

Abstract
Human African trypanosomiasis (HAT, also called sleeping sickness), a neglected tropical disease endemic to sub-Saharan Africa, is caused by the parasites Trypanosoma brucei gambiense and T. brucei rhodesiense. Current drugs against this disease have significant limitations, including toxicity, increasing resistance, and/or a complicated parenteral treatment regimen. DB829 is a novel aza-diamidine that demonstrated excellent efficacy in mice infected with T. b. rhodesiense or T. b. brucei parasites. The current study examined the pharmacokinetics, in vitro and in vivo activity against T. b. gambiense, and time of drug action of DB829 in comparison to pentamidine. DB829 showed outstanding in vivo efficacy in mice infected with parasites of T. b. gambiense strains, despite having higher in vitro 50% inhibitory concentrations (IC50s) than against T. b. rhodesiense strain STIB900. A single dose of DB829 administered intraperitoneally (5 mg/kg of body weight) cured all mice infected with different T. b. gambiense strains. No cross-resistance was observed between DB829 and pentamidine in T. b. gambiense strains isolated from melarsoprol-refractory patients. Compared to pentamidine, DB829 showed a greater systemic exposure when administered intraperitoneally, partially contributing to its improved efficacy. Isothermal microcalorimetry and in vivo time-to-kill studies revealed that DB829 is a slower-acting trypanocidal compound than pentamidine. A single dose of DB829 (20 mg/kg) administered intraperitoneally clears parasites from mouse blood within 2 to 5 days. In summary, DB829 is a promising preclinical candidate for the treatment of first- and second-stage HAT caused by both Trypanosoma brucei subspecies.
INTRODUCTION
Human African trypanosomiasis (HAT), also known as sleeping sickness, is a neglected tropical disease threatening the health of millions of people in the poorest regions of sub-Saharan Africa (1). HAT is fatal if left untreated. The disease is caused by two subspecies of the unicellular parasite Trypanosoma brucei, T. b. gambiense and T. b. rhodesiense, and is transmitted through the bite of an infected tsetse fly. Two clinical stages are defined. In the first stage, the parasites reside in the blood and lymph system, whereas in the second stage, the parasites cross the blood-brain barrier (BBB) and infect the central nervous system (CNS). The disease was almost eliminated around 1960, but it reemerged as a result of political instability and lack of an adequate health care and control system (2). The most recent epidemic occurred in the 1990s, when the number of estimated cases in 1998 was alarmingly high, with about 300,000 cases per year (3). Since the turn of the millennium, the number of infections has declined due to interventions to control the disease, such as trapping tsetse flies, surveillance, and appropriate treatment of patients (2, 4). The number of actual cases in 2009 was estimated at 30,000 annually by the World Health Organization. Control measures have to be maintained by all means, otherwise the disease will reemerge, as history has shown (2, 4).
The availability of effective drugs is crucial for HAT control and elimination, as chemotherapy is the only option for cure. Vaccines are not available for HAT and are unlikely to be developed, mainly because of the antigenic variation of the surface glycoproteins of the parasites (5–7). There are four drugs available as monotherapies (two for the first stage and two for the second stage) to treat the disease, and all require parenteral administration, which is a significant problem in the remote rural areas of Africa, where HAT primarily occurs. Moreover, drugs used to treat second-stage HAT (i.e., melarsoprol and eflornithine) are unsatisfactory due to severe adverse reactions, increasing treatment failures, and/or complicated treatment schedules requiring frequent infusion. Hence, new or improved chemotherapies are needed, especially for second-stage HAT.
One of the compounds used to treat first-stage HAT is the aromatic diamidine drug pentamidine (Fig. 1). It was discovered in 1941 and has been used against T. b. gambiense infections for 7 decades. It is still highly efficacious, as shown by the fact that no increase in treatment failures has been reported (6). The diamidine structure of pentamidine seems to be essential for its antiparasitic activity, which is primarily based on binding to the parasite DNA (8). The exact mechanism of action, however, is still unknown. During the last 4 decades, our research has focused on the identification of novel diamidines with improved efficacy, oral bioavailability, and BBB penetration. One of the first lead compounds was furamidine (DB75), which is highly active in vitro as well as in vivo but has poor oral bioavailability. Its methamidoxime prodrug, pafuramidine (DB289 maleate), has good oral bioavailability (9) and good oral efficacy in T. b. rhodesiense-infected mice (10). Pafuramidine was selected by the Consortium for Parasitic Drug Development (CPDD) in 2000 to enter clinical trials as the first oral drug to be tested for first-stage HAT. During a phase III clinical trial, this compound was as effective as pentamidine and its safety profile was inconspicuous (11). However, during a phase I clinical trial for safety with an extended treatment regimen, reversible hepatotoxicity and delayed-onset renal insufficiency were observed, which led to the discontinuation of pafuramidine development (12).
Fig 1.
Chemical structures of DB829 and pentamidine.
Further screening of novel diamidines and prodrugs has continued in order to identify compounds that are safer, offer the possibility of oral administration, and are in particular active in the CNS for second-stage HAT (10, 13, 14). The prodrug approach was originally employed to improve oral absorption by masking the positive charge of amidine moieties but also had potential to improve drug delivery across the BBB, a prerequisite to cure patients with second-stage infections. Several diamidine prodrugs have been tested in the GVR35 CNS mouse model, in which DB844 and DB868 administered orally cured CNS infections in mice (10). DB844 and DB868 have been further tested in the vervet monkey model of second-stage HAT, and results have been reported recently (15, 16). DB868 was significantly better tolerated in vervet monkeys than was DB844 and could be a promising oral drug candidate, especially for first-stage disease (17).
The active metabolite of the prodrug DB868 is DB829 (or CPD-0801 [Fig. 1]). It is a cationic aza-diamidine, potent in vitro against T. b. rhodesiense STIB900 parasites (50% inhibitory concentration [IC50], 20 nM). DB829 is of special interest, because when administered intraperitoneally it cured mice with second-stage infections (with T. b. brucei) (10). This indicated that it was able to cross the BBB. DB829 therefore represents a new class of cationic diamidines that are CNS active and holds promise for the direct use of active diamidines to treat second-stage HAT. This potentially avoids any detrimental effects of the prodrug strategy, such as variable prodrug activation between different patients, which would simplify pharmacokinetic predictions and dosage selection.
The aim of this study was to characterize the pharmacokinetics, in vitro and in vivo activity against T. b. gambiense, and time of drug action of the CNS-active diamidine DB829. Since more than 98% of HAT cases are due to T. b. gambiense infections currently (18), it is imperative to determine the effectiveness of DB829 against T. b. gambiense parasites. Isothermal microcalorimetry (19) was employed to monitor in vitro parasite killing rate (time of drug action) of DB829 on a real-time basis. Lastly, the in vivo parasite clearance time and pharmacokinetics of DB829 in mice were determined. These results are important factors to be considered for further development of DB829 and related compounds to treat African sleeping sickness.
MATERIALS AND METHODS
Materials.
Pentamidine isethionate was purchased from Sigma-Aldrich (St. Louis, MO). Synthesis of DB829 (or CPD-0801; hydrochloride salt form) and deuterium-labeled DB75 (DB75-d8; deuterated phenyl rings; internal standard) has been previously reported (20, 21). CPD-0801 and CPD-0802 are both synonyms for DB829. The different codes indicate their different salt forms. CPD-0801 is the hydrochloride salt form, and CPD-0802 is the diacetate salt form of DB829. Only the hydrochloride salt form of DB829 was used in this study.
Animals for pharmacokinetic studies.
Protocols for the animal studies were approved by the Institutional Animal Care and Use Committee of the University of Kansas. Male Swiss Webster mice (weighing 20 to 25 g) were purchased from the Charles River Laboratories (O'Fallon, MO). Mice were housed in a clean room under filtered, pathogen-free air, in a 12-h light/dark cycle, and with food pellets and water available ad libitum.
Pharmacokinetics of DB829 and pentamidine after i.v. and i.p. administration in mice.
DB829 and pentamidine were first dissolved in sterile water and further diluted with saline (1:1, vol/vol) to the desired concentrations. This prevented the precipitation that occurred if the compounds were added directly to saline. The dose level for intravenous (i.v.) administration was 7.5 μmol/kg body weight (approximately 2.3 mg/kg for DB829 and 2.6 mg/kg for pentamidine), which was well tolerated in a preliminary overt toxicity study in mice. The dose level for intraperitoneal (i.p.) administration was 20 mg/kg (approximately 65 μmol/kg for DB829 and 59 μmol/kg for pentamidine), which was the higher of two doses used in the following efficacy study and was selected to directly evaluate if pharmacokinetics played a role in determining in vivo efficacy of the two compounds. The dose volume was 5 ml/kg. Blood samples were collected via submandibular bleeding (∼0.04 ml per bleed) for serial sampling or cardiac puncture (∼0.8 ml of blood) for terminal sampling. To accurately determine the concentration-time curve near and shortly after the time to maximum concentration of drug in plasma (Tmax) and terminal elimination half-life (t1/2), the time points for blood sampling included 0.0167, 0.0833 (two additional time points for i.v. administration), 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h following drug administration, and three mice were used for blood sampling at each time point. Plasma was separated by centrifugation in lithium heparin-coated Microvet tubes (Sarstedt Inc., Newton, NC) or Microtainer tubes (BD Biosciences, Franklin Lakes, NJ). Plasma samples were stored at −20°C until ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) analysis.
Unbound fraction in mouse plasma.
The unbound fraction of DB829 and pentamidine in mouse plasma was determined using rapid equilibrium dialysis devices (Thermo Scientific Pierce, Rockford, IL) as previously described (22). Blank mouse plasma was spiked with DB829 or pentamidine to give a final concentration of 1 μM, below which they spent the majority of time after a single i.v. or i.p. administration of the tested doses. Spiked plasma (in triplicate) was incubated for 6 h at 37°C to allow equilibration with a phosphate-buffered saline (100 mM, pH 7.4) and then analyzed for total and unbound concentrations by UPLC-MS/MS.
UPLC-MS/MS analysis. (i) Sample preparation.
Plasma samples (2 μl) were mixed with 200 μl of 7:1 (vol/vol) methanol-water containing 0.1% trifluoroacetic acid and internal standard (1 nM DB75-d8) and then vortex mixed for 30 s, followed by centrifugation (2,800 × g) to pellet proteins. The supernatant was transferred to a new tube and dried using a 96-well microplate evaporator (Apricot Designs Inc., Covina, CA) under N2 at 50°C and reconstituted with 100 μl of 15% methanol containing 0.1% trifluoroacetic acid.
(ii) Determination of drug concentration.
The reconstituted samples (5-μl injection volume) were analyzed for drug concentration using a Waters Xevo TQ-S mass spectrometer (Foster City, CA) coupled with a Waters Acquity UPLC I-Class system. Compounds were separated on a Waters UPLC BEH C18 column (2.1 by 50 mm, 1.7 μm) equilibrated at 50°C. UPLC mobile phases consisted of water containing 0.1% formic acid (A) and methanol containing 0.1% formic acid (B). After a 0.15-min initial holding period at 5% B, the mobile phase composition started with 5% B and was increased to 60% B over 2.2 min with a flow rate of 0.4 ml/min. Then, the column was washed with 90% B for 0.5 min and was reequilibrated with 5% B for 1.1 min before the next sample was injected. The characteristic single-reaction monitoring (SRM) transitions for DB829, pentamidine, and DB75-d8 were m/z 307.1→273.1, 341.4→120.1, and 313.2→296.3, respectively, under positive electrospray ionization mode. The calibration curves for DB829 and pentamidine ranged from 0.5 nM to 50 μM and 2.5 nM to 50 μM, respectively, for plasma samples. The interday coefficient of variation (CV) and accuracy were determined by measuring the same preparation of three standards three times on different days. At concentrations of 5, 500, and 10,000 nM, the intraday CV and average accuracy of DB829 quantification were 8.4% and 91%, 9.4% and 104%, and 3.6% and 99%, respectively. The interday CV and average accuracy of pentamidine quantification were 7.4% and 108% (at 5 nM), 4.7% and 93% (at 500 nM), and 8.1% and 97% at (at 10,000 nM), respectively.
(iii) Pharmacokinetic analysis.
The area under the plasma concentration-time curve (AUC), terminal elimination half-life (t1/2), maximum concentration of drug in plasma (Cmax), and time to reach Cmax (Tmax) were calculated using the trapezoidal rule-extrapolation method and noncompartmental analysis (WinNonlin Version 5.0; Pharsight, Mountain View, CA).
Antitrypanosomal activity of DB829 and pentamidine in vitro. (i) Preparation of compounds.
Compounds were dissolved in 100% dimethyl sulfoxide (DMSO) and finally diluted in culture medium prior to the in vitro assay. The DMSO concentration never exceeded 1% in the in vitro alamarBlue assays at the highest drug concentration. For microcalorimetry assays, the DMSO concentration was kept at 0.1% in all samples to avoid any DMSO effect on the membrane permeability of the trypanosomes. Melarsoprol was diluted in water instead of DMSO. For in vivo experiments, the compounds were dissolved in DMSO and further diluted with distilled water to a final DMSO concentration of 10% prior to administration to the animals.
(ii) Parasites.
The T. b. rhodesiense strain STIB900 is a derivative of strain STIB704 (23), which was isolated from a patient in Ifakara, Tanzania, in 1982. STIB900 was used for routine in vitro screening and for the standard acute mouse model, which mimics the first stage of HAT. Strain STIB799, also known as KETRI 243, is a T. b. rhodesiense strain isolated from a patient from Busoga in Uganda in 1961 after four treatment failures with melarsoprol (24).
The T. b. gambiense strains used within this study were STIB930, ITMAP141267, 130R, 45R, 349R, DAL898R, K03048, and 40R (Table 1). We included more isolates from the Democratic Republic of the Congo (DRC) in our panel of different strains, as most of the T. b. gambiense infections occur in this central African country (25, 26). The strains labeled with an “R” were isolated from patients after a melarsoprol treatment failure. STIB930 (also named STIB754) is a derivative of the strain TH1/78E(031),which was isolated from a patient in Cote d'Ivoire in 1978 (27). ITMAP141267, was isolated from a patient in DRC in 1960 (28). Strain 130R was isolated from a patient in DRC in 2005 after two treatment failures with melarsoprol. The first relapse was detected 8 months after a 3-day melarsoprol treatment, and the second relapse was detected 1 year after a melarsoprol-nifurtimox combination therapy (29). Strain 45R was isolated from a patient in DRC in 2005 after two treatment failures with melarsoprol. The first relapse was detected 1 year after a 3-day melarsoprol treatment, and the second relapse was detected 8 months after a melarsoprol-nifurtimox combination therapy (29). Strain 40R was also isolated from a patient in DRC in 2005, 6 months after failure of the 10-day melarsoprol treatment (29). The strain 349R was isolated from a patient in DRC in 2006 after a treatment failure with a 10-day melarsoprol treatment (29). DAL898R was isolated from a patient in Cote d'Ivoire in 1985 after melarsoprol treatment failure (23). DAL898R was the only strain that had previously shown an increased IC50 for melarsoprol among the panel of tested DAL (Daloa in Cote d'Ivoire) isolates (23). Strain K03048 was isolated from a patient in South Sudan in 2003 (30).
Table 1.
T. b. gambiense isolates
| Trypanosome straina | Yr of isolation | Origin |
|---|---|---|
| STIB930/STIB754 | 1978 | Cote d'Ivoire |
| ITMAP141267 | 1960 | DRC |
| 130R | 2005 | DRC |
| 40R | 2005 | DRC |
| 45R | 2005 | DRC |
| 349R | 2006 | DRC |
| DAL898R | 1985 | Cote d'Ivoire |
| K03048 | 2003 | South Sudan |
Strains labeled with an “R” were isolated from patients after a relapse with a melarsoprol treatment.
(iii) In vitro growth inhibition assays using T. brucei subspecies.
The 50% inhibitory concentrations were determined using the alamarBlue assay as described by Raz et al. (31), with minor modifications. Assays were carried out at least three times independently, set up on different days and each time in duplicate. Coefficients of variation of these assays were less than 50%. Different culture and assay media were used for T. b. gambiense and for T. b. rhodesiense. For T. b. rhodesiense, the compounds were tested in minimum essential medium with Earle's salts, supplemented according to the protocol of Baltz et al. (32) with the following modifications: 0.2 mM 2-mercaptoethanol, 1 mM Na-pyruvate, 0.5 mM hypoxanthine, and 15% heat-inactivated horse serum. For T. b. gambiense, a modified protocol of Hirumi and Hirumi (33) was used: Iscove's modified Dulbecco's medium (IMDM) was supplemented with 0.2 mM 2-mercaptoethanol, 1 mM Na-pyruvate, 0.5 mM hypoxanthine, 0.05 mM bathocuproindisulfate, 1.5 mM l-cysteine-HCl, 2 mM l-glutamine, and 5% heat-inactivated human serum plus 15% heat-inactivated fetal calf serum.
Serial drug dilutions were prepared in 96-well microtiter plates, and each well was inoculated with 2,000 bloodstream forms for the T. b. rhodesiense isolates and with 2,500 bloodstream forms for the T. b. gambiense isolates. Drug treatment was carried out for 70 h for T. b. rhodesiense and 68 h for T. b. gambiense. Ten microliters of the viability marker alamarBlue (12.5 mg resazurin dissolved in 100 ml phosphate-buffered saline) was then added to each well, and the plates were incubated for an additional 2 to 6 h until the signal/background fluorescence ratio was about 10 for T. b. rhodesiense isolates or about 5 for T. b. gambiense isolates. The plates were read in a SpectraMax Gemini XS microplate fluorescence scanner (Molecular Devices, Sunnyvale, CA) using an excitation wavelength of 536 nm and an emission wavelength of 588 nm. The IC50s were calculated from the sigmoidal inhibition curves using SoftmaxPro software.
(iv) Microcalorimetry studies using T. brucei subspecies.
In vitro time of drug action was monitored using isothermal microcalorimetry. Previous experiments have shown that heat flow data can be used as a proxy for the number of viable cells (34), defined as trypanosomes still moving under the microscope. With this method, the time to the onset of drug action and the time to kill the parasite population can be determined on a real-time basis (19). The same culture media were used for the T. b. rhodesiense strain and for the T. b. gambiense strains as for the in vitro growth inhibition assays. Strain STIB900 was used as a representative for T. b. rhodesiense and STIB930 (and sometimes ITMAP141267) for T. b. gambiense.
For experiments with permanent drug exposure, bloodstream trypanosomes (2 ml at 5 × 104 organisms/ml per ampoule) were placed in 4-ml ampoules and spiked with DB829 or pentamidine at different concentrations with a final DMSO concentration of 0.1% (vol/vol). The trypanocidal drug melarsoprol was dissolved in water and included as a nondiamidine control for the first experiments with STIB900. Trypanosome-free negative controls contained only culture medium. After the ampoules had been inserted into the isothermal microcalorimetry instrument (Thermal Activity Monitor, model 249 TAM III; TA Instruments, New Castle, DE), heat flow was continuously measured (1 reading/second) at 37°C. Each experiment with permanent drug exposure was set up in triplicate and repeated 3 times.
(v) Inoculum studies.
For studies of drug effects on different parasite densities in the inoculum, the parasite density was reduced to 1 × 104/ml bloodstream trypanosomes and supplemented with DB829 or pentamidine at the desired concentrations. Inhibition of growth and viability was compared with the samples containing the standard inoculum of 5 × 104/ml bloodstream trypanosomes and the same drug concentrations. Inoculum experiments with higher parasite densities were omitted, as the heat flow oscillations, which appear at high initial densities, interfere with drug action (19).
(vi) Drug washout experiments.
For the 24-h exposure experiment, trypanosomes (T. b. rhodesiense strain STIB900 and T. b. gambiense strain STIB930) were incubated with DB829 for 24 h at 37°C and then washed twice at 1,850 × g for 10 min to remove the compound. Subsequently, the washed trypanosomes were resuspended in drug-free culture medium and transferred to ampoules and inserted into the isothermal microcalorimetry instrument. The drug dilution factor after two washing steps was estimated to be >1,800-fold. The drug-free control samples (drug free, washed) were washed identically to the drug-containing samples. Each 24-h exposure experiment was set up in triplicate for each assay and repeated 2 times.
(vii) Analysis of microcalorimetry heat flow data.
The metabolic heat production rate in each ampoule was recorded continuously. The electric signal was calibrated to obtain a reading in watts. To facilitate data handling, the recorded data were resampled to obtain an effective sampling frequency of 1 data point per 90 s, using the manufacturer's software (TAM assistant version v1.2.4.0), and exported to Microsoft Excel. A time correction was needed for the preparation of the ampoules and the transfer from the bench to the calorimeter prior to equilibration in the calorimeter and the measurement. The correction was achieved by adding 45 min to the time recorded by the microcalorimeter. Resulting data were plotted as heat flow (in μW) over time. Each single curve for heat flow (in W) over time was analyzed using software R (The R Foundation for Statistical Computing, Vienna, Austria). Due to metabolic oscillations clearly visible in the heat flow curves, a cubic spline was applied to smooth the data prior to parameter determination (i.e., onset of drug action, time to peak, and time to kill). The time to onset of drug action was determined as the time at which a divergence between the heat flow rate of the drug-free controls and the drug-containing specimens could be observed (19). The time to peak was determined as the time point at which the maximum heat flow was observed for each sample. The reversal shows the time after which a decrease of the parasite density is expected. The time to kill the parasite population was defined as the time point at which the heat production was reduced to the level of the sterile medium control (base level). At this time, the metabolic heat production of the culture was below the detection limit, suggesting that most of the parasites were inactivated. Typically, a heat flow of 8 μW was recorded at the maximum density of 1 × 106 to 1.5 × 106 trypanosomes/ml, and the microcalorimeter had a detection limit around 100 to 200 nW. The surviving population at the detection limit was about 1.25% to 2.5% of the maximum density, representing a density of <4 × 104 trypanosomes/ml.
Finally, the growth rate (μ) of each culture was calculated using the data of heat over time (i.e., the integrated heat flow data over time). For this calculation, the Gompertz growth model (35) was fitted over the whole curve using software R and the Grofit package (36) as described previously (37, 38). Although both heat and heat flow data can be used for the determination of the growth rate, the long thermal equilibration period and the metabolic oscillations observed previously (19) can interfere with the curve-fitting process when using the heat flow data. Therefore, in our case, the calculated growth rate is more precisely determined when using the heat data. The growth rates calculated by this approach cannot be directly compared to the growth rate of exponential phase determined by a conventional approach, because processes such as lysis or chemical processes due to the nature of the medium might also contribute to the overall heat signal. The growth rate calculated based on the heat released by the metabolic activity in the culture should be considered a proxy for the conventional growth rate (34).
In vivo assays. (i) Measurement of efficacy and time to kill.
Adult female NMRI mice were obtained from Janvier, France. They weighed between 20 and 25 g at the beginning of the study and were housed under standard conditions with food pellets and water ad libitum. All protocols and procedures used in the current study were reviewed and approved by the local veterinary authorities of the Canton Basel-Stadt, Switzerland.
(ii) Mouse model to measure drug efficacy against T. b. rhodesiense STIB900.
Experiments were performed as previously reported (10) with minor modifications. Briefly, 72 h prior to drug administration, female NMRI mice (n = 4 per group) were infected i.p. with 5 × 103 STIB900 bloodstream forms. Drug administration (i.p.) began on day 3 postinfection and continued for four consecutive days (for a total of 4 doses) for a standard treatment regimen. Since a full dose-response curve had been previously determined for DB829 (10), only an effective dose (4 × 5 mg/kg i.p.) was tested here for DB829. An additional, higher dose (4 × 20 mg/kg i.p.) was tested for pentamidine due to its inferior in vivo efficacy. For single-dose treatment, drug administration took place on day 3 only. A control group was infected but remained untreated. Since the minimum curative dose of DB829 was 4 × 5 to 10 mg/kg i.p. (10), we chose 20 mg/kg i.p. for the single-dose study, expecting that this dose would give a decent activity without eliciting any toxicity. An additional, lower dose at 5 mg/kg was also tested to probe the minimal curative single dose. The same dose regimen was used for pentamidine. All mice were monitored for parasitemia by microscopic examination of tail blood until 60 days after infection when surviving, and aparasitemic mice were considered cured. Mice with a parasitemia relapse prior to 60 days were not considered cured, and the time to parasite relapse was recorded to calculate the mean relapse time (MRD) in days postinfection.
(iii) Mouse model to measure drug efficacy against T. b. gambiense.
Female NMRI mice (n = 4 per group) were immunosuppressed with 200 mg/kg cyclophosphamide (Endoxan; Baxter, Deerfield, IL) 2 days prior to infection with 105 bloodstream forms with one of the four T. b. gambiense strains (i.e., ITMAP141267, STIB930, 130R, or 45R) (Table 1). Immunosuppression with cyclophosphamide followed once a week until the end of the experiment. Drug administration (i.p.) began on day 3 postinfection and continued for four consecutive days (for a total of 4 doses) for a standard treatment regimen. For single-dose treatment, drug administration took place on day 3 only. A control group was infected but remained untreated. The same dose regimens as those for T. b. rhodesiense STIB900 infections were evaluated for acute T. b. gambiense infections. Surviving and aparasitemic mice at day 90 postinfection were considered cured, and the time to parasite relapse was recorded to calculate MRD in days postinfection.
(iv) In vivo time to kill.
Trypanosome infection and immunosuppression (for T. b. gambiense-infected mice) were carried out as described above for the efficacy experiments. The parasite load varied between the different mouse models, which was also observed in the efficacy experiments. The parasitemia was higher in STIB900-infected and STIB930-infected mice than in ITMAP141267-infected mice due to better adaptation of the parasites in mice. Mice were treated with a single dose of 20 mg/kg i.p. 3 days postinfection, the same dose as that used in the pharmacokinetic studies. At this dose, DB829 cured all acute T. b. rhodesiense and gambiense infections in mice, whereas pentamidine initially cleared parasites, but 2/4 mice infected with T. b. rhodesiense STIB900 later relapsed. The first microscopic parasitemia examination was 24 h after treatment (as DB829 is a slow-acting compound) and was followed by two examinations per day until all parasites had disappeared. When parasite detection by microscopy was not sensitive enough to detect any parasites, a hematocrit buffy coat examination was performed. The first time point at which no trypanosomes were detected on the microscopic slide or in the buffy coat was considered the clearance time (or time to kill) in mice. The detection limit by buffy coat examination was <102 trypanosomes/ml of blood. Mice were kept and further observed by tail blood examination until the end of the experiment to verify whether the administered dose was curative in the infected mice.
RESULTS
Pharmacokinetics of DB829 and pentamidine after i.p. and i.v. administration in mice.
The mean concentration in plasma over time profiles of DB829 and pentamidine after a single i.p. or i.v. dose were determined to compare the systemic exposure and other pharmacokinetic properties of the two compounds (Fig. 2 and Table 2). The profiles exhibited a biphasic decline with an initial distribution phase and a terminal elimination phase. About 1 min after i.v. administration at 7.5 μmol/kg (or approximately 2.3 mg/kg for DB829 and 2.6 mg/kg for pentamidine), concentration in plasma reached 36.1 and 14.3 μM and then declined to below 0.1 μM within 4 to 8 h for DB829 and 0.5 to 1 h for pentamidine. DB829 and pentamidine stayed above their IC50s of 20 and 3 nM (against T. b. rhodesiense strain STIB900) for at least 12 h (60 nM for DB829 and 18 nM for pentamidine at 12 h) before decreasing slightly below the IC50s by 24 h (19 and 2 nM at 24 h, respectively). The total body clearance of DB829 was considerably smaller (by 84%) than that of pentamidine. Both compounds appeared to be extensively tissue bound, as indicated by large volumes of distribution (Table 2). The observed terminal plasma half-lives were 18.1 h for DB829 and 6.8 h for pentamidine, and the systemic exposure (as determined by AUC) of DB829 was 6.4-fold greater than that of pentamidine. Furthermore, the unbound fraction of DB829 in mouse plasma was 4.8-fold greater than that of pentamidine (Table 2).
Fig 2.
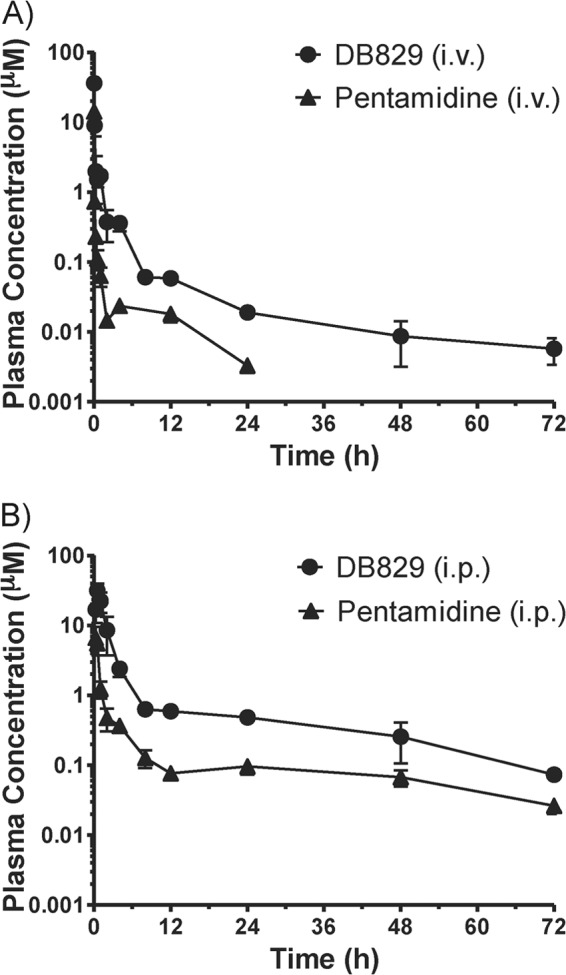
Concentration-time profiles of DB829 (circles) and pentamidine (triangles) in plasma following intravenous (A) or intraperitoneal (B) administration to uninfected mice. A single dose of DB829 (or pentamidine) was administered at 7.5 μmol/kg i.v. or 20 mg/kg i.p. Symbols and error bars represent means and standard errors (SEs), respectively, of triplicate determinations.
Table 2.
Pharmacokinetics of DB829 and pentamidine in uninfected mice
| Route | Parameter,a unit | DB829 | Pentamidine |
|---|---|---|---|
| i.v. | Dose, μmol/kg (mg/kg) | 7.5 (2.3) | 7.5 (2.6) |
| Cmax, μM | 36.1 | 14.3 | |
| AUClast, μmol/liter · h | 7.5 | 1.2 | |
| AUC0–∞, μmol/liter · h | 7.6 | 1.2 | |
| t1/2, h | 18 | 6.8 | |
| CL, liter/h/kg | 0.99 | 6.3 | |
| Vz, liter/kg | 26 | 66 | |
| MRT, h | 7.0 | 3.7 | |
| i.p. | Dose, μmol/kg (mg/kg) | 65 (20) | 58 (20) |
| Cmax, μM | 31.6 | 6.66 | |
| Tmax, h | 0.5 | 0.25 | |
| AUClast, μmol/liter · h | 72 | 11 | |
| AUC0–∞, μmol/liter · h | 74 | 12 | |
| t1/2, h | 21 | 34 | |
| fu, P, % | 40 ± 8.2 | 8.4 ± 1.2 |
Abbreviations: Cmax, maximum concentration; Tmax, time to reach Cmax; AUClast, area under the curve from time zero to the last measurable concentration; AUC0–∞, area under the curve from time zero to infinite time; t½, terminal elimination half-life; CL, total body clearance; Vz, volume of distribution; MRT, mean residence time; fu, P, unbound fraction in mouse plasma (mean ± standard deviation).
After i.p. administration at 20 mg/kg, DB829 and pentamidine were absorbed into the systemic circulation. DB829 reached a Cmax of 31.6 μM at 0.5 h and pentamidine a Cmax of 6.7 μM at 0.25 h postdosing (Table 2). Although both compounds stayed above their IC50s against strain STIB900 for the entire 72-h experimental period and had similar terminal half-lives, DB829 achieved significantly greater exposure; AUC was 6.2-fold greater and Cmax 4.7-fold greater than for pentamidine.
In vitro activity of DB829 and pentamidine against different trypanosome isolates.
In vitro activity was originally determined against bloodstream trypanosomes of T. b. rhodesiense reference strain STIB900 using the alamarBlue assay and a 72-h drug exposure time (10). As most sleeping sickness cases are due to T. b. gambiense infections, here we tested DB829 and pentamidine against a panel of different T. b. gambiense isolates. Among them were isolates from different regions, older and more recent isolates, and isolates from relapse patients after melarsoprol treatment (Table 1). DB829 was less potent against T. b. gambiense isolates (IC50 in the range of 69 to 314 nM) than against the T. b. rhodesiense reference strain STIB900 (IC50, 20 nM) (Table 3 and Fig. 3), as well as T. b. rhodesiense strain STIB799 (IC50, 27 nM) (unpublished data) and T. b. brucei strain BS221 (IC50,14 nM) (10). This indicates a reduced in vitro potency of DB829 against the T. b. gambiense subspecies. Among the T. b. gambiense isolates, there was no clear relationship between melarsoprol treatment failure (strains marked with “R”) and reduced potency of DB829, whereas pentamidine appeared to be less active against recently isolated strains (Table 3 and Fig. 3). For example, DB829 was more potent against three “R” strains (130R, 349R, and DAL898R) than the reference melarsoprol-sensitive strain STIB930. Furthermore, pentamidine exhibited reduced trypanocidal activity (IC50, >40 nM) against strains recently isolated (after the year 2000), i.e., 40R, 45R, 130R, 349R, and K03048. ITMAP141267 was the oldest strain (isolated in 1960 in DRC) tested in this study, and it was the strain most sensitive to DB829 and among those most sensitive to pentamidine. However, no correlation in reduced trypanocidal activity among T. b. gambiense strains was observed between DB829 and pentamidine (Pearson r2 = 0.14, P = 0.37; Fig. 3), suggesting lack of cross-resistance.
Table 3.
In vitro and in vivo antitrypanosomal activity of DB829 and pentamidine against different T. brucei strains
| Organism and drug regimen (no. of doses × mg/kg i.p.) | DB829 |
Pentamidine |
||
|---|---|---|---|---|
| Mean IC50 ± SD (nM) | No. cured/no. infected (MRDa) | Mean IC50 ± SD (nM) | No. cured/no. infected (MRD) | |
| T. b. rhodesiense STIB900b | 20 ± 4 | 3 ± 0.8 | ||
| 1 × 5 | 2/4 (53.5) | NDd | ||
| 1 × 20 | 4/4 | 2/4 (22.5) | ||
| 4 × 5 | 4/4 | 1/4 (23) | ||
| 4 × 20 | ND | 2/4 (28.5) | ||
| T. b. gambiense ITMAP141267 | 69 ± 17 | 6 ± 1.2 | ||
| 1 × 5 | 3/3c | 3/4c (48) | ||
| 1 × 20 | 4/4 | 4/4 | ||
| 4 × 5 | 4/4 | 4/4 | ||
| 4 × 20 | ND | ND | ||
| T. b. gambiense STIB930 | 304 ± 87 | 2 ± 1.1 | ||
| 1 × 5 | 4/4 | 3/4 (59) | ||
| 1 × 20 | 4/4 | 4/4 | ||
| 4 × 5 | 4/4 | 4/4 | ||
| 4 × 20 | ND | ND | ||
| T. b. gambiense 130R | 99 ± 23 | 53 ± 28 | ||
| 1 × 5 | ND | ND | ||
| 1 × 20 | 4/4 | 4/4 | ||
| 4 × 5 | 4/4 | 2/3 (60) | ||
| 4 × 20 | ND | 4/4 | ||
| T. b. gambiense 45R | 302 ± 113 | 81 ± 8 | ||
| 1 × 5 | ND | ND | ||
| 1 × 20 | 4/4 | 4/4 | ||
| 4 × 5 | 4/4 | 3/3 | ||
| 4 × 20 | ND | ND | ||
MRD, mean relapse time in days.
DB829 also cured 3/4 mice at 4 × 50 mg/kg administered orally, 4/4 mice at 1 × 10 mg/kg i.p. or 1 × 10 mg/kg i.v., and 1/4 mice at 4 × 5 mg/kg i.v. for STIB900-infected animals.
The experiment was terminated on day 60 postinfection, instead of day 90 for other experiments with T. b. gambiense.
ND, not done.
Fig 3.
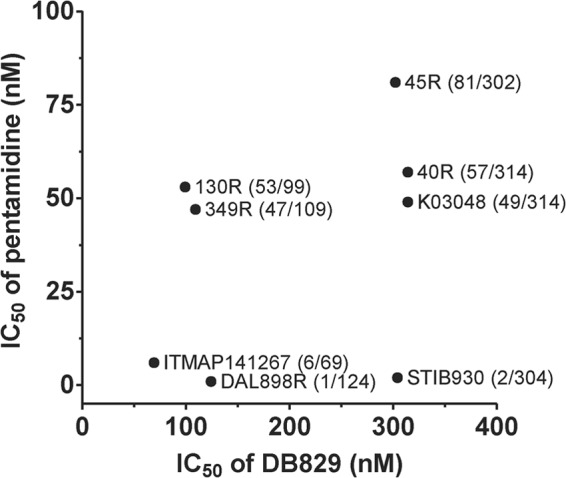
In vitro activities of DB829 and pentamidine against different T. b. gambiense strains. Symbols represent the average IC50s of three independent determinations. The first number in parentheses represents IC50 for pentamidine, and the second number is the IC50 for DB829. No significant correlation between pentamidine and DB829 activities was observed (Pearson r2 = 0.14; P = 0.37).
In vivo efficacy in T. b. gambiense-infected mice.
To investigate whether potency against T. b. gambiense was lower than against T. b. rhodesiense in vivo as well as in vitro, we tested DB829 and pentamidine in mice infected with four different T. b. gambiense isolates and compared their activities with those obtained in the T. b. rhodesiense STIB900 mouse model. Both compounds were more efficacious in mice infected with T. b. gambiense strains than with T. b. rhodesiense strain STIB900 (Table 3). In contrast to the trend observed in vitro, DB829 cured T. b. gambiense-infected mice at lower doses than it cured T. b. rhodesiense STIB900-infected mice. DB829 was curative (3/3 or 4/4 mice) at 1 × 5 mg/kg i.p. in the T. b. gambiense-infected mice (ITMAP141267 or STIB930), whereas the same dose was not sufficient to cure all (only 2/4) T. b. rhodesiense STIB900-infected mice. Similarly, pentamidine was curative (4/4 mice) at 1 × 20 mg/kg i.p. in all mice infected with any of the four T. b. gambiense strains, whereas this dose cured only 2/4 mice infected with T. b. rhodesiense STIB900. Both compounds were efficacious in mice infected with T. b. gambiense strains associated with melarsoprol treatment failure (i.e., 130R and 45R; Table 3), suggesting lack of cross-resistance with melarsoprol in vivo.
Comparison of the two drugs showed that despite being less potent in vitro, DB829 was more active than pentamidine in vivo. The lowest curative dose of DB829 in STIB900-infected mice was 4 × 5 mg/kg i.p. or 1 × 20 mg/kg i.p., but pentamidine was only partially curative (2/4 mice) at 4 × 20 mg/kg i.p. (Table 3). For T. b. gambiense-infected mice, DB829 was completely curative at all doses tested, even with a single dose of 5 mg/kg i.p. In contrast, it took a higher dose of 1 × 20 mg/kg i.p. for pentamidine to achieve a complete cure. These in vivo experiments highlight the high in vivo potency of DB829, especially against T. b. gambiense infections.
In addition to testing i.p. administration, we also evaluated the efficacy of DB829 via i.v. or oral (p.o.) administration based on the observed pharmacokinetics of the compound. The maximum concentration of DB829 in plasma following an i.v. dose of 7.5 μmol/kg (or 2.3 mg/kg) was similar (36.1 versus 31.6 μM) to that after an i.p. dose of 65 μmol/kg (or 20 mg/kg) (Table 2). Previous experiments had demonstrated that DB829 could be absorbed after oral administration, reaching a Cmax of 83 nM following a single dose of 100 μmol/kg (or 31 mg/kg) (14). Hence, we tested DB829 efficacy in the standard mouse model with T. b. rhodesiense STIB900 using an i.v. dose of 4 × 5 mg/kg and an oral dose of 4 × 50 mg/kg. DB829 cured 1/4 mice after i.v. administration, so it was less active than with i.p. administration using the same dose regimen (Table 3). However, to our surprise, oral DB829 cured 3/4 mice. This was the first time that a cationic diamidine was shown to have oral efficacy in this first-stage HAT model.
In addition to being less active, DB829 administered i.v. also showed a narrower therapeutic window, as signs of acute toxicity (lethal hypotension) were observed above 20 mg/kg of DB829 in a single dose. In contrast, i.p. administration was much better tolerated up to >50 mg/kg. The slow release by the i.p. route seems to improve tolerability without any loss of efficacy. Pentamidine exhibited a similar tolerability pattern in relation to the site of administration, but in general, DB829 was more efficacious than pentamidine.
Time of drug action in vitro. (i) Parameters to describe time of drug action.
The time of drug action of DB829 and that of pentamidine in vitro against T. b. rhodesiense strain STIB900 and T. b. gambiense strains ITMAP141267 and STIB930 were recorded and analyzed using isothermal microcalorimetry analysis. We originally proposed to use three parameters (i.e., onset of drug action, time to peak, and time to kill) to describe time of drug action (19). Here, another parameter, the growth rate (μ), was added to describe how fast the culture grows (if any growth is observed) and hence the level of growth rate inhibition, i.e., 1 − (μdrug/μdrug free).
(ii) Time of drug action against T. b. rhodesiense strain STIB900.
The onset of drug action on T. b. rhodesiense strain STIB900 ranged from 5 to 19 h for DB829 and from <4 to 14 h for pentamidine, depending on effective drug concentrations (≥0.5× IC50). The onset of drug action of melarsoprol was more concentration dependent than that of DB829 and pentamidine (Table 4). At 200 nM (∼50× IC50), melarsoprol instantly inhibited parasite growth, resulting in a heat flow curve comparable to that of trypanosome-free control samples (Fig. 4). The time to peak did not vary much for DB829 (17 to 32 h) at different concentrations (20 to 20,000 nM). For melarsoprol and pentamidine, the time to peak was considerably shorter, and at the high drug concentration of 200 nM (50× to 80× IC50) it could not be calculated because of complete or almost-complete inhibition. The time to kill the parasite population ranged from approximately 3 to 4 days for DB829, showing little variation with drug concentrations (ranging from 20 to 20,000 nM). In contrast, for pentamidine the time to kill decreased over 6-fold (89 h versus 14 h) as the drug concentration increased. Melarsoprol (at 200 nM) killed all parasites in so short a time that time to kill could not be measured (Table 4 and Fig. 4). The inhibition of growth rate (μ) by DB829 was dependent on drug concentration up to 2,000 nM and did not reach more than 72% even at the highest drug concentration tested (20,000 nM, ∼1,000× IC50) (Table 4). However, pentamidine and melarsoprol nearly completely inhibited parasite growth at 200 nM. From visual inspection, the overall shapes of the isothermal curves of DB829 and pentamidine were similar, but the shape was different for melarsoprol, especially at 20 nM (Fig. 4C), presumably as a result of distinct modes of action for cationic diamidines and organoarsenicals.
Table 4.
Drug action analysis of antitrypanosomal compounds by isothermal microcalorimetry
| T. brucei strainb and drug regimen and concn (nM) | Mean (SD) |
Inhibitione (%) | |||
|---|---|---|---|---|---|
| Onset of action (h) | Time to peak (h) | Time to kill (h) | Growth rate (μ) (h−1/1,000) | ||
| STIB900 | |||||
| Drug free | 36 (4) | 119 (13) | 29 (2) | ||
| DB829 | |||||
| 2c | 19 (8)c | 35 (4) | 115 (10) | 28 (1) | 5 |
| 20 | 19 (4) | 32 (3) | 98 (5) | 25 (1) | 15 |
| 200 | 11 (2) | 26 (2) | 77 (13) | 16 (4) | 46 |
| 2,000 | 7 (2) | 17 (4) | 58 (17) | 10 (3) | 66 |
| 20,000 | 5 (1) | 21 (3) | 67 (10) | 8 (3) | 72 |
| Pentamidine | |||||
| 0.2c | 23 (3)c | 39 (6) | 138 (10) | 25 (4) | 12 |
| 2 | 14 (4) | 31 (3) | 89 (14) | 19 (4) | 34 |
| 20 | <4 | 9 (2) | 25 (2) | <1 | ∼98 |
| 200d | <4 | NM | 14 (0) | <1 | ∼98 |
| Melarsoprol | |||||
| 2c | NMa | 40 (5) | 135 (14) | 27 (2) | 3 |
| 20 | 7 (3) | 25 (11) | 107 (56) | 7 (4) | 74 |
| 200d | NM | NM | NM | NM | ∼100 |
| STIB930 | |||||
| Drug free | 53 (2) | 139 (3) | 24 (2) | ||
| DB829 | |||||
| 20c | NM | 51 (4) | 136 (19) | 25 (2) | ∼0 |
| 200 | 26 (7) | 52 (2) | 124 (4) | 23 (1) | 2 |
| 2,000 | 17 (7) | 41 (5) | 99 (11) | 16 (3) | 33 |
| 20,000 | 10 (2) | 41 (2) | 97 (8) | 14 (2) | 43 |
| Pentamidine | |||||
| 0.2c | NM | 53 (2) | 140 (3) | 23 (2) | 4 |
| 2 | 8 (1) | 30 (2) | 75 (8) | 11 (2) | 54 |
| 20b | <4 | 12 (1) | 22 (2) | <1 | ∼96 |
| 200b | <4 | NM | NM | <1 | ∼96 |
| ITMAP141267 | |||||
| Drug free | 58 (4) | 170 (12) | 18 (3) | ||
| DB829 | |||||
| 20c | NM | 57 (5) | 167 (4) | 17 (3) | 6 |
| 200 | 28 (9) | 50 (3) | 124 (7) | 17 (4) | 5 |
| 2,000 | 14 (5) | 33 (12) | 100 (13) | 12 (2) | 33 |
| 20,000 | 12 (5) | 34 (14) | 113 (10) | 10 (2) | 42 |
| Pentamidine | |||||
| 2c | 28 (7) | 57 (2) | 159 (3) | 18 (3) | ∼0 |
| 20 | 6 (1) | 10 (1) | 41 (8) | 3.5 (0.7) | 80 |
| 200 | 4 (1) | 8 (2) | 23 (9) | 1.2 (0.4) | 94 |
NM, not measurable.
Initial inoculum density was 5 × 104 trypanosomes/ml.
Inhibition was too small to accurately recover parameters from every experiment performed.
Inhibition was too strong to accurately recover parameters from every experiment performed.
Inhibition is measured by the formula [1 − (μdrug/μdrugfree)] × 100.
Fig 4.

Microcalorimetry growth profiles of T. b. rhodesiense strain STIB900 in the presence of various concentrations of DB829 (A), pentamidine (B), and melarsoprol (C). The drug-free experiment included parasites (5 × 104/ml inoculum) without drug treatment, and the trypanosome-free experiment did not include parasites or drug treatment. Each curve represents the mean of three incubations.
(iii) Time of drug action against T. b. gambiense strains.
In drug-free control cultures, the growth rate of the two T. b. gambiense strains was lower than that of the T. b. rhodesiense reference strain STIB900 at identical starting trypanosome densities (5 × 104 trypanosomes/ml) (Fig. 5 and Table 4). The time to peak was 53 to 58 h versus 36 h for STIB900, the growth rate (μ) was 0.024 to 0.018 h−1 versus 0.029 h−1 for STIB900, and the time to death by overgrowth was also longer, 139 to 170 h compared to 119 h for STIB900. In cultures treated with drugs, the onset of drug action, the time to peak, and the time to kill of DB829 and pentamidine were generally longer for the two T. b. gambiense strains (except pentamidine against STIB930) than for the T. b. rhodesiense reference strain STIB900 (Table 4). In particular, the time to kill the two T. b. gambiense strains with DB829 ranged from 97 to 124 h (or 4 to 5 days) at effective concentrations of 20,000 to 200 nM. This is considerably longer than the standard 72 h observed in the in vitro growth inhibition assay. The slow trypanocidal action could be one reason for the greatly increased in vitro IC50s of DB829 observed in the growth inhibition assays with the T. b. gambiense strains (Table 3 and Fig. 3). However, pentamidine appeared to be faster acting, with a time to kill of <2 days at 20 nM or higher concentrations (Table 4).
Fig 5.

Microcalorimetry growth profiles of T. b. gambiense strains STIB930 (A and C) and ITMAP141267 (B and D) in the presence of various concentrations of DB829 (A and B) and pentamidine (C and D). The drug-free experiment included parasites (5 × 104/ml inoculum) without drug treatment and the trypanosome-free experiment did not include parasites or drug treatment. Each curve represents the mean of three incubations.
(iv) Effects of drug washout on parasites.
To determine whether continuous drug exposure for several days is required for DB829 to kill the trypanosomes, experiments were carried out in which the drug was washed out after 24 h of drug incubation, and heat flow was compared with that for parasites exposed to the drug throughout (permanent) the isothermal microcalorimetry assay. These measurements were compared for two strains (STIB900 and STIB930). The time to peak of the heat flow over time for the washed, drug-free samples was delayed by 11 to 22 h compared to that for the drug-free samples that were not washed (subjected to permanent exposure) (Fig. 6 and Table 5). As both samples were set up and run simultaneously, we attribute this delay to the loss of some trypanosomes during the washing steps. Trypanosomes were still viable, as seen by heat production right after the 24-h DB829 exposure at 200 or 2,000 nM, but they died later on when cultured in the drug-free medium (Fig. 6). The times to kill after drug washout were longer than with permanent drug exposure (96 versus 72 h for the T. b. rhodesiense reference strain STIB900 at 200 nM and 130 versus 89 h for T. b. gambiense strain STIB930 at 2,000 nM) (Table 5). A 24-h exposure with DB829 at 200 nM was finally sufficient to kill the T. b. rhodesiense strain STIB900 and at 2,000 nM to kill the T. b. gambiense strain STIB930. A revival of the parasite strain STIB930 occurred in a single experiment at 200 nM. These results suggest that the trypanosomes took up lethal concentrations of DB829 at 200 nM (STIB900) or 2,000 nM (STIB930) in the first 24 h, and this was sufficient to kill them at a later time.
Fig 6.
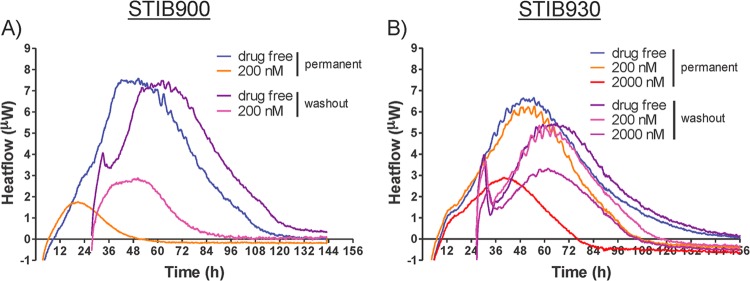
Effects of drug washout on microcalorimetry growth profiles of T. b. rhodesiense strain STIB900 (A) and T. b. gambiense strain STIB930 (B) in the presence of DB829. Drug washout was performed after a 24-h exposure to DB829. The drug-free experiment included parasites (5 × 104/ml inoculum) without drug treatment. Each curve represents the mean of three incubations.
Table 5.
Effects of inoculum and drug washout on drug action of DB829 and pentamidine by isothermal microcalorimetry
| T. brucei strain and inoculum | Compound | Drug concn (nM) | Mean (SD) |
Inhibitionf (%) | |||
|---|---|---|---|---|---|---|---|
| Onset of action (h) | Time to peak (h) | Time to kill (h) | Growth rate (μ) (×1,000 h−1) | ||||
| STIB900 | 0 (drug free) | 37 (4) | 123 (12) | 28 (3) | |||
| 5 × 104/ml | DB829 | 20 | 22 (5) | 34 (2) | 98 (4) | 22 (4) | 15 |
| 200 | 10 (2) | 24 (3) | 72 (8) | 13 (2) | 52 | ||
| Pentamidine | 0.2 | 20 (1) | 36 (4) | 134 (11) | 27 (2) | 4 | |
| 2 | 12 (2) | 30 (2) | 84 (14) | 19 (5) | 35 | ||
| 20b | 3 (1) | 9 (2) | 21 (9) | <1 | ∼98 | ||
| 1 × 104/ml | 0 (drug free) | 52 (5) | 135 (13) | 27 (1) | |||
| DB829 | 20 | 17 (4) | 33 (2) | 69 (7) | 8 (3) | 72 | |
| 200b | 14 (2) | 16 (4) | 36 (12) | 2 (2) | >90 | ||
| Pentamidine | 0.2a,c | 20 (1) | 42 (3) | 152 (9) | 28 (1) | 3 | |
| 2b | 12 (3) | 20 (4) | 40 (5) | <1 | ∼97 | ||
| 20b | NMe | NM | NM | <1 | ∼99 | ||
| 5 × 104/ml | Washout | 0 (drug free) | 59 (5) | 132 (5) | 26 (2) | ||
| DB829 | 200c | <27 | 46 (6) | 96 (6) | 9 (0.4) | 64 | |
| STIB930 | 0 (drug free) | 52 (2) | 151 (9) | 25 (2) | |||
| 5 × 104/ml | DB829 | 200 | 27 (6) | 51 (1) | 121 (4) | 24 (1) | 4 |
| 2,000c | 16 (5) | 39 (1) | 89 (2) | 12 (1) | 52 | ||
| Pentamidine | 0.2d | <4 | 53 (1) | 163 (19) | 23 (1) | 8 | |
| 2 | 6 (1) | 29 (7) | 66 (12) | 8 (3) | 67 | ||
| 1 × 104/ml | 0 (drug free) | 69 (2) | 159 (11) | 24 (2) | |||
| DB829 | 200 | 34 (10) | 56 (4) | 111 (10) | 14 (3) | 42 | |
| 2,000b,c | 15 (5) | 37 (4) | 66 (3) | 2 (0.3) | 91 | ||
| Pentamidine | 0.2d | <6 | 74 (2) | 161 (5) | 22 (4) | 12 | |
| 2b | <6 | NM | NM | NM | ∼100 | ||
| 5 × 104/ml | Washout | 0 (drug free) | 63 (5) | 154 (11) | 21 (1) | ||
| DB829 | 200c | <27 | 60 (2) | 128 (9) | 21 (0.5) | ∼0 | |
| 2,000d | <27 | 58 (2) | 130 (12) | 15 (2) | 28 | ||
Inhibition was too small to accurately recover parameters from every experiment performed.
Inhibition was too strong to accurately recover parameters from every experiment performed.
Two independent experiments were performed (each in triplicate).
One independent experiment was performed (in triplicate).
NM, not measurable.
Inhibition is measured by the formula [1 − (μdrug/μdrugfree)] × 100.
(v) Effects of inoculum on time of drug action.
Inhibition of trypanosome growth by DB829 was dependent on the initial parasite density (inoculum effect). With an inoculum of 5 × 104/ml, the growth rate of T. b. rhodesiense strain STIB900 was inhibited by 15% and 52% with DB829 concentrations of 20 nM and 200 nM, respectively. Considerably stronger inhibition, 72% with 20 nM drug and 90% with 200 nM drug, was observed with an initial inoculum of 1 × 104/ml (Table 5). However, the two parameters, onset of action and time to peak of the heat flow curves, remained similar at different inocula (Table 5 and Fig. 7). We also examined inoculum effects with T. b. gambiense strain STIB930. At a drug concentration of 200 nM DB829, parasite growth rate was inhibited by 4% and with 2,000 nM by 52% with an initial inoculum of 5 × 104/ml. Significantly stronger inhibition was observed (42% at 200 nM and 91% at 2,000 nM) when the initial inoculum was 1 × 104/ml (Table 5). As with STIB900, the other two parameters of the heat flow curves remained similar at different inocula (Table 5 and Fig. 7). The same inoculum effects were also observed for pentamidine (Table 5 and Fig. 7).
Fig 7.

Inoculum effect on microcalorimetry growth profiles of T. b. rhodesiense strain STIB900 (A and C) and T. b. gambiense strain STIB930 (B and D) in the presence of various concentrations of DB829 (A and B) and pentamidine (C and D). Two initial trypanosome densities were tested: standard inoculum at 5 × 104/ml and low inoculum at 1 × 104/ml. The drug-free experiment included parasites without drug treatment, and the trypanosome-free experiment did not include parasites or drug treatment. Each curve represents the mean of three incubations.
(vi) Time of drug action in vivo.
The in vivo time of drug action of DB829 and that of pentamidine were determined, following a single i.p. dose of 20 mg/kg to mice infected with T. b. rhodesiense strain STIB900 or T. b. gambiense strains ITMAP141267 and STIB930, to compare the time of drug action in vitro with that in vivo. For mice infected with T. b. rhodesiense strain STIB900, the in vivo onset of drug action of DB829 was less than 24 h, which is in accordance with the in vitro time of the onset of action (5 to 19 h at DB829 concentrations of 20 to 20,000 nM) (Table 4). The in vivo time to kill (parasite clearance time) was on average 78 h (i.e., the first mouse was parasite free 72 h after drug administration and the last mouse after 80 h) (Table 6). This is in the range of the in vitro time to kill (58 to 98 h; Table 4) at the different DB829 concentrations (20 to 20,000 nM). The time to kill in mice infected with T. b. gambiense strain ITMAP141267 was on average 102 h (96 to 120 h), and in STIB930-infected mice, it was 62 h (56 to 80 h) (Table 6). This is similar to or slightly faster than the in vitro time to kill for these two parasite strains (100 to 124 h and 99 h, respectively, at approximately 10× IC50) (Table 4).
Table 6.
In vivo time to kill (parasite clearance time) in infected mice administered DB829 or pentamidinea
| T. brucei strains | Mean (range) time to kill (h) |
|
|---|---|---|
| DB829 | Pentamidine | |
| STIB900 | 78 (72–80) | 26 (24–32) |
| ITMAP141267 | 102 (96–120) | 30 (24–48) |
| STIB930 | 62 (56–80) | 48 (48) |
Mean data for four mice per group; range, time elapsed between the first mouse and the last mouse becoming aparasitemic. For each drug, a single dose of 20 mg/kg was administered i.p. on day 3 postinfection.
In addition, the in vivo times to kill of pentamidine were shorter than those of DB829 in all experiments, although some mice infected with the T. b. rhodesiense strain STIB900 relapsed later (Table 3). The in vivo times to kill of pentamidine were on average 26 h, 30 h, and 48 h for mice infected with T. b. rhodesiense strain STIB900 and T. b. gambiense strains ITMAP141267 and STIB930, respectively. These results are similar to the in vitro time to kill at relevant drug concentrations (around 20 nM) (Table 4). The parasite load in STIB930-infected mice was higher than in mice with ITMAP141267 infections due to better adaptation of STIB930 in mice.
DISCUSSION
Recent efforts to enhance the CNS activity of antiparasitic diamidines led to the discovery of aza analogues of furamidine. One of these, DB829, cured all mice in the second-stage HAT model using T. b. brucei strain GVR35 when administered intraperitoneally (10), in spite of the dogma that cationic diamidines cannot traverse cellular membranes via passive diffusion. This liability limits their oral bioavailability and brain penetration, so that they are generally considered to be effective only against first-stage HAT and only through parenteral administration. Our discovery of aza-diamidines led to the further testing of DB829 in a second-stage HAT monkey model (results will be published in due course) and in the present study to the comprehensive pharmacological characterization of DB829 and the comparator drug pentamidine with two human-infective T. brucei subspecies. Effectiveness against T. b. gambiense is of special interest since most HAT patients are infected with this trypanosome subspecies.
In comparison to the activity against the reference strain T. b. rhodesiense STIB900, the potency of DB829 in vitro against T. b. gambiense strains was reduced, ranging from 3.5- to 15-fold (Table 3). Pentamidine also displayed a reduction in potency (2- to 27-fold) against most T. b. gambiense strains, except for STIB930 and DAL898R strains (Fig. 3). No cross-resistance between DB829 and pentamidine was observed in this study, which included isolates from relapsed patients, in contrast to the cross-resistance previously reported for pentamidine and melarsoprol (39, 40). Transporters are known to be an important determinant for developing resistance in trypanosomes to pentamidine and melarsoprol (41–44). The P2 aminopurine transporter plays a predominant role in the uptake of aza analogues of furamidine into T. b. brucei s427 trypanosomes (45). The limited cross-resistance of DB829 to pentamidine is presumably because different transporters (41, 46)—in addition to P2—are involved in drug uptake of both pentamidine and melarsoprol but do not transport DB829. The existence of less efficient transporters in the T. b. gambiense strains may also explain the observed lower in vitro potency of DB829 against these parasite strains.
Despite being less active against T. b. gambiense strains in vitro, DB829 was surprisingly more efficacious in vivo, curing all mice infected with different T. b. gambiense isolates at all dose regimens tested (Table 3). In particular, a single dose of DB829 administered intraperitoneally was highly effective, curing all T. b. gambiense-infected animals at a dose as low as 5 mg/kg of body weight, whereas the same dose of DB829 cured only 2/4 mice infected with T. b. rhodesiense strain STIB900. The high efficacy of the lowest DB829 doses used in our present study meant that the minimal curative dose of DB829 for T. b. gambiense-infected mice has not yet been established. In comparison, pentamidine was less efficacious than DB829 in T. b. gambiense-infected mice (3/4 cures at 5 mg/kg, single dose, compared with 4/4 or 3/3 cures with DB829) and in T. b. rhodesiense-infected mice (1/4 cures compared to 4/4 cures at 4 × 5 mg/kg or 2/4 versus 4/4 cures at 20 mg/kg, single dose, respectively).
Two factors may have contributed to the observed difference in the activity of DB829 against T. b. gambiense in vitro and in vivo. First, downregulation of diamidine uptake transporter(s) (e.g., P2 aminopurine transporter) under the in vitro culturing conditions could have reduced the susceptibility of the T. b. gambiense isolates to DB829, whereas in vivo bloodstream trypanosomes in infected mice maintain a normal expression and function of diamidine uptake transporter(s) and hence would be more susceptible to DB829 than trypanosomes in in vitro culture. A striking decrease (from 100% to 4.2%) in the functional activity of the P2 aminopurine transporter in T. b. brucei s427 cells over a 24-h culture period after fresh isolation from an infected rat was observed by Ward et al. (45). However, it is yet to be demonstrated that such downregulation of the P2 aminopurine transporter also occurred in the T. b. gambiense isolates tested in vitro in our study. The observed decrease in in vitro potency (IC50) of the T. b. gambiense isolates did not seem to have a major effect on the time to kill. At approximately 10× IC50 the in vitro and in vivo times to kill were similar (Tables 4 and 6), which indicates that the drug uptake in vitro was high enough to kill the parasites in a period similar to that in vivo.
A second reason for the greater potency of DB829 against T. b. gambiense isolates in vivo compared with T. b. rhodesiense could have been the levels of parasitemia. Mice infected with T. b. gambiense strains generally have a lower parasitemia (<107 trypanosomes/ml of blood) than those infected with T. b. rhodesiense strain STIB900 (≥108 trypanosomes/ml of blood) (47). An inoculum effect (i.e., initial parasite density-dependent growth inhibition) could result in a better-than-expected in vivo activity due to lower parasitemia in mice infected with T. b. gambiense strains. We assessed inoculum effects on time of drug action by DB829 and pentamidine in vitro using isothermal microcalorimetry and confirmed that inhibition of trypanosome growth rate by DB829 and pentamidine depended on the initial inoculum: the lower the density of parasites, the stronger the inhibition of growth of either T. b. rhodesiense or gambiense strains (Fig. 7 and Table 5). Although the causes of the inoculum effect are still unclear to us (see additional discussion in the supplemental material), the observed inoculum effect is not expected to have any untoward effect on efficacy in HAT patients treated with pentamidine (or potentially DB829), since parasitemia in human patients, especially those with T. b. gambiense infections, is very low (<105 trypanosomes/ml).
Using isothermal microcalorimetry, we were able to make a real-time comparison of the effects of different drugs on different parasite strains. It provided us with a convenient tool to monitor parasite growth and to visualize and quantify the level of growth rate inhibition by antiparasitic drugs on a real-time basis. Wenzler et al. (19) first described the use of this technology to characterize the parasite growth and effects of drug treatments for T. brucei and Plasmodium falciparum. Here we used mathematical models and included an additional parameter, growth rate (μ). The new four-parameter model (onset of drug action, time to peak, time to kill, and growth rate) describes time of drug action in more detail.
The inoculum effect was revealed by using the same drug concentration but different initial inocula (Table 5). The effect on the inhibition of growth rate is discussed above. In contrast, inoculum effects were not as clearly apparent on onset of drug action or time to peak. This indicates that the drug action starts almost simultaneously at identical drug concentrations for DB829 and pentamidine at the different inocula. Inhibition is considerably stronger in the samples with lower trypanosome densities, when their drug-free control samples are still in the exponential growth phase. On the other hand, at higher parasite densities inhibition may be less observable because the onset of drug action and/or time to peak is already close to the stationary phase, when growth of the drug-free control samples is reduced as well—not by drug inhibition but due to overgrowth of the parasite culture. Growth inhibition by slow-acting drugs was less visible at higher inocula since a decreased metabolic activity may coincide with the overgrowing culture.
The time of onset of drug action is a pharmacodynamic parameter that is important when considering potential use for treatment. A main feature of isothermal microcalorimetry is real-time monitoring of organism growth, which can be used to readily calculate in vitro time of drug action. Traditionally, time of drug action was obtained through laborious multiple single time point antiparasitic inhibition assays with variable drug exposure times. Onset of action, time to peak, and time to kill could only be estimated, and the level of inhibition was calculated at the few tested time points (19). Microcalorimetry offers a more reliable and less laborious method. However, as the microcalorimeter typically requires a few hours to fully equilibrate 4-ml ampoules containing 2 ml of culture medium, the analysis of onset of drug action is still difficult, especially for fast-acting compounds. Further technical improvements (e.g., adding an injection system) will be desirable to allow preequilibration of ampoules prior to injecting compound solutions, thus reducing the equilibration time following drug addition (48).
DB829 was shown to be a slow-acting trypanocidal compound. It was possible to obtain reliable results for the in vitro time to kill at a range of concentrations. This ranged from 3 to 4 days for T. b. rhodesiense strain STIB900 and from 4 to 5 days for the T. b. gambiense strains, depending on effective (≥0.5× IC50) drug concentrations (Table 4). In comparison, pentamidine is faster acting, reducing parasites to below the detection limit within 1 day after drug addition at 200 nM concentration for all three strains and at 20 nM for STIB900 and ITMAP141267. This is probably due to differences in the parasite uptake kinetics of the two compounds, which have been previously characterized using T. b. brucei strain s427 (45, 49). It is noteworthy that parasites of T. b. rhodesiense strain STIB900 grew faster than those of the two T. b. gambiense strains in the absence of drug treatment, resulting in a shorter time to death of the parasite culture (approximately 5 days versus 6 to 7 days, respectively) as a result of overgrowing. Since our standard in vitro growth inhibition assays utilize a 72-h of incubation period, we believe that the slow trypanocidal action of DB829 contributed, in part, to the increases in measured IC50s against the T. b. gambiense strains.
Activity of DB829 in vitro was not increased with drug concentrations higher than 2,000 nM (Fig. 4A and 5A and B). This is most likely due to the saturation of the P2 transporter, which is the main route of DB829 uptake (45). In a P2 transporter-mediated uptake assay, the Michaelis-Menten constant, Km, of DB829 is 1.1 μM, indicating that saturation (maximum uptake rate, Vmax) is almost reached after 2 μM and therefore no significant higher inhibition can be expected with this compound at higher drug concentrations.
Parasite clearance time in vivo, following a single intraperitoneal injection of DB829 at a dose of 20 mg/kg of body weight, ranged from 3 to 3.3 days for mice infected with T. b. rhodesiense strain STIB900 and from 2.3 to 5 days for mice infected with the different T. b. gambiense strains (Table 6). These times were in good agreement with the in vitro times to kill, given the high concentration in plasma of DB829 (Cmax = 31.6 μM) reached at this dose level (Table 2). As predicted based on in vitro time to kill, pentamidine is also faster acting in vivo than DB829, clearing parasites from the bloodstream within 1 to 1.3 days for mice infected with T. b. rhodesiense strain STIB900 and 1 to 2 days for mice infected with the T. b. gambiense strains. In summary, it generally takes more time for DB829 to clear T. brucei parasites than for pentamidine.
An interesting feature of the action of DB829 is that despite being slow acting, it was able to kill parasites in vitro after a short (24-h) drug exposure, as demonstrated by drug washout experiments (Fig. 6 and Table 5). This is consistent with the observation that a single dose of DB829 could effectively cure infected mice (Table 3), which was partially due to desirable pharmacokinetics, with a high maximum drug concentration in plasma, relatively long terminal elimination half-life at the dose tested, and relatively low plasma protein binding (Table 2).
Based on the outstanding efficacy of DB829 in the mouse models with acute infections and its desirable pharmacokinetic properties, it is of interest to consider the potential value of this compound in the treatment of HAT. A single dose of DB829 administered intramuscularly could be suggested as a new short treatment for first-stage HAT. An even more important property of DB829 is that it was shown to be efficacious in mice with second-stage infection when administered intraperitoneally (10). This has prompted further testing of parenteral DB829 (as well as its methamidoxime oral prodrug, DB868) in the vervet monkey model for second-stage HAT with promising results (16).
New, safe, and effective drugs are urgently needed for treatment of both stages of HAT. It is also desirable that they be easy to administer, with a simple dosing regimen. With improved drugs, it should be possible to control HAT effectively and perhaps even eliminate the disease. DB829 is a promising candidate for further development. It is CNS active (by itself, without bioconversion), so it could be used against second-stage HAT. In mouse models, it showed outstanding efficacy against both first and second stages of infection. Furthermore, it cured both T. b. gambiense and T. b. rhodesiense infections at low and well-tolerated doses. Parenteral DB829 with a short dosing regimen such as 5 days or less for the second stage and a single dose for the first stage should be seriously considered for further development as a new treatment for sleeping sickness.
Supplementary Material
ACKNOWLEDGMENTS
We thank Pati Pyana and Anne Clarisse Lekane Likeufack for isolating T. b. gambiense strains from patients in the Democratic Republic of the Congo, Jennifer Jenkins for critical reading and inputs to the manuscript, Guy Riccio and Christiane Braghiroli for carrying out experiments on in vivo efficacy and time to kill in mice, and Kirsten Gillingwater for time-to-kill experiments in mice.
This work was supported by the Bill and Melinda Gates Foundation through the Consortium for Parasitic Drug Development (CPDD).
Footnotes
Published ahead of print 19 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00398-13.
REFERENCES
- 1.Simarro PP, Cecchi G, Franco JR, Paone M, Fevre EM, Diarra A, Postigo JA, Mattioli RC, Jannin JG. 2011. Risk for human African trypanosomiasis, Central Africa, 2000–2009. Emerg. Infect. Dis. 17:2322–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simarro PP, Jannin J, Cattand P. 2008. Eliminating human African trypanosomiasis: where do we stand and what comes next? PLoS Med. 5:e55. 10.1371/journal.pmed.0050055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO 1998. Control and surveillance of African trypanosomiasis. Report of a WHO Expert Committee. World Health Organ. Tech. Rep. Ser. 881(I–VI):1–114 [PubMed] [Google Scholar]
- 4.Barrett MP. 2006. The rise and fall of sleeping sickness. Lancet 367:1377–1378 [DOI] [PubMed] [Google Scholar]
- 5.Barrett MP, Gilbert IH. 2006. Targeting of toxic compounds to the trypanosome's interior. Adv. Parasitol. 63:125–183 [DOI] [PubMed] [Google Scholar]
- 6.Brun R, Blum J, Chappuis F, Burri C. 2010. Human African trypanosomiasis. Lancet 375:148–159 [DOI] [PubMed] [Google Scholar]
- 7.McCulloch R. 2004. Antigenic variation in African trypanosomes: monitoring progress. Trends Parasitol. 20:117–121 [DOI] [PubMed] [Google Scholar]
- 8.Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, Boykin DW. 2005. Dications that target the DNA minor groove: compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr. Med. Chem. Anticancer Agents 5:389–408 [DOI] [PubMed] [Google Scholar]
- 9.Midgley I, Fitzpatrick K, Taylor LM, Houchen TL, Henderson SJ, Wright SJ, Cybulski ZR, John BA, McBurney A, Boykin DW, Trendler KL. 2007. Pharmacokinetics and metabolism of the prodrug DB289 (2,5-bis[4-(N-methoxyamidino)phenyl]furan monomaleate) in rat and monkey and its conversion to the antiprotozoal/antifungal drug DB75 (2,5-bis(4-guanylphenyl)furan dihydrochloride). Drug Metab. Dispos. 35:955–967 [DOI] [PubMed] [Google Scholar]
- 10.Wenzler T, Boykin DW, Ismail MA, Hall JE, Tidwell RR, Brun R. 2009. New treatment option for second-stage African sleeping sickness: in vitro and in vivo efficacy of aza analogs of DB289. Antimicrob. Agents Chemother. 53:4185–4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burri C. 2010. Chemotherapy against human African trypanosomiasis: is there a road to success? Parasitology 137:1987–1994 [DOI] [PubMed] [Google Scholar]
- 12.Paine MF, Wang MZ, Generaux CN, Boykin DW, Wilson WD, De Koning HP, Olson CA, Pohlig G, Burri C, Brun R, Murilla GA, Thuita JK, Barrett MP, Tidwell RR. 2010. Diamidines for human African trypanosomiasis. Curr. Opin. Investig. Drugs 11:876–883 [PubMed] [Google Scholar]
- 13.Maser P, Wittlin S, Rottmann M, Wenzler T, Kaiser M, Brun R. 2012. Antiparasitic agents: new drugs on the horizon. Curr. Opin. Pharmacol. 12:562–566 [DOI] [PubMed] [Google Scholar]
- 14.Brun R, Don R, Jacobs RT, Wang MZ, Barrett MP. 2011. Development of novel drugs for human African trypanosomiasis. Future Microbiol. 6:677–691 [DOI] [PubMed] [Google Scholar]
- 15.Thuita JK, Wang MZ, Kagira JM, Denton CL, Paine MF, Mdachi RE, Murilla GA, Ching S, Boykin DW, Tidwell RR, Hall JE, Brun R. 2012. Pharmacology of DB844, an orally active aza analogue of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl. Trop. Dis. 6:e1734. 10.1371/journal.pntd.0001734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thuita JK. 2012. Biological and pharmacological investigations of novel diamidines in animal models of human African trypanosomiasis. Ph.D. thesis University of Basel, Basel, Switzerland [Google Scholar]
- 17.Thuita JK, Wolf KK, Murilla GA, Liu Q, Mutuku JN, Chen Y, Bridges AS, Mdachi RE, Ismail MA, Ching S, Boykin DW, Hall JE, Tidwell RR, Paine MF, Brun R, Wang MZ. 2013. Safety, pharmacokinetic, and efficacy studies of oral DB868 in a first stage vervet monkey model of human African trypanosomiasis. PLoS Negl. Trop. Dis. 7(6):e2230. 10.1371/journal.pntd.0002230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.WHO Updated June 2013. Media Centre, fact sheet N°259. Trypanosomiasis, human African (sleeping sickness). http://www.who.int/mediacentre/factsheets/fs259/en/ Accessed 25 July 2013
- 19.Wenzler T, Steinhuber A, Wittlin S, Scheurer C, Brun R, Trampuz A. 2012. Isothermal microcalorimetry, a new tool to monitor drug action against Trypanosoma brucei and Plasmodium falciparum. PLoS Negl. Trop. Dis. 6:e1668. 10.1371/journal.pntd.0001668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das BP, Boykin DW. 1977. Synthesis and antiprotozoal activity of 2,5-bis(4-guanylphenyl)furans. J. Med. Chem. 20:531–536 [DOI] [PubMed] [Google Scholar]
- 21.Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. 2003. Synthesis and antiprotozoal activity of aza-analogues of furamidine. J. Med. Chem. 46:4761–4769 [DOI] [PubMed] [Google Scholar]
- 22.Yan GZ, Brouwer KL, Pollack GM, Wang MZ, Tidwell RR, Hall JE, Paine MF. 2011. Mechanisms underlying differences in systemic exposure of structurally similar active metabolites: comparison of two preclinical hepatic models. J. Pharmacol. Exp. Ther. 337:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brun R, Schumacher R, Schmid C, Kunz C, Burri C. 2001. The phenomenon of treatment failures in Human African Trypanosomiasis. Trop. Med. Int. Health 6:906–914 [DOI] [PubMed] [Google Scholar]
- 24.Bacchi CJ, Nathan HC, Livingston T, Valladares G, Saric M, Sayer PD, Njogu AR, Clarkson AB., Jr 1990. Differential susceptibility to DL-alpha-difluoromethylornithine in clinical isolates of Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 34:1183–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simarro PP, Cecchi G, Paone M, Franco JR, Diarra A, Ruiz JA, Fevre EM, Courtin F, Mattioli RC, Jannin JG. 2010. The atlas of human African trypanosomiasis: a contribution to global mapping of neglected tropical diseases. Int. J. Health Geogr. 9:57. 10.1186/1476-072X-9-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simarro PP, Diarra A, Ruiz Postigo JA, Franco JR, Jannin JG. 2011. The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000–2009: the way forward. PLoS Negl. Trop. Dis. 5:e1007. 10.1371/journal.pntd.0001007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Felgner P, Brinkmann U, Zillmann U, Mehlitz D, Abu-Ishira S. 1981. Epidemiological studies on the animal reservoir of gambiense sleeping sickness. Part II. Parasitological and immunodiagnostic examination of the human population. Tropenmed. Parasitol. 32:134–140 [PubMed] [Google Scholar]
- 28.Likeufack AC, Brun R, Fomena A, Truc P. 2006. Comparison of the in vitro drug sensitivity of Trypanosoma brucei gambiense strains from West and Central Africa isolated in the periods 1960–1995 and 1999–2004. Acta Trop. 100:11–16 [DOI] [PubMed] [Google Scholar]
- 29.Pyana PP, Ngay Lukusa I, Mumba Ngoyi D, Van Reet N, Kaiser M, Karhemere Bin Shamamba S, Buscher P. 2011. Isolation of Trypanosoma brucei gambiense from cured and relapsed sleeping sickness patients and adaptation to laboratory mice. PLoS Negl. Trop. Dis. 5:e1025. 10.1371/journal.pntd.0001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maina N, Maina KJ, Maser P, Brun R. 2007. Genotypic and phenotypic characterization of Trypanosoma brucei gambiense isolates from Ibba, South Sudan, an area of high melarsoprol treatment failure rate. Acta Trop. 104:84–90 [DOI] [PubMed] [Google Scholar]
- 31.Raz B, Iten M, Grether-Buhler Y, Kaminsky R, Brun R. 1997. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 68:139–147 [DOI] [PubMed] [Google Scholar]
- 32.Baltz T, Baltz D, Giroud C, Crockett J. 1985. Cultivation in a semi-defined medium of animal infective forms of Trypanosoma brucei, T. equiperdum, T. evansi, T. rhodesiense and T. gambiense. EMBO J. 4:1273–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirumi H, Hirumi K. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75:985–989 [PubMed] [Google Scholar]
- 34.Braissant O, Wirz D, Gopfert B, Daniels AU. 2010. Use of isothermal microcalorimetry to monitor microbial activities. FEMS Microbiol. Lett. 303:1–8 [DOI] [PubMed] [Google Scholar]
- 35.Zwietering MH, Jongenburger I, Rombouts FM, van't Riet K. 1990. Modeling of the bacterial growth curve. Appl. Environ. Microbiol. 56:1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kahm M, Hasenbrink G, Lichtenberg-Frate H, Ludwig J, Kschischo M. 2010. Grofit: fitting biological growth curves with R. J. Statistical Software 33:1–21 [Google Scholar]
- 37.Astasov-Frauenhoffer M, Braissant O, Hauser-Gerspach I, Daniels AU, Wirz D, Weiger R, Waltimo T. 2011. Quantification of vital adherent Streptococcus sanguinis cells on protein-coated titanium after disinfectant treatment. J. Mater. Sci. Mater. Med. 22:2045–2051 [DOI] [PubMed] [Google Scholar]
- 38.Braissant O, Bonkat G, Wirz D, Bachmann A. 2013. Microbial growth and isothermal microcalorimetry: growth models and their application to microcalorimetric data. Thermochimica Acta 555:64–71 [Google Scholar]
- 39.Bernhard SC, Nerima B, Maser P, Brun R. 2007. Melarsoprol- and pentamidine-resistant Trypanosoma brucei rhodesiense populations and their cross-resistance. Int. J. Parasitol. 37:1443–1448 [DOI] [PubMed] [Google Scholar]
- 40.Williamson J, Rollo IM. 1959. Drug resistance in trepanosomes: cross-resistance analyses. Br. J. Pharmacol. Chemother. 14:423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baker N, Glover L, Munday JC, Aguinaga Andres D, Barrett MP, de Koning HP, Horn D. 2012. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. U. S. A. 109:10996–11001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Koning HP. 2001. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters: implications for cross-resistance with arsenicals. Mol. Pharmacol. 59:586–592 [DOI] [PubMed] [Google Scholar]
- 43.Maser P, Sutterlin C, Kralli A, Kaminsky R. 1999. A nucleoside transporter from Trypanosoma brucei involved in drug resistance. Science 285:242–244 [DOI] [PubMed] [Google Scholar]
- 44.Matovu E, Stewart ML, Geiser F, Brun R, Maser P, Wallace LJ, Burchmore RJ, Enyaru JC, Barrett MP, Kaminsky R, Seebeck T, de Koning HP. 2003. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot. Cell 2:1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward CP, Wong PE, Burchmore RJ, de Koning HP, Barrett MP. 2011. Trypanocidal furamidine analogues: influence of pyridine nitrogens on trypanocidal activity, transport kinetics, and resistance patterns. Antimicrob. Agents Chemother. 55:2352–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teka IA, Kazibwe AJ, El-Sabbagh N, Al-Salabi MI, Ward CP, Eze AA, Munday JC, Maser P, Matovu E, Barrett MP, de Koning HP. 2011. The diamidine diminazene aceturate is a substrate for the high-affinity pentamidine transporter: implications for the development of high resistance levels in trypanosomes. Mol. Pharmacol. 80:110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maina NW, Oberle M, Otieno C, Kunz C, Maeser P, Ndung'u JM, Brun R. 2007. Isolation and propagation of Trypanosoma brucei gambiense from sleeping sickness patients in south Sudan. Trans. R. Soc. Trop. Med. Hyg. 101:540–546 [DOI] [PubMed] [Google Scholar]
- 48.Manneck T, Braissant O, Haggenmuller Y, Keiser J. 2011. Isothermal microcalorimetry to study drugs against Schistosoma mansoni. J. Clin. Microbiol. 49:1217–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathis AM, Bridges AS, Ismail MA, Kumar A, Francesconi I, Anbazhagan M, Hu Q, Tanious FA, Wenzler T, Saulter J, Wilson WD, Brun R, Boykin DW, Tidwell RR, Hall JE. 2007. Diphenyl furans and aza analogs: effects of structural modification on in vitro activity, DNA binding, and accumulation and distribution in trypanosomes. Antimicrob. Agents Chemother. 51:2801–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.