

Abstract
GSK2485852 (referred to here as GSK5852) is a hepatitis C virus (HCV) NS5B polymerase inhibitor with 50% effective concentrations (EC50s) in the low nanomolar range in the genotype 1 and 2 subgenomic replicon system as well as the infectious HCV cell culture system. We have characterized the antiviral activity of GSK5852 using chimeric replicon systems with NS5B genes from additional genotypes as well as NS5B sequences from clinical isolates of patients infected with HCV of genotypes 1a and 1b. The inhibitory activity of GSK5852 remained unchanged in these intergenotypic and intragenotypic replicon systems. GSK5852 furthermore displays an excellent resistance profile and shows a <5-fold potency loss across the clinically important NS5B resistance mutations P495L, M423T, C316Y, and Y448H. Testing of a diverse mutant panel also revealed a lack of cross-resistance against known resistance mutations in other viral proteins. Data from both the newer 454 sequencing method and traditional population sequencing showed a pattern of mutations arising in the NS5B RNA-dependent RNA polymerase in replicon cells exposed to GSK5852. GSK5852 was more potent than HCV-796, an earlier inhibitor in this class, and showed greater reductions in HCV RNA during long-term treatment of replicons. GSK5852 is similar to HCV-796 in its activity against multiple genotypes, but its superior resistance profile suggests that it could be an attractive component of an all-oral regimen for treating HCV.
INTRODUCTION
Infection with hepatitis C virus (HCV) often leads to chronic infection, resulting in an estimated 160 million people infected worldwide (1). HCV is the leading cause of death from liver disease and the leading indication for liver transplantation in the United States (2). Recently, 2 protease inhibitors, boceprevir and telaprevir, were approved for the treatment of HCV when used in combination with pegylated alpha interferon (IFN-α) and ribavirin (3). Although the addition of these direct-acting antivirals (DAAs) to IFN-α and ribavirin has resulted in a substantial increase in the sustained viral response (SVR) rate and in some cases a shortening of the duration of IFN-α and ribavirin treatment from 48 weeks to 28 or 24 weeks, there is still a need for newer therapies because of the side effects associated with the currently approved regimens. IFN-α therapy is associated with fatigue, headache, myalgia, fever, and nausea (4–6), while ribavirin can cause hemolytic anemia (7, 8). Patients taking telaprevir had increased incidences of rash and anemia (9, 10), while boceprevir usage was associated with anemia and dysgeusia (11, 12) when used in combination with IFN-α and ribavirin. Additional DAAs are being combined in clinical trials to explore treatment regimens that no longer require IFN-α or ribavirin (13, 14).
HCV was first identified as the major etiological agent of parenteral non-A, non-B hepatitis in 1989 (15). The HCV genome is a single-stranded, positive-sense RNA encoding a single polyprotein precursor (16, 17) that is processed by host and viral proteases into structural and nonstructural proteins responsible for viral RNA replication and assembly into viral particles (for a review, see reference 18). The NS5B protein is an RNA-dependent RNA polymerase (RDRP) that is responsible for replicating the viral genome. The high nucleotide misincorporation rate of NS5B (19) has led to the genetic diversity seen in HCV. A phylogenetic analysis of these diverse HCV sequences resulted in their classification into 6 major genotypes that differ by 30 to 40% at the nucleotide level (20). The response to therapies or their ability to be used at all may depend upon the HCV genotype that is infecting the patient. Pegylated IFN-α and ribavirin treatment can achieve SVR rates of 70 to 80% in genotype 2- or 3-infected patients, but the SVR rate is less than 50% for genotype 1- or 4-infected patients (4, 5). Telaprevir is not active against genotypes 3 and 4 of HCV (21, 22) and is approved only for treatment of genotype 1-infected patients. There is a clear need for agents with pangenotypic activity due to the diversity of HCV genotypes and their differential responses to current therapies.
NS5B is the HCV RDRP and is a clinically validated target for therapeutic intervention, with many compounds in development (23, 24). These agents can be broadly classified into nucleoside and nonnucleoside inhibitors (NIs and NNIs). NIs are an attractive class because they are active against all of the HCV genotypes and appear to have a high genetic barrier to resistance, although several have failed in the clinic due to adverse events (25). NNIs are classified by their binding to one of 4 allosteric sites on NS5B identified through structural studies and associated with distinctive but sometimes overlapping resistance mutations (24, 26). NNIs are generally regarded as having a low genetic barrier to resistance because single nucleotide changes can result in viable resistant variants that emerge rapidly during monotherapy. They are also at a disadvantage compared to NIs because most NNIs do not inhibit all HCV genotypes. However, due to the conservation of the binding site residues across HCV genotypes, NNI palm site 2 inhibitors have the best potential to have pangenotypic activity among the NNIs, as was demonstrated in the clinic by HCV-796 (Fig. 1) (27). Unfortunately, safety issues resulted in the termination of its development (28). Substantial decreases in potency were also observed for amino acid variants at position 316 (29). We have developed NNI palm site 2 inhibitors with improvements in activity against HCV-796-resistant mutations (30). We describe here the characterization of GSK2485852 (referred to here as GSK5852) as an NNI palm site 2 inhibitor that has a high genetic barrier to resistance and has the potential to be a pangenotypic HCV inhibitor.
Fig 1.
Structures of GSK5852 and HCV-796.
MATERIALS AND METHODS
Cell lines.
Stable cell lines carrying a bicistronic genotype 1a (H77), genotype 1b (Con1), or genotype 2a (JFH-1) replicon were created in-house, licensed from ReBLikon GmbH (Mainz, Germany), or created in-house, respectively (31–33). All three replicons express luciferase, neomycin phosphotransferase, and HCV NS3-5B. The cysteine at position 316 of NS5B in Con1 was changed to asparagine by site-directed mutagenesis for generating the stable cell line genotype 1b (C316N). Cells were maintained subconfluent and were split 1:4 to 1:6 twice a week in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), GlutaMAX-1, nonessential amino acids, and 500 μg/ml Geneticin. ET cured cells are a derivative of Con1 cells generated by treating Con1 cells with IFN-α for several passages until HCV RNA levels are undetectable. Huh-7 Lunet cells were licensed from ReBLikon GmbH (Mainz, Germany). Huh7.5 cells were licensed from Apath LLC (Brooklyn, NY). Non-replicon-containing cell lines were maintained as subconfluent monolayers, split 1:4 to 1:6 twice a week, in DMEM supplemented with 10% FBS, nonessential amino acids, glutamine, and penicillin-streptomycin (complete media). MT-4 cells were from B. Larder. HepG2 and Vero cell lines were obtained from ATCC.
Preparation of replicon mutants.
A modified genotype 1a replicon, referred to as the 1a fit replicon (34), and the Con1 replicon were mutagenized by site-directed mutagenesis. Single-point mutations were generated by using the QuikChange mutagenesis kit (Stratagene) according to the manufacturer's instructions or by GenScript USA Inc. (Piscataway, NJ), an external contract organization, using standard molecular biology techniques. Plasmid isolation was performed using mini- or maxiprep kits (Qiagen, Valencia, CA) according to the manufacturer's instructions.
The replication capacity was determined by transient transfection of NS5B mutant replicons and by measuring the replication level as the ratio of the firefly luciferase signal at 96 h posttransfection to the firefly luciferase signal at 4 h posttransfection normalized to the replication levels of the corresponding genotype 1a or 1b reference replicons.
Cloning of NS5B sequences for chimeras containing sequences from genotypes 1 to 6.
The identification of NS5B sequences for making chimeric replicons is described in the supplemental material. GenBank accession numbers for the patient sequences used are included in Table S1 in the supplemental material.
Full-length NS5B patient or consensus sequences were synthesized and used to replace the NS5B sequence of a plasmid containing the bicistronic genotype 1b Con1 NS3-5B subgenomic replicon by an external contractor (GenScript) using standard DNA synthesis and cloning techniques. All constructs were sequence verified.
Replicon activity assays.
Stable replicon cell lines were seeded at a density of 2 × 104 cells per well in a final volume of 200 μl of assay medium (DMEM supplemented with 5% FBS, penicillin-streptomycin, and nonessential amino acids) in 96-well assay plates containing compounds or dimethyl sulfoxide (DMSO). Alternatively, stable replicon cell lines were seeded at a density of 5 × 103 cells per well in a final volume of 50 μl of assay medium (DMEM supplemented with 5% FBS, penicillin-streptomycin, and nonessential amino acids) in 384-well assay plates containing compounds or DMSO. Cells were then incubated at 37°C and 5% CO2 for 48 h. HCV replication was monitored by determining firefly luciferase activity using Steady-Glo (Promega) and measurement of luminescence in an EnVision 2103 Multilabel Reader (PerkinElmer) or ViewLux (PerkinElmer). Cytotoxicity was measured on parallel plates using CellTiter-Glo (Promega). Replicon 50% effective concentrations (EC50s) and CC50s (the concentration of compound required to inhibit 50% of the assay response), were calculated by curve fitting data to the Hill equation, using XLFit for the 96-well assays. ActivityBase (IDBS Software) with XE Runner for curve fitting was used to plot the curve of percentage of inhibition against compound concentration and derive the 50% effective concentration (EC50) for the compounds assayed in the 384-well format. Cytotoxicity in HepG2, MT4, or Vero cells was also measured using CellTiter-Glo after 4 to 5 days of compound treatment.
Transient-transfection assays used plasmid constructs containing wild-type, mutant, or chimeric replicons as the templates for in vitro transcription reactions using the T7 Express kit (Promega). The in vitro transcripts were aliquoted and stored at −80°C before use. For genotype 1a constructs, 15 μg of RNA was electroporated into 5 × 106 Huh7 Lunet cells in Cytomix supplemented with 2 mM ATP and 5 mM glutathione (35). Electroporation was in 0.4-cm cuvettes using a Bio-Rad Gene Pulser II at 270 V, 950 μF, and infinite resistance. Transient transfections with genotype 1b constructs were performed similarly, except that 5 μg of RNA was electroporated into 5 × 106 ET cured cells in phosphate-buffered saline (PBS). Electroporated cells were resuspended in growth medium, and 2 × 104 cells were transferred to wells of a 96-well plate containing compounds or DMSO. Cells were incubated at 37°C and 5% CO2 for 3 days, and inhibition of HCV replication was measured as for the stable replicon cells.
HCVcc assay.
HCVcc was constructed from a plasmid licensed from Apath, LLC (Brooklyn, NY). HCVcc, also known as Jc1p7Fluc2a, is a genotype 2a/2a chimera consisting of J6CF- and JFH-1-derived sequences fused at a junction after the first transmembrane domain in NS2. The licensed plasmid was modified such that the Gaussia luciferase gene was replaced with a DNA cassette encoding the firefly luciferase gene in tandem with the foot-and-mouth disease virus (FMDV) 2a peptide using standard cloning techniques (36). RNA transcripts were prepared using standard techniques, and 10 μg was used to transfect 1 × 107 Huh7.5 cells. Supernatant from the transfected cells was collected every 12 h for 5 days and pooled, and the titers were determined. Huh7.5 cells (1 × 104 cells) and Jc1p7Fluc2a virus (multiplicity of infection [MOI], 0.05) were mixed in assay medium (total volume, 200 μl) and added to the 96-well assay plates containing compound. Cells were incubated at 37°C and 5% CO2 for 4 days. HCV replication was monitored by measuring firefly luciferase activity using SteadyGlo (Promega) and measuring luminescence in an EnVision 2103 Multilabel Reader (PerkinElmer).
Antiviral assays.
The antiviral activity of GSK5852 against a panel of RNA and DNA viruses was determined by Southern Research Institute (Birmingham, AL). The cytoprotective effect of GSK5852 was determined at a single concentration of 10 μM.
Resistance screens.
Genotype 1a and 1b replicon cells were plated in T150 flasks with the appropriate amount of compound in complete medium plus 1 mg/ml Geneticin. The cells were incubated at 37°C and 5% CO2 with medium changes every 3 to 4 days. If the cells reached confluence within the first week of selection, they were split 1:3 into the same medium plus compound. Generally, we observed a few days of growth after addition of compound and Geneticin, followed by an extended period of cell death. Following this period, the surviving cells proliferated and repopulated the flasks, generally taking 3 to 4 weeks from beginning to end of selection. After sufficient cells were present, the cells were trypsinized and RNA was isolated from the pooled cells using TRIzol (Invitrogen) according to the manufacturer's instructions.
Sequence analysis of resistant mutants.
Standard methods for isolating HCV RNA and sequencing by population sequencing or 454 sequencing were used to determine nucleotide changes. Detailed information is provided in the supplemental material.
Combination studies.
Compounds used in the combination studies were commercially available or were synthesized within GlaxoSmithKline. The NS3 protease inhibitor is BILN-2061 (37), the NS4B inhibitor is GSK5337 (compound +(−)28a in reference 38), the NS5A inhibitor is GSK6805 (39), the NS5B nucleoside inhibitor is 2′-C-methylcytidine (40), NS5B NNI thumb pocket 1 is a Merck NNI (compound A in reference 41), NS5B NNI thumb pocket 2 is filibuvir (42), and the NS5B NNI palm site 1 is SB-711845 (compound 2 in reference 43). The starting concentration for each compound was ≅4 times (4×) the EC50 for the genotype 1b replicon. Separate compound dilution plates were prepared to create a matrix of both compounds at concentrations comprising 2-fold serial dilutions from the starting concentration. Genotype 1b replicon cells were maintained subconfluent prior to the assay. Cells were trypsinized and resuspended in assay medium to 2 × 105 cells/ml. A 92-μl volume of resuspended cells was added to all wells of triplicate 96-well assay plates, and 4 μl from each compound plate was added. Assay plates were incubated at 37°C and 5% CO2 for 48 h before measuring inhibition of HCV replication. The data were analyzed by the dose-wise additivity model (44), which requires estimation of the EC50s for each compound in combination or alone to calculate the combination index (CI). At 50% inhibition, it is calculated as follows: CI = (dA/EC50A) + (dB/EC50B), where EC50A and EC50B are the concentrations of compounds A and B that result in 50% inhibition for each respective compound alone, and dA and dB are the concentrations of each compound in the mixture that yield 50% inhibition. CI measures the type and amount of interaction between two compounds, A and B. A CI value of <1 implies dose-wise synergism between compounds A and B, a CI value of 1 implies dose-wise additivity, and a CI value of >1 implies dose-wise antagonism between compounds A and B. For each fixed concentration of compound A in the plate layout, we calculate the concentration of compound B required to give 50% inhibition and calculate the combination index for these component concentrations. A similar calculation is repeated for each fixed concentration of compound B. The CI value that is being reported here is the average across all individual CIs.
Twenty-day HCV RNA reduction assay.
HCV RNA reduction assays were run using target concentrations 3× and 10× the EC90 values for GSK5852 and 10× the EC90 value of HCV-796 in the absence of Geneticin. A total of 1.25 × 106 genotype 1a or 1b replicon cells were added to T75 flasks, supplemented with medium containing the target concentration of compound, and then incubated at 37°C in 5% CO2. On days 5, 10, 15, and 20, the cells were trypsinized, resuspended, and reseeded with 1.25 ×106 cells/flask in medium containing the target concentration of compound. Media were replaced on days 3, 7, 12, and 17. A volume of 0.5 ml to 1.5 ml of resuspended cells was collected for analysis each time the cells were split.
RNA was isolated from cell pellets using an RNeasy kit (Qiagen) according to the manufacturer's instructions and adjusted to a concentration of 40 ng/μl in water. cDNA was prepared from RNA using the High Capacity cDNA reverse transcription (RT) kit (Applied Biosystems) in MicroAmp 96-well reaction plates. The RT reaction was run as follows in a GeneAmp PCR System 9700: 25°C for 10 min, 37°C for 30 min, followed by 85°C for 1 min. Following this sequence, the block was cooled to 4°C. The TaqMan master mix was prepared as follows: for each reaction mixture, 2 μl of water was mixed with 10 μl of TaqMan fast universal PCR master mix (2×; Applied Biosystems), 3 μl neomycin primer probe mix, and 1 μl GAPDH primer probe mix (Applied Biosystems). The neomycin primer probe mix contains 200 μl 100 μM neomycin gene forward primer (5′-TGGATTGCACGCAGGTTCT-3′), 200 μl 100 μM neomycin reverse primer (5′-GTGCCCAGTCATAGCCGAAT-3′), 55 μl 100 μM neomycin TaqMan probe (5′-(FAM)-CGGCCGCTTGGGTGGAGAGG-(TAMRA)-3′; Biosearch Technologies), and 2,245 μl of RNase-free water. The neomycin primer probe mix was aliquoted and stored at −20°C. To set up the quantitative PCRs, 16 μl of TaqMan master mix and 4 μl cDNA were added to each well of a MicroAmp Fast Optical 96-well reaction plate. The TaqMan reaction was carried out in a 7900HT Sequence Detection System in Fast Real Time mode. The reduction in HCV RNA was determined by relative quantitation of HCV RNA levels (neomycin is assayed as a proxy for HCV) compared to cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase) RNA levels using calculations adapted from an Applied Biosystems tutorial (Guide to performing relative quantitation of gene expression using real-time quantitative PCR,” part number 4371095, rev. B).
Statistical analysis.
The mean and 95% confidence intervals of EC50 and CC50 values were determined using log-transformed data. When the number of replicates was less than four, then the range rather than the 95% confidence interval was shown.
RESULTS
In vitro antiviral activity and cytotoxicity of GSK5852.
Compounds with a benzofuran core structure have been shown to be potent and selective inhibitors of allosteric palm site 2 of NS5B. Chemical optimization of this class of molecules led to unique structure activity relationships between a boronic acid moiety and potent inhibition of subgenomic replicons and resulted in the discovery of GSK5852, a molecule with a benzofuran core and a benzyl boronic acid (Fig. 1). The mechanism of action was confirmed to be inhibition of NS5B by inhibition of its enzymatic activity and cocrystallization with NS5B (30). The antiviral activity of GSK5852 was evaluated on subgenomic replicons derived from the genotype 1a H77, genotype 1b Con1, and genotype 2a JFH-1 strains in replicon assays using the luciferase reporter as a readout as well as the genotype 2a chimeric virus. The compound exhibited low nanomolar activity against all of the replicons as well as the chimeric virus (Table 1). These potencies were about 10-fold higher than those of HCV-796. The antiviral activity of GSK5852 was further tested on the clinically relevant polymorphic variant containing asparagine at amino acid 316 in the genotype 1b NS5B polymerase, representing 35% of the sequences in the European HCV database. The C316N polymorph resulted in loss of potency to HCV-796, confirming previously published reports (29), while GSK5852 showed no loss of potency (Table 1). The cellular cytotoxicities (CC50) of GSK5852 in the genotype 1b replicon, HepG2, MT-4, and Vero cells were >50 μM, >50 μM, >17 μM, and 8.1 μM, respectively, which led to a selectivity index of >1,100 for GSK5852.
Table 1.
Comparison of GSK5852 and HCV-796 in stable replicon and HCVcc assays
| Assay system | GSK5852 |
HCV-796 |
||||
|---|---|---|---|---|---|---|
| EC50 (nM) | 95% confidence interval (nM) | n | EC50 (nM) | 95% confidence interval (nM) | n | |
| Genotype 1a replicon (H77) | 3.0 | 2.8–3.3 | 82 | 26.1 | 24.2–28.3 | 139 |
| Genotype 1b replicon (Con1) | 1.6 | 1.5–1.8 | 125 | 16.9 | 16.0–17.7 | 581 |
| Genotype 1b replicon (C316N) | 1.9 | 1.6–2.2 | 26 | 257 | 242–274 | 241 |
| Genotype 2a replicon (JFH1) | 7.6 | 6.4–8.9 | 16 | 92.6 | 80.7–106 | 10 |
| HCVcc virus (Jc1p7Fluc2a) | 47 | 34–61a | 2 | 95 | 93–98a | 2 |
Range for values with n of <4.
The specificity of GSK5852 for HCV was determined by testing for antiviral activity against a panel of RNA and DNA viruses (see Table S2 in the supplemental material). Minimal inhibition was seen at a concentration of 10 μM against adenovirus (types 1, 3, and 40), bovine viral diarrhea virus (BVDV), coxsackie virus (groups A7 and B4), dengue virus (strains 1, 2, 3, and 4), enterovirus, herpes simplex virus 1 (HSV-1), HSV-2, influenza virus (A and B), measles virus, parainfluenza virus, poliovirus, respiratory syncytial virus (RSV), rhinovirus, rotavirus, and yellow fever virus.
To determine the effect of protein binding on the in vitro antiviral activity of GSK5852, the genotype 1b replicon assay was performed with cell culture medium supplemented with 40 mg/ml human serum albumin and the fold shift in EC50s was determined. There was an average fold shift of 17 ± 3 in the presence of human serum albumin (n = 5). For HCV-796, the fold shift was 1.5 ± 0.3 (n = 19) (data not shown).
Resistance profiling of GSK5852 on genotype 1a and 1b replicon cells revealed a resistance pattern in allosteric palm site 2 of NS5B.
Two independent resistance screens were run for GSK5852 using both genotype 1a and 1b replicon cells. In each experiment, selection was performed at compound concentrations equivalent to 5× and 20× EC50s. RNA isolated from resistant cells was sequenced by 454 and/or population sequencing. When both sequencing methods were used, concordant results were obtained for mutations present in >20% of the RNA population. As expected, the 454 technology identified several mutants present in <20% of the population.
Previously reported resistance selection with HCV-796 utilized only genotype 1b replicon cells and identified mutations conferring a >5-fold loss of potency at amino acid positions 316, 365, 414, and 445 (29, 45). Changes at these amino acid positions were also found in our resistance selection with GSK5852 in genotypes 1a and 1b, with additional changes detected at low frequency by 454 sequencing (Table 2).
Table 2.
Frequency of amino acid changes detected by 454 sequencing during selection with GSK5852 in genotype 1a and 1b replicon cells
| Genotype 1a |
Genotype 1b |
||||
|---|---|---|---|---|---|
| Substitution (codon change) | Frequency (%) |
Substitution (codon change) | Frequency (%) |
||
| 5× EC50 | 20× EC50 | 5× EC50 | 20× EC50 | ||
| I23V (ATC→GTC) | 8 | L127Q (CTG→CAG) | 3 | ||
| V131A (GTA→GCA) | 3 | Q309K (CAG→AAG) | 11 | ||
| N273S (AAC→AGC) | 4 | C316F (TGC→TTC) | 1 | ||
| C316F (TGT→TTT) | 7 | C316Y (TGC→TAC) | 39 | 90 | |
| C316Y (TGT→TAT) | 62 | S365L (TCA→TTA) | 12 | 6 | |
| S365L (TCA→TTA) | 24 | 79 | S365T (TCA→ACA) | 3 | |
| S368F (TCC→TTC) | 2 | 17 | A395G (GCT→GGT) | 2 | |
| S368Y (TCC→TAC) | 2 | 3 | M414I (ATG→ATA) | 1 | |
| A377T (GCT→ACT) | 4 | M414L (ATG→TTG) | 1 | ||
| L392F (CTC→TTC) | 2 | M414V (ATG→GTG) | 3 | ||
| N411Y (AAC→TAC) | 1 | C445F (TGT→TTT) | 38 | 62 | |
| N411S (AAC→AGC) | 1 | W574R (TGG→CGG) | 4 | ||
| M414I (ATG→ATA) | 3 | I585T (ATC→ACC) | 1 | ||
| M414V (ATG→GTG) | 1 | I585V (ATC→GTC) | 4 | ||
For genotype 1a, population sequencing detected a C316Y change in the 5× EC50 selected replicon cells, an S365L change in both the 5× and 20× EC50 selected replicon cells, and an S368F change in the 20× EC50 selected replicon cells. A second resistance screen also detected the C316Y and S365L change in the 5× EC50 selected replicon cells and the S368F change in the 20× EC50 selected replicon cells. A fourth amino acid change was detected in the samples at position 392 in the 5× EC50 selection. The L392F change was also detected by 454 sequencing from the first resistance screen. Sequencing of samples from the first resistance screen by the 454 method also detected a C316F change and an S368Y change not detected by population sequencing. There were also changes at other positions detected at low frequencies. The frequencies of the changes in the first genotype 1a resistance screen are presented in Table 2.
To determine the impact of these changes, single point mutations were made in genotype 1a replicon plasmids and then tested in transient-transfection assays (Table 3). Two substitutions were tested at amino acid position 316, resulting in moderate shifts in potency for GSK5852. The wild-type EC50 of 0.73 nM shifted to 3.25 nM and 16.4 nM when the cysteine at position 316 was changed to a tyrosine and a phenylalanine, respectively. Mutation S365L resulted in a 310-fold shift in potency to 226 nM, while EC50s for replicons containing S368F or S368Y increased to 670 nM or 278 nM, respectively. The L392F mutation detected in both resistance screens resulted in only a modest shift in potency. The effect of these mutations on the activity of HCV-796 was also tested. Table 3 shows that HCV-796 is more sensitive to these changes than GSK5852. Changes in NS5B of I23V, V131A, N273S, A377T, N411Y, and M414I/V were detected at low frequencies by 454 sequencing, but replicons containing these changes had no shifts in GSK5852 potency (data not shown).
Table 3.
Activity of GSK5852 and HCV-796 against genotype 1a and 1b NS5B variants
| Genotype and amino acid change | Mean replication capacity ± SD (n) | GSK5852 EC50 (95% confidence interval) (nM) | n | HCV-796 EC50 (95% confidence interval) (nM) | n |
|---|---|---|---|---|---|
| Genotype 1a | |||||
| None | 100% | 0.73 (0.65–0.82) | 13 | 11.9 (10.0–14.0) | 126 |
| C316F | NDb | 16.4 (14.6–18.6) | 8 | 8,035 (7,080–9,120)a | 2 |
| C316Y | 7% ± 3% (3) | 3.25 (2.61–4.04) | 8 | 1,900 (1,300–2,800) | 38 |
| S365L | 2% ± 1% (3) | 226 (193–266) | 4 | 400 (2.4–66,000) | 4 |
| S368F | 1% ± 1% (3) | 670 (557–806) | 8 | 9,000 (7,400–11,000)a | 2 |
| S368Y | 3% ± 3% (3) | 278 (260–299) | 4 | 310 (23–4,100) | 6 |
| L392F | 180% ± 10% (3) | 1.83 (1.47–2.27) | 4 | 10.8 (9.0–13.0) | 4 |
| Genotype 1b | |||||
| None | 100% | 0.83 (0.76–0.90) | 47 | 7.1 (6.6–7.6) | 69 |
| C316F | 7% ± 2% (3) | 3.78 (1.64–8.73) | 4 | 2,460 (2,000–3,030) | 8 |
| C316Y | 7% ± 8% (3) | 1.4 (0.81–2.40) | 8 | 400 (320–510) | 16 |
| S365A | 72% ± 32% (3) | 0.31 (0.25–0.38)a | 2 | 440 (340–560) | 10 |
| S365F | 14% ± 10% (3) | 28,300 (12,000–66,700) | 4 | ND | 0 |
| S365L | 6% ± 2% (3) | 110 (97.1–126) | 8 | >5,000 | 1 |
| S365T | 9% ± 4% (3) | 4.2 (2.91–6.19) | 8 | 1,100 (420–2,900) | 11 |
| C445F | 171% ± 123% (3) | 1.85 (1.07–3.19) | 4 | 43 (34–54) | 10 |
| C316Y/C445F | ND | 22.4 (19.4–25.8) | 4 | 1,600 (590–4,200) | 6 |
Range is for values with n of <4.
ND, not determined.
For genotype 1b selected replicon cells, two independent resistance selections with GSK5852 were performed. Population sequencing from the first resistance screen detected C316Y and C445F in samples from both the 5× and 20× EC50 selected replicon cells. The C316F mutation was also detected by 454 sequencing, as were the S365L and S365T mutations. The frequencies of these changes from the first resistance screen determined by 454 sequencing, as well as changes in other amino acids, are presented in Table 2. The second resistance screen had similar findings. The 454 sequencing method was applied on a smaller region of NS5B that spanned positions 269 to 449 for the second resistance screen. In addition to residue changes found in the first resistance screen, an S365F change (codon change, TCA→TTT) was detected in 9% of the RNA populations selected at 20× EC50. The 454 sequencing also showed that C316Y was linked to C445F in 29% of the 5× EC50 selected sequences.
Single-point mutations were made in genotype 1b replicon plasmids, and the GSK5852 EC50s are shown in Table 3. The cysteine at position 316 of NS5B was mutated to either phenylalanine or tyrosine, resulting in minimal loss of potency compared to the wild type. The wild-type EC50 was 0.83 nM in the transient-transfection assay, and the C316F and C316Y mutants were 3.78 nM and 1.4 nM, respectively. In addition to the three mutations at amino acid position 365 detected in the GSK5852 resistance screen, a fourth mutation of S365A identified previously for HCV-796 (29) was also made. The S365F mutant showed a substantial loss of potency with an EC50 of 28.3 μM. The mutations S365A, S365L, and S365T had EC50s of 0.31 nM, 110 nM, and 4.2 nM, respectively. The M414I/V, L127Q, Q309K, A395G, W574R, and I585T/V mutations had no shifts in GSK5852 potency (data not shown). The C445F mutation did not result in a large change in potency; however, the EC50 for the C316Y/C445F double mutant was shifted to 22.4 nM. The impact of these mutations was greater for HCV-796 than for GSK5852.
Cross-resistance profiling of GSK5852.
Single amino acid substitutions associated with resistance to other DAAs were engineered into the genotype 1b replicon. The EC50s were determined and used to calculate the fold change in comparison to the wild-type EC50s for GSK5852 and HCV-796 (Fig. 2). GSK5852 showed no cross-resistance to mutations in NS5B that result in resistance to nonnucleoside inhibitors targeting the thumb pocket 1, thumb pocket 2, palm site 1, nucleoside inhibitors, or the nonnucleoside inhibitor GS-9190 (Fig. 2). The activity of GSK5852 is also not affected by resistance mutations to HCV NS3 protease, NS4B, or NS5A inhibitors. These results are consistent with GSK5852 being an NS5B palm site 2 inhibitor and highlight its potential to be used in combination with other agents for treating HCV infection.
Fig 2.

Cross-resistance profiling of GSK5852 or HCV-796 on HCV mutants. Genotype 1b replicons containing the indicated amino acid changes in NS3, NS4B, NS5A, and NS5B were constructed by site-directed mutagenesis. The mean EC50s for GSK5852 or HCV-796 were determined by transient-transfection assays (n ≥ 4) and compared to the Con1 EC50 to calculate fold changes.
In vitro antiviral activity of GSK5852 against chimeric replicons containing genotype 1a,1b, and non-genotype 1 NS5B sequences.
To evaluate the activity of GSK5852 against a panel of different HCV strains within genotype 1, chimeric replicons containing NS5B sequences from clinical isolates were constructed. To efficiently cover the breadth of diversity within the genotype, we identified clinical isolates as representatives for genotype 1a and 1b (see Fig. S1 and S2 in the supplemental material). The potencies were not shifted from the genotype 1a or 1b reference strains for any of the chimeric replicons (Fig. 3). Similarly, the potencies for HCV-796 did not shift drastically for the chimeric replicons, except for a genotype 1b clinical isolate that had an EC50 of 1665 nM due to an asparagine at position 316 of NS5B (accession number EU256088). As expected, all of the chimeric replicons showed similar susceptibilities to the control compound, an NS5A inhibitor (data not shown).
Fig 3.

Inter- and intragenotypic activity of GSK5852. The mean EC50s for genotype 1a, 1b, 4a, and 5a patient sequence replicons and genotype 2a, 2b, and 3a consensus sequence replicons are represented by open circles (○), and the Con1 replicon is represented by the filled circles (●). EC50s were determined by transient transfection for genotype 1a and 1b and with stable cell lines for the non-genotype 1 chimeric replicons (n ≥ 4).
HCV isolates exhibit substantial diversity leading to classification of an isolate into one of six genotypes. To ascertain the spectrum of HCV activity, we constructed stable chimeric replicon cell lines that reflected the genotypic diversity of NS5B and determined their susceptibility to GSK5852. Full-length NS5B sequences for the genotype 2a, 2b, 3a, 4a, 5a, and 6 consensus and the patient sequence closest to the consensus were assembled and cloned into the genotype 1b replicon. Only genotype 2a consensus, 3a consensus, 4a patient, and 5a patient chimeric replicons yielded stable cell lines, which were then used to determine the antiviral activity of GSK5852 (Fig. 3). The potency of GSK5852 remained in the single-digit nanomolar range on all chimeric replicons, with EC50s ranging from 0.83 nM to 6.4 nM. HCV-796 also showed activity that was not affected by genotype.
Combination studies with GSK5852 and representative inhibitors of various classes.
In order to assess the ability of GSK5852 to work in combination with other inhibitors of HCV replication, we used the genotype 1b replicon system. GSK5852 was tested in combination with representative NS3 protease, NS4B, NS5B, and NS5A inhibitors as well as cyclosporine, IFN-α, and ribavirin. The compound was also tested in combination with itself.
Analysis using the dose-wise additivity model (44) to determine the combination index showed that GSK5852 is nearly additive or slightly synergistic when combined with ribavirin, IFN-α, cyclosporine, an NS3 protease inhibitor (BILN-2061), an NS4B inhibitor (GSK5337), an NS5A inhibitor (GSK6805), an NS5B nucleoside inhibitor (2′-C-methylcytidine), and inhibitors targeting the allosteric site 1 (Merck NNI), site 2 (filibuvir), or site 3 (SB-711845) of the NS5B polymerase, as well as with itself (see Table S3 in the supplemental material). These data support the use of GSK5852 in combination with other DAAs for the treatment of HCV infection.
Twenty-day HCV RNA reduction assay of GSK5852 on genotype 1a and 1b replicons.
HCV RNA reduction assays were performed to investigate the reduction in HCV RNA levels resulting from GSK5852 inhibition of NS5B. In this assay, cells were cultured in the presence of compound for 20 days in the absence of Geneticin, and HCV RNA levels were determined at 5-day intervals. Declines in this assay may be limited due to the emergence of resistance in NS5B resulting in a plateau level or a rebound of HCV RNA. Exposure of genotype 1a replicon cells to 3× and 10× EC90 concentrations of GSK5852 led to reductions in HCV RNA levels that ranged between 3 and 4 log (Fig. 4A). When genotype 1b replicon cells were treated, we observed a decline between 4.5 and 5 log for both 3× and 10× EC90 concentrations of GSK5852 (Fig. 4B). No colonies were recovered when the cells were cultured in medium containing Geneticin but no GSK5852 after the 20-day treatment period, suggesting that the cells had been cured of replicon RNA. In both replicon systems, the 3× and the 10× EC90 concentrations of GSK5852 resulted in a 1-log-greater reduction of HCV RNA levels than the 10× EC90 concentration of HCV-796.
Fig 4.
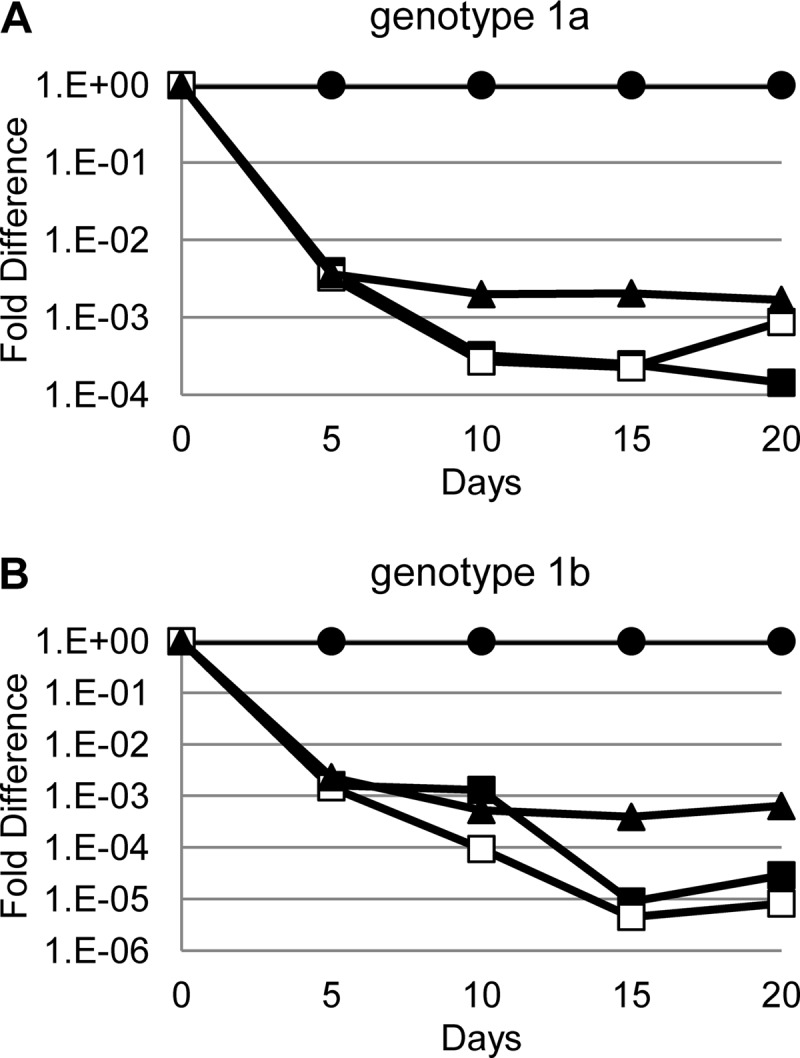
HCV RNA reduction assay. Genotype 1a (A) or 1b (B) replicon cells were treated with compounds and reductions in replicon RNA determined as described in Materials and Methods. ●, DMSO control; ■, 3× EC90 GSK5852; □, 10× EC90 GSK5852; ▲, 10× EC90 HCV-796.
DISCUSSION
The treatment paradigm for chronic HCV infections is changing from IFN-α-based regimens to orally bioavailable DAAs that will have more convenient dosing, shorter treatment duration, and reduced side effects resulting in greater access to treatment. However, most DAAs in clinical development are active only against genotype 1 HCV, so non-genotype-1-infected patients will have limited access to all DAA regimens. NNIs that bind to palm site 2 of NS5B have the potential to inhibit all 6 genotypes of HCV, and their development with other pangenotypic inhibitors would extend the availability of interferon-free regimens to all HCV-infected patients.
GSK5852 inhibits HCV replication by targeting the RDRP activity of NS5B (30). It binds to the NNI allosteric palm site 2 of NS5B as shown by single amino acid changes at positions 316, 365, and 368 of NS5B identified during selection of resistance to GSK5852. The C316Y mutation was the major resistance mutation to emerge during monotherapy treatment with HCV-796 (46). In contrast to HCV-796, GSK5852 is still active against C316Y replicons as well as several other mutations that severely impacted HCV-796 activity (Table 3). GSK5852 was found to form a direct hydrogen bond between the boronic acid residue and the tyrosine hydroxyl group in genotype 1b crystal structures, potentially accounting for the retention of activity against C316Y (30). For genotype 1b, most of the single amino acid changes did not result in significant loss of potency except for some changes at position 365. However, the S365F change requires 2 nucleotide changes, as does the C316Y/C445F double mutation, which results in a substantial shift in potency. The requirement for multiple mutations for high levels of resistance suggests that GSK5852 has a higher genetic barrier to resistance than HCV-796. The higher genetic barrier to resistance may explain the larger reduction in HCV RNA achieved by GSK5852 at 3× EC90 compared to HCV-796 at 10× EC90 in both genotype 1a and 1b replicons (Fig. 4).
Preexisting variants that are resistant to NNIs have been detected in clinical isolates from untreated HCV-infected patients (47, 48). If the variants are a minor proportion of the quasispecies present in a patient, then treatment with a combination of DAAs may still be efficacious. However, if the resistant variant is the dominant strain, then the decrease in the number of active components in the regimen makes treatment failure due to viral breakthroughs more likely. The resistance mutations selected by GSK5852 at amino acid positions 316, 365, and 368 of NS5B are not present as polymorphs in the sequences for genotypes 1 to 6 available in the European HCV database. The activity of GSK5852 was determined on a representative panel of chimeric replicons containing NS5B from 9 genotype 1a- and 9 genotype 1b-infected patients in order to ensure that polymorphisms occurring outside NNI palm site 2 would not result in resistance. The EC50s for these genotype 1a and genotype 1b chimeric replicons were all very close to those of the reference strains, suggesting that preexisting resistance-associated variants were not present in these patient sequences and that GSK5852 will be broadly active in patients infected with genotype 1 HCV.
The pangenotypic activity of GSK5852 was determined using chimeric replicons containing NS5B sequences from other genotypes. Viable chimeric replicons were obtained from NS5B sequences derived from the consensus of all available genotype 2a and 3a sequences and the patient sequence closest to the consensus sequences for genotypes 4a and 5a. The genotype 3a, 4a, and 5a chimeric replicons had EC50s very close to that of the genotype 1b replicon. The activity of GSK5852 against the consensus genotype 2a chimeric replicon was similar to the JFH1 replicon in being slightly shifted relative to the genotype 1b replicon, but the EC50 was still in the single-digit nanomolar range. The genotype 6 chimeric replicons that were constructed were not viable. However, GSK5852 inhibited genotype 6a NS5B in an enzymatic assay with a 50% inhibitory concentration (IC50) very close to that for genotype 1b NS5B (data not shown). GSK5852 therefore has the potential to be equally active against HCV of all genotypes. This pangenotypic activity is highly specific for HCV because of its lack of cytotoxicity and lack of antiviral activity against a large panel of RNA and DNA viruses.
In summary, the characterization of GSK5852 presented here demonstrates that it is a highly selective inhibitor of HCV that targets NNI palm site 2 of NS5B. It has a high genetic barrier to resistance and an improved resistance profile to HCV-796, an earlier NNI palm site 2 inhibitor that showed antiviral activity in the clinic. Profiling against a panel of chimeric replicons containing NS5B patient or consensus sequences suggests that GSK5852 will be an attractive candidate for inclusion in oral interferon-free regimens because of its ability to inhibit all HCV genotypes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Luz Helena Kryn for her assistance with the replicon assays and Andrew Spaltenstein for his valuable support of the program.
All authors are current or former employees of GlaxoSmithKline.
Footnotes
Published ahead of print 12 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00874-13.
REFERENCES
- 1.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115 [DOI] [PubMed] [Google Scholar]
- 2.Kim WR. 2002. The burden of hepatitis C in the United States. Hepatology 36:S30–S34 [DOI] [PubMed] [Google Scholar]
- 3.Jacobson IM, Pawlotsky JM, Afdhal NH, Dusheiko GM, Forns X, Jensen DM, Poordad F, Schulz J. 2012. A practical guide for the use of boceprevir and telaprevir for the treatment of hepatitis C. J. Viral Hepat. 19(Suppl 2):1–26 [DOI] [PubMed] [Google Scholar]
- 4.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, Haussinger D, Diago M, Carosi G, Dhumeaux D, Craxi A, Lin A, Hoffman J, Yu J. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347:975–982 [DOI] [PubMed] [Google Scholar]
- 5.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Jr, Bernstein D, Rizzetto M, Zeuzem S, Pockros PJ, Lin A, Ackrill AM. 2004. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 140:346–355 [DOI] [PubMed] [Google Scholar]
- 6.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht B. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958–965 [DOI] [PubMed] [Google Scholar]
- 7.Lindahl K, Stahle L, Bruchfeld A, Schvarcz R. 2005. High-dose ribavirin in combination with standard dose peginterferon for treatment of patients with chronic hepatitis C. Hepatology 41:275–279 [DOI] [PubMed] [Google Scholar]
- 8.Sulkowski MS. 2003. Anemia in the treatment of hepatitis C virus infection. Clin. Infect. Dis. 37(Suppl 4):S315–S322 [DOI] [PubMed] [Google Scholar]
- 9.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 10.Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, Fried MW, Adler M, Reesink HW, Martin M, Sankoh AJ, Adda N, Kauffman RS, George S, Wright CI, Poordad F. 2011. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 365:1014–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R. 2011. Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1207–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee LY, Tong CY, Wong T, Wilkinson M. 2012. New therapies for chronic hepatitis C infection: a systematic review of evidence from clinical trials. Int. J. Clin. Pract. 66:342–355 [DOI] [PubMed] [Google Scholar]
- 14.Poordad F, Chee GM. 2012. Interferon free hepatitis C treatment regimens: the beginning of another era. Curr. Gastroenterol. Rep. 14:74–77 [DOI] [PubMed] [Google Scholar]
- 15.Choo QL, Kuo G, Jeiner A, Rerby L, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362 [DOI] [PubMed] [Google Scholar]
- 16.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby A, Barr PJ, Weiner AJ, Bradley DW, Kuo G, Houghton M. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 88:2451–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, Shimotohno K. 1990. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc. Natl. Acad. Sci. U. S. A. 87:9524–9528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poenisch M, Bartenschlager R. 2010. New insights into structure and replication of the hepatitis C virus and clinical implications. Semin. Liver Dis. 30:333–347 [DOI] [PubMed] [Google Scholar]
- 19.Powdrill MH, Tchesnokov EP, Kozak RA, Russell RS, Martin R, Svarovskaia ES, Mo H, Kouyos RD, Gotte M. 2011. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. U. S. A. 108:20509–20513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B, Beall E, Yap PL, Kolberg J, Urdea MS. 1993. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J. Gen. Virol. 74:2391–2399 [DOI] [PubMed] [Google Scholar]
- 21.Foster GR, Hezode C, Bronowicki JP, Carosi G, Weiland O, Verlinden L, van Heeswijk R, Vangeneugden T, Picchio G, Beumont-Mauviel M. 2009. Activity of telaprevir alone or in combination with peginterferon alfa-2a and ribavirin in treatment naive genotype 2 and 3 hepatitis-C patients: interim results of study C209. J. Hepatol. 50(Suppl 2):s22. 10.1016/S0168-8278(09)60052-0 [DOI] [Google Scholar]
- 22.Benhamou JP, Moussali J, Ratziu V, Lebray P, Gysen V, de Backer K, Ghys A, van Heeswijk R, Vangeneugden T, Picchio G, Beumont-Mauviel M. 2009. Results of a proof of concept study (C210) of telaprevir monotherapy and in combination with peginterferon alfa-2a and ribavirin in treatment-naive genotype 4 HCV patients. J. Hepatol. 50(Suppl 1):s6. 10.1016/S0168-8278(09)60012-X [DOI] [Google Scholar]
- 23.Beaulieu PL. 2009. Recent advances in the development of NS5B polymerase inhibitors for the treatment of hepatitis C virus infection. Expert Opin. Ther. Pat. 19:145–164 [DOI] [PubMed] [Google Scholar]
- 24.Powdrill MH, Bernatchez JA, Gotte M. 2010. Inhibitors of the hepatitis C virus RNA-dependent RNA polymerase NS5B. Viruses 2:2169–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furman PA, Lam AM, Murakami E. 2009. Nucleoside analog inhibitors of hepatitis C viral replication: recent advances, challenges and trends. Future Med. Chem. 1:1429–1452 [DOI] [PubMed] [Google Scholar]
- 26.Pauwels F, Mostmans W, Quirynen LM, Van der Helm L, Boutton CW, Rueff AS, Cleiren E, Raboisson P, Surleraux D, Nyanguile O, Simmen KA. 2007. Binding-site identification and genotypic profiling of hepatitis C virus polymerase inhibitors. J. Virol. 81:6909–6919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kneteman NM, Howe AY, Gao T, Lewis J, Pevear D, Lund G, Douglas D, Mercer DF, Tyrrell DL, Immermann F, Chaudhary I, Speth J, Villano SA, O'Connell J, Collett M. 2009. HCV796: a selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 49:745–752 [DOI] [PubMed] [Google Scholar]
- 28.Franciscus A. September 2007. HCV-796 dosing discontinued. HCV Advocate Newsl. http://www.hcvadvocate.org/news/newsLetter/2007/advocate0907.html
- 29.Howe AY, Cheng H, Johann S, Mullen S, Chunduru SK, Young DC, Bard J, Chopra R, Krishnamurthy G, Mansour T, O'Connell J. 2008. Molecular mechanism of hepatitis C virus replicon variants with reduced susceptibility to a benzofuran inhibitor, HCV-796. Antimicrob. Agents Chemother. 52:3327–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maynard A, Crosby R, Ellis B, Hamatake R, Hong Z, Johns B, Kahler K, Koble C, Banka A, Leivers M, Mathis A, Peat A, Pouliot J, Roberts C, Samano V, Schmidt R, Smith G, Spaltenstein A, Stewart E, Thommes P, Turner E, Voitenleitner C, Walker J, Waitt G, Weatherhead J, Weaver K, Williams S, Wright L, Xiong P, Haigh D, Shotwell JB. 2013. Discovery of a potent boronic acid derived inhibitor of the HCV RNA-dependent RNA polymerase. J. Med. Chem. [Epub ahead of print.] 10.1021/jm400317w [DOI] [PubMed] [Google Scholar]
- 31.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2003. Efficient replication of hepatitis C virus genotype 1a RNAs in cell culture. J. Virol. 77:3181–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817 [DOI] [PubMed] [Google Scholar]
- 34.Voitenleitner C, Bechtel J, Arfsten A, Hamatake R. 2012. Hepatitis C genotype 1a replicon improved through introduction of fitness mutations. Biotechniques 52:273–275 [DOI] [PubMed] [Google Scholar]
- 35.van den Hoff MJ, Moorman AF, Lamers WH. 1992. Electroporation in ‘intracellular' buffer increases cell survival. Nucleic Acids Res. 20:2902. 10.1093/nar/20.11.2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blackham S, Baillie A, Al-Hababi F, Remlinger K, You S, Hamatake R, McGarvey MJ. 2010. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 84:5404–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, Laplante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189 [DOI] [PubMed] [Google Scholar]
- 38.Miller JF, Chong PY, Shotwell JB, Catalono JG, Tai VWF, Fang J, Banka AL, Roberts CD, Youngman M, Zhang H, Xiong Z, Mathis A, Pouliot JJ, Hamatake RK, Price DJ, Seal JW, Stroup LL, Creech KL, Carballo LH, Todd D, Spaltenstein A, Furst S, Hong Z, Peat AJ. 2013. Hepatitis C replication inhibitors that target the viral NS4B protein. J. Med. Chem. [Epub ahead of print.] 10.1021/jm400125h [DOI] [PubMed] [Google Scholar]
- 39.Bechtel J, Crosby R, Wang A, Woldu E, Van Horn S, Horton J, Creech K, Carballo LH, Voitenleitner C, Vamathevan J, Duan M, Spaltenstein A, Kazmierski W, Roberts C, Hamatake R. 2013. In vitro profiling of GSK2336805, a potent and selective inhibitor of HCV NS5A. J. Hepatol. 54:S307 http://www.natap.org/2011/EASL/EASL_74.htm [Google Scholar]
- 40.Pierra C, Benzaria S, Amador A, Moussa A, Mathieu S, Storer R, Gosselin G. 2005. Nm 283, an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. Nucleosides Nucleotides Nucleic Acids 24:767–770 [DOI] [PubMed] [Google Scholar]
- 41.Giuliano C, Fiore F, Di MA, Padron VJ, Bishop A, Bonelli F, Gonzalez-Paz O, Marcucci I, Harper S, Narjes F, Pacini B, Monteagudo E, Migliaccio G, Rowley M, Laufer R. 2005. Preclinical pharmacokinetics and metabolism of a potent non-nucleoside inhibitor of the hepatitis C virus NS5B polymerase. Xenobiotica 35:1035–1054 [DOI] [PubMed] [Google Scholar]
- 42.Shi ST, Herlihy KJ, Graham JP, Nonomiya J, Rahavendran SV, Skor H, Irvine R, Binford S, Tatlock J, Li H, Gonzalez J, Linton A, Patick AK, Lewis C. 2009. Preclinical characterization of PF-00868554, a potent nonnucleoside inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 53:2544–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaw AN, Tedesco R, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Duffy KJ, Fitch DM, Gates A, Johnston VK, Keenan RM, Lin-Goerke J, Liu N, Sarisky RT, Wiggall KJ, Zimmerman MN. 2009. Substituted benzothiadizine inhibitors of Hepatitis C virus polymerase. Bioorg. Med. Chem. Lett. 19:4350–4353 [DOI] [PubMed] [Google Scholar]
- 44.Selleseth DW, Talarico CL, Miller T, Lutz MW, Biron KK, Harvey RJ. 2003. Interactions of 1263W94 with other antiviral agents in inhibition of human cytomegalovirus replication. Antimicrob. Agents Chemother. 47:1468–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Flint M, Mullen S, Deatly AM, Chen W, Miller LZ, Ralston R, Broom C, Emini EA, Howe AY. 2009. Selection and characterization of hepatitis C virus replicons dually resistant to the polymerase and protease inhibitors HCV-796 and boceprevir (SCH 503034). Antimicrob. Agents Chemother. 53:401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villano SA, Howe AY, Rabile D, Harper D, Speth J, Bichier G. 2012. Analysis of HCV NS5B genetic variants following monotherapy with HCV-796, a non-nucleoside polymerase inhibitor, in treatment-naive HCV-infected patients. Hepatology 44:607A–608A [Google Scholar]
- 47.Le Pogam S, Seshaadri A, Kosaka A, Chiu S, Kang H, Hu S, Rajyaguru S, Symons J, Cammack N, Najera I. 2008. Existence of hepatitis C virus NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J. Antimicrob. Chemother. 61:1205–1216 [DOI] [PubMed] [Google Scholar]
- 48.Dryer PD, Limketkai BN, Martin CM, Ma G, Sherman KE, Taylor LE, Mayer KH, Jamieson DJ, Blackard JT. 2009. Screening for hepatitis C virus non-nucleotide resistance mutations in treatment-naive women. J. Antimicrob. Chemother. 64:945–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.