczer D, Pokrovsky V, Pulido F, Almond S, Margolis D, Brennan C, Min S, SPRING-2 Study Group 2013. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet 381:735–743 [DOI] [PubMed] [Google Scholar]
- 2.Eron JJ, Clotet B, Durant J, Katlama C, Kumar P, Lazzarin A, Poizot-Martin I, Richmond G, Soriano V, Ait-Khaled M, Fujiwara T, Huang J, Min S, Vavro C, Yeo J, VIKING Study Group 2013. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING study. J. Infect. Dis. 207:740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, Richmond G, Buendia CB, Fourie J, Ramgopal M, Hagins D, Felizarta F, Madruga J, Reuter T, Newman T, Small CB, Lombaard J, Grinsztejn B, Dorey D, Underwood M, Griffith S, Min S. 2013. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet pii:S0140–6736(13)61221–0 [Epub ahead of print]. 10.1016/S0140-6736(13)61221-0 [DOI] [PubMed] [Google Scholar]
- 4.Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E, Webster LO, Harmon KA, Clarke JD, Polli JW. 2013. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab. Dispos. 41:353–361 [DOI] [PubMed] [Google Scholar]
- 5.Spreen W, Min S, Ford SL, Chen S, Lou Y, Bomar M, St. Clair M, Piscitelli S, Fujiwara T. Pharmacokinetics, safety, and monotherapy antiviral activity of GSK1265744, an HIV integrase strand transfer inhibitor. HIV Clin. Trials, in press [DOI] [PubMed] [Google Scholar]
- 6.Spreen W, Ford SL, Chen S, Gould E, Wilfret D, Subich D, Taishi T, Hong Z. 2012. Pharmacokinetics, safety and tolerability of the HIV integrase inhibitor S/GSK1265744 long acting parenteral nanosuspension following single dose administration to healthy adults, poster A-452-0100-10191. Abstr. 19th Int. AIDS Conf. Int. AIDS Soc., Washington, DC [Google Scholar]
- 7.Ford SL, Gould E, Chen S, Lou Y, Dumont E, Spreen W, Piscitelli S. 2013. Effects of etravirine on the pharmacokinetics of the integrase inhibitor S/GSK1265744. Antimicrob. Agents Chemother. 57:277–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baert L, van 't Klooster G, Dries W, François M, Wouters A, Basstanie E, Iterbeke K, Stappers F, Stevens P, Schueller L, Van Remoortere P, Kraus G, Wigerinck P, Rosier J. 2009. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur. J. Pharm. Biopharm. 72:502–508 [DOI] [PubMed] [Google Scholar]
- 9.Tibotec 2011. Edurant (rilpivirine) prescribing information. Tibotec, Raritan, NJ [Google Scholar]
- 10.Song I, Borland J, Chen S, Lou Y, Peppercorn A, Wajima T, Min S, Piscitelli SC. 2011. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next generation HIV integrase inhibitor, S/GSK1349572. Br. J. Clin. Pharmacol. 72:103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song I, Min SS, Borland J, Lou Y, Chen S, Patel P, Ischibashi T, Piscitelli SC. 2011. The effect of lopinavir/ritonavir and darunavir/ritonavir on the HIV integrase inhibitor S/GSK1349572 in healthy participants. J. Clin. Pharmacol. 51:237–242 [DOI] [PubMed] [Google Scholar]
- 12.Song I, Min SS, Borland J, Lou Y, Chen S, Ishibashi T, Wajima T, Piscitelli SC. 2010. Lack of interaction between the HIV integrase inhibitor S/GSK1349572 and tenofovir in healthy subjects. J. Acquir. Immune Defic. Syndr. 55:365–367 [DOI] [PubMed] [Google Scholar]
- 13.Patel P, Song I, Borland J, Patel A, Lou Y, Chen S, Wajima T, Peppercorn A, Min SS, Piscitelli SC. 2011. Pharmacokinetics of the HIV integrase inhibitor S/GSK1349572 co-administered with acid-reducing agents and multivitamins in healthy volunteers. J. Antimicrob. Chemother. 66:1567–1572 [DOI] [PubMed] [Google Scholar]
- 14.Song I, Borland J, Min S, Lou Y, Chen S, Patel P, Wajima T, Piscitelli SC. 2011. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob. Agents Chemother. 55:3517–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.FDA February 2012. Food and Drug Administration guidance for industry drug interaction studies—study design data analysis implications for dosing and labeling recommendations. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf
- 16.Cohen CJ, Molina J-M, Cahn P, Clotet B, Fourie J, Grinsztejn B, Wu H, Johnson MA, Saag M, Supparatpinyo K, Crauwels H, Lefebvre E, Rimsky LT, Vanveggel S, Williams P, Boven K, ECHO and THRIVE Study Groups 2012. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive, HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE trials. J. Acquir. Immune Defic. Syndr. 60:33–42 [DOI] [PubMed] [Google Scholar]