

Abstract
Major surface glycoprotein (Msg), the most abundant cell surface protein of Pneumocystis, plays an important role in the interaction of this opportunistic pathogen with host cells, and its potential for antigenic variation may facilitate evasion of host immune responses. In the present study, we have identified and characterized the promoter region of msg in 3 species of Pneumocystis: P. carinii, P. jirovecii, and P. murina. Because Pneumocystis cannot be cultured, promoter activity was measured in Saccharomyces cerevisiae, a related fungus, using a yeast vector modified to utilize the gene coding for Renilla luciferase as a reporter gene. The 5′-flanking sequences of msg from all three Pneumocystis species showed considerable promoter activity, with increases in luciferase activity up to 15- to 44-fold above baseline. Progressive deletions helped define an ∼13-bp sequence in each Pneumocystis species that appears to be critical for promoter activity. Electrophoretic mobility shift analysis using P. carinii-specific msg promoter sequences demonstrated binding of nuclear proteins of S. cerevisiae. The 144-bp 5′-flanking region of P. murina msg showed 72% identity to that of P. carinii. The 5′-flanking region of P. jirovecii msg showed 58 and 61% identity to those of P. murina and P. carinii, respectively. The msg promoter is a good candidate for inclusion in a construct designed for genetic manipulation of Pneumocystis species.
INTRODUCTION
Pneumocystis jirovecii is a pathogen that causes pneumonia in AIDS and other immunocompromised patients (1, 2). Members of the genus Pneumocystis infecting different mammalian hosts are unique and genetically divergent (3–6) and are designated different species, with P. jirovecii infecting humans, P. carinii infecting rats, and P. murina infecting mice (7–9). Major surface glycoprotein (Msg) is the most abundant protein on the cell surface of Pneumocystis. Msg plays an important role in host-organism interactions and potentially facilitates evasion of host immune responses through antigenic variation. Msg is encoded by a multicopy family of related but unique genes (an estimated 25 to 80 copies per genome) that are clustered in tandem arrays near the telomeres of most chromosomes (10–16). Available data suggest that only a single msg gene is expressed in an individual cell and that this expressed msg gene is located downstream of a region termed the upstream conserved sequence (UCS) (17–21).
Although expression of Msg gene variants has been well studied, little is known about how msg expression is regulated. In P. carinii and P. jirovecii, there is an ∼1,500-bp region upstream of the Msg expression site that does not contain any identifiable coding regions, which is unusual for Pneumocystis since intergenic regions are usually ∼300 to 500 bp long (unpublished observations). This suggests that this region may be important in regulating msg expression, possibly through promoter elements (22).
Although Pneumocystis cannot currently be cultured for sustained periods, development of vectors that can be used to transfect the organism may facilitate studies of its biology. Because Msg is the most highly expressed protein of Pneumocystis (23), its regulatory regions may provide an ideal promoter sequence to include in such a vector.
In this study, we undertook to identify the regulatory regions important for Msg expression in three different species of Pneumocystis: P. carinii, P. murina, and P. jirovecii. Because of the difficulty in culturing Pneumocystis, we used a heterologous system in Saccharomyces cerevisiae, an organism that is phylogenetically related to Pneumocystis, for promoter-reporter analysis.
MATERIALS AND METHODS
Yeast strains and culture.
S. cerevisiae strain YPH 499 (MATa ura3-52 lys2-801amber ade2-101ochre trp1-Δ63 his3-Δ200 leu2-Δ1) was obtained from Stratagene (Santa Clara, CA). Yeast cells were grown at 30°C in YPDA medium (0.0075% l-adenine hemisulfate salt, 1% yeast extract, 2% Bacto peptone, 2% dextrose). For screening of yeast transformants, synthetic dextrose medium without uracil (SD dropout medium) containing 0.67% yeast nitrogen base without amino acids and 2% dextrose was used.
Transformation of S. cerevisiae.
Transformation was carried out using lithium acetate as described in the “pESC epitope tagging vectors” instruction manual (Stratagene). The yeast cells grown in 50 ml of YPDA medium were collected in mid-log phase and washed with LTE buffer (0.1 M LiOAc, 10 mM Tris-HCl [pH 7.5], 1 mM EDTA). The cells were resuspended in 500 μl LTE buffer. Plasmid DNA (3 μg) was added to 50 μl of cell suspension, followed by the addition of 300 μl of transformation mix (40% polyethylene glycol 3350 [PEG 3350], 0.1 M LiOAc, 10 mM Tris-HCl [pH 7.5], 1 mM EDTA). The mixture was incubated at 30°C for 30 min, followed by 42°C for 15 min. The transformation mix was plated on Ura SD dropout plates and cultured for 3 to 4 days, after which colonies were picked and expanded.
Pneumocystis DNA preparation.
P. carinii or P. murina organisms were isolated from the lungs of immunosuppressed rats or scid mice, respectively, by Ficoll-Hypaque density gradient centrifugation (24). P. jirovecii-infected autopsy lung tissue was obtained from patients with Pneumocystis pneumonia. Genomic DNA was isolated using QIAamp DNA minikit (Qiagen, Valencia, CA). The guidelines of the National Institutes of Health were followed in the conduct of these studies. Animal studies were approved by the NIH Clinical Center Animal Care and Use Committee.
PCR amplification.
PCR was performed using Accupfx master mix (Invitrogen, Carlsbad, CA). The general PCR conditions used were as follows: an initial denaturation cycle of 5 min at 95°C, followed by 35 cycles of 30 s at 94°C, 30 s at 53°C, and 2 min at 72°C and a final extension of 10 min at 72°C. The annealing temperature was optimized for each set of primers. The sequences of the primers used for amplification are listed in Table 1.
Table 1.
Oligonucleotides utilized in this study
| Oligonucleotide | Sequence (5′→3′)a | Descriptionb |
|---|---|---|
| GKtef1 | ATATAGGATCCACGGCTCTAAATGTTTCTACTCCT | Corresponds to bp 2794–2808 of pBC542 vector |
| GKtef2 | TATATGCGGCCGCAAACTTAGATTAGATTG | Complementary to bp 3169–3185 of pBC542 vector |
| GKflyluc1 | ATATAGCGGCCGCATGGAAGACGCCAAA | Corresponds to bp 88–102 of PGL3 enhancer vector |
| GKflyluc6 | TATATGAGCTCTTACTTTCCGCCCTTCTTG | Complementary to bp 1713–1728 of PGL3 enhancer vector |
| GKrenilluc6 | ATATACTCGAGATGACTTCGAAAGTTTA | Corresponds to bp 694–710 of pRL-SV40 vector |
| GKrenilluc7 | TATATAAGCTTTTGTTCATTTTTGAGAAC | Complementary to bp 1609–1626 of pRL-SV40 vector |
| GKjiromsgpro7 | ATATAGGATCCGATATATTTGCGACATGTCG | Corresponds to bp 22–41 of P. jirovecii msg gene (AF367050) |
| GKjiromsgpro8 | TATATCTCGAGGATTGCACAAATGAAATAT | Complementary to bp 1311–1329 of P. jirovecii msg gene (AF367050) |
| GKcarimsgpro7 | ATATAAGATCTGTACTAGTCCCTCGTCGTC | Corresponds to bp 526–544 of P. carinii msg gene (U53921) |
| GKcarimsgpro8 | TATATCTCGAGTATTGCACAAACAAACTGAAGA | Complementary to bp 1918–1939 of P. carinii msg gene (U53921) |
| GKmurimsgpro18 | ATATAGGATCCCGGGAGTTTATGAG | Corresponds to bp 1–14 of P. murina msg promoter |
| GKmurimsgpro19 | TATATCTCGAGAATTGCACAAATTTGAG | Complementary to bp 128–144 of P. murina msg promoter |
| GKcarimsgpro42 | CCATCCGGAAATTGGCTTTTC | Corresponds to bp 1744–1764 of P. carinii msg gene (U53921) |
| GKcarimsgpro44 | TACAGTCCAGAAACAGTCC | Complementary to bp 1834–1852 of P. carinii msg gene (U53921) |
| GK582 | GCAATCCTCATAATTGCACA | Complementary to bp 43–62 of P. murina msg gene (AF043102) |
| GK581 | CAAGACATAACTAAGTTGCGCA | Corresponds to bp 78–99 of P. murina msg gene (AF043102) |
| GK584 | ATGAGGAAGATGTTTATGGTTAT | Corresponds to bp 131–153 of P. murina msg gene (AF043102) |
The underlined portion of each sequence corresponds to the description. The beginning regions of the first 12 oligonucleotides include filler nucleotides and a restriction enzyme site to facilitate subcloning.
GenBank accession numbers are given in parentheses.
We had previously identified the genomic sequences upstream of the P. carinii and P. jirovecii msg translation initiation site (17, 19). For P. carinii, ∼1.4 kb spanning bp 526 to 1939 of the published sequence (GenBank accession no. U53921), which corresponds to bp −1414 to −1 relative to the ATG start codon, was amplified from genomic DNA extracted from partially purified P. carinii organisms, using primers GKcarimsgpro7 (with a BglII site) and GKcarimsgpro8 (with an XhoI site). For P. jirovecii, ∼1.3 kb spanning bp 22 to 1329 of the published sequence (GenBank accession no. AF367050), which corresponds to bp −1308 to −1 relative to the ATG start codon, was amplified using GKjiromsgpro7 (with a BamHI site) and GKjiromsgpro8 (with an XhoI site), using DNA extracted from a P. jirovecii-infected autopsy lung sample. For P. murina, the 5′-flanking sequence of msg was obtained by inverse PCR using primers designed from the known sequence (GenBank accession no. AF043102) (21). The region corresponding to bp −144 to −1 relative to the ATG start codon was amplified with primers GKmurimsgpro18 (with a BamHI site) and GKmurimsgpro19 (with an XhoI site), using genomic DNA extracted from partially purified P. murina organisms.
The constitutive tef1 promoter region was amplified from the pBC542 vector (a gift from Brendan Cormack) (25) using primers GKtef1 (with a BamHI site) and GKtef2 (with a NotI site). The firefly luciferase gene was amplified from PGL3 enhancer vector plasmid (Promega, Madison, WI), using GKflyluc1 (with a NotI site) and GKflyluc6 (with a SacI site), while the Renilla luciferase gene was amplified from pRL-SV40 vector plasmid DNA (Promega), using GKrenilluc6 (with an XhoI site) and GKrenilluc7 (with a HindIII site).
Construction of promoter-reporter construct.
pESC-URA vector (Stratagene, Santa Clara, CA) was used to make the promoter-reporter construct. Gal1 and Gal10 promoter sequences were deleted from the vector plasmid, using the QuikChange II site-directed mutagenesis kit (Stratagene), and replaced with Tef1 and Pneumocystis msg promoter sequences (Fig. 1). The tef1 promoter was cloned into the BamHI and NotI sites, while the P. jirovecii and P. murina msg promoter sequences were cloned into the BamHI and XhoI sites, in the opposite direction. Since the P. carinii msg promoter sequence has an internal BamHI site, it was cloned into BglII and XhoI sites after a BglII restriction enzyme site was added to the vector upstream of the BamHI site. The Renilla luciferase gene was cloned downstream of the Pneumocystis promoter between the XhoI and HindIII restriction sites, while the firefly luciferase gene was cloned into the NotI and SacI sites downstream of the tef1 promoter. A construct without any insert upstream of the Renilla luciferase gene was used as a control for basal expression of this gene.
Fig 1.
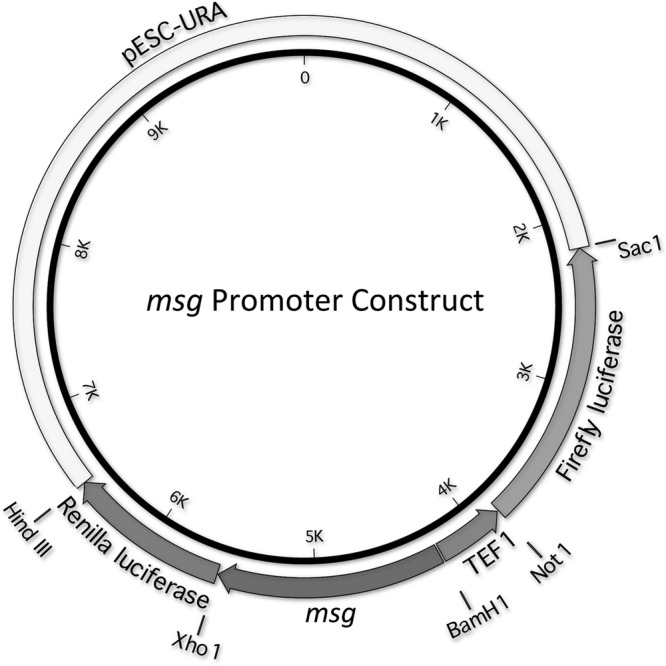
Schematic diagram of the Pneumocystis msg promoter construct. Gal1 and Gal10 promoter sequences were deleted from pESC-URA vector and replaced with Tef1 and Pneumocystis msg promoter sequences (in opposing directions). The sequence encoding Renilla luciferase was cloned downstream of the Pneumocystis promoter, while that of firefly luciferase was cloned downstream of the tef1 promoter. The vector was modified with the respective full-length or deleted promoter constructs specific for P. carinii, P. jirovecii, and P. murina.
Inverse PCR.
The genomic sequence upstream of the P. murina msg ATG start codon was obtained by inverse PCR, which was performed as previously described (26). Genomic DNA extracted from partially purified P. murina organisms was digested with the HindIII restriction enzyme (New England BioLabs, Beverly, MA), ligated using T4 DNA ligase (New England BioLabs, Beverly, MA), purified using the QIAquick PCR purification kit (Qiagen), and amplified by nested PCR using primers GK584 and GK581 for the first round and GK584 and GK582 for the second round, which were designed from the published sequence (GenBank accession no. AF043102).
Site-directed mutagenesis.
All deletions were done with the QuikChange II site-directed mutagenesis kit (Stratagene), using primers designed according to the guidelines of the manufacturer. The PCR conditions used were as follows: an initial denaturation cycle of 2 min at 95°C, followed by 18 cycles of 30 s at 94°C, 30 s at 42°C, 8 min at 68°C and a final extension of 10 min at 68°C. The annealing temperature was optimized for each set of primers. DpnI-digested DNA was transformed into TOP10 cells, and then the cells were plated on LB plates containing the appropriate antibiotics.
Sequencing.
PCR products purified using the QuickStep 2 PCR purification kit (Edge Biosystem, Gaithersburg, MD) and plasmid DNA preparations made using the QIAprep spin miniprep kit (Qiagen) were sequenced using an ABI 3100 genetic analyzer (Applied Biosystems, Carlsbad, CA). All constructs were sequenced prior to use in promoter assays.
Luciferase assay.
Yeast transformants were grown overnight at 30°C, after which the cultures were diluted in dextrose medium to 0.2 optical density at 600 nm (OD600) units/ml and incubated at 30°C for 3 h. The cultures were again diluted to 0.2 OD600 U/ml, and 1-ml aliquots were centrifuged. The cells were then washed with phosphate-buffered saline (PBS), disrupted with glass beads in 750 μl PBS containing 15 μl protease inhibitor (P8849; Sigma-Aldrich, St. Louis, MO), and centrifuged; 10 μl of the supernatant was used for the luciferase assay, which was performed using the Promega dual-luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer's instructions. The Renilla luciferase activity was normalized to firefly luciferase activity to account for biologic variation associated with performance of the assay. While the absolute firefly luciferase activity varied among constructs, it was not suppressed with increased Renilla luciferase activity. Each experiment was repeated at least 3 times for each construct, using a separate clone from the Ura dropout plate.
Preparation of nuclear extracts.
Yeast cells grown in YPDA medium were collected in mid-log phase, washed with cold water containing phosphatase inhibitors (nuclear extract kit; Active Motif, Carlsbad, CA), resuspended in 5 volumes of Zymolyase buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 1 M sorbitol) containing 30 mM dithiothreitol (DTT), and incubated at room temperature for 15 min. Following centrifugation, the cells were resuspended in 3 volumes of Zymolyase buffer containing 1 mM DTT, 10 mg/ml Zymolyase 20T (Zymo Research Corporation, Irvine, CA), and protease inhibitor (P8215; Sigma-Aldrich, St. Louis, MO), and incubated at 30°C with shaking until spheroplast formation was complete. Spheroplasts were centrifuged at 9,300 × g for 5 min and washed gently 3 times with cold Zymolyase buffer containing 1 mM DTT. The nuclear extracts were prepared using the nuclear extract kit (Active Motif) following the manufacturer's instructions. The spheroplasts were incubated on ice for 15 min after resuspension in 1× hypotonic buffer plus detergent and then vortexed for 10 s at the highest setting. After centrifugation at 14,000 × g for 5 min at 4°C, the pellet (nuclei) was resuspended in lysis buffer and incubated at 4°C while shaking for 30 min. The extract was then centrifuged at 14,000 × g for 10 min at 4°C, and the supernatant (nuclear extract) was collected.
EMSA.
A 107-bp probe corresponding to bp −196 to −90 of the P. carinii promoter was prepared by PCR using primers GKcarimsgpro42 (biotinylated at the 5′ end) and GKcarimsgpro44. The electrophoretic mobility shift assay (EMSA) was performed using the Lightshift chemiluminescent EMSA kit (Thermo Scientific, Rockford, IL). The nuclear extract was incubated with biotin-labeled probe (20 fmol) in binding buffer containing 10 mM Tris, 50 mM KCl, 1 mM DTT (pH 7.5), 2.5% glycerol, 0.05% NP-40, and 50 ng/μl poly(dI-dC) for 30 min at room temperature. For competition, a 400-fold excess of unlabeled probe (either the same PCR product that was not biotinylated or a PCR product corresponding to positions −144 to −49 of the P. murina promoter) was included in the binding reactions. DNA-protein complexes were separated on 6% DNA retardation gels by electrophoresis, transferred to a nylon membrane (Biodyne B; Thermo Scientific), and UV cross-linked. The membrane was then incubated with streptavidin-horseradish peroxidase conjugate, and biotin-labeled DNA-protein complexes were detected by chemiluminescence.
RESULTS
Pneumocystis msg 5′-flanking sequences have promoter activity.
To determine whether the 5′-flanking regions of msg genes of different Pneumocystis species have promoter activity, we made promoter reporter constructs using a modified yeast pESC-URA vector (Fig. 1). The Gal1 and Gal10 promoter sequences in the vector were replaced with tef1 and Pneumocystis msg promoter sequences, and the Renilla luciferase coding sequence was then inserted downstream of the Pneumocystis msg promoter, while the firefly luciferase coding sequence was inserted downstream of the tef1 promoter. Renilla luciferase activity was normalized to firefly luciferase activity, and msg promoter activity was determined by comparing it to Renilla activity in a construct with no msg promoter. All Pneumocystis msg promoter constructs terminated at −1 relative to the ATG start codon, and all numbering is relative to this start codon; none of the UCS coding sequence was included in any plasmid.
A plasmid containing ∼1.4 kb of sequence upstream of the ATG start codon of P. carinii msg (17) showed ∼5.5-fold-higher Renilla luciferase activity than the control (Fig. 2, left). For P. jirovecii, ∼1.3 kb of msg promoter sequence (19) showed ∼12-fold-higher activity than that of the control (Fig. 2, center), while for P. murina, a 144-bp 5′-flanking sequence of msg (5′-CGGGAGTTTA TGAGAATGTT AATTATTATT TTCGTATTAA AGGGGTTATT TCCGGACTGT GTATTTGCTT TACCCGTTAT TTGTTTTAGC GTTGAAAAAT ATATTTTTCT TGATATCCGT CTCTTTTCTC AAATTTGTGC AATT-3′) showed promoter activity that was ∼44-fold higher than that of the control (Fig. 2, right). Thus, the 5′-flanking sequences of msg from all three Pneumocystis species showed considerable promoter activity after transformation into S. cerevisiae.
Fig 2.
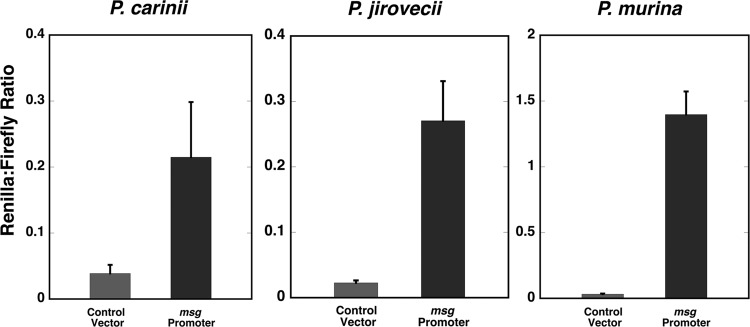
Promoter activity of the 5′-flanking region of Pneumocystis msg. The 5′-flanking regions of msg genes were cloned into modified pESC-URA vector and transformed into S. cerevisiae; promoter activity was assessed using a dual-luciferase system. (Left panel) An ∼1.4-kb 5′-flanking region of P. carinii msg showed ∼5.5-fold-higher promoter activity than the control vector. (Center panel) An ∼1.3-kb 5′-flanking region of P. jirovecii msg showed ∼12-fold-higher promoter activity than the control vector. (Right panel) A 144-bp 5′-flanking sequence of P. murina msg showed ∼44-fold-higher activity than the control vector. The y axis shows the ratio of Renilla luciferase activity to firefly luciferase activity, as measured by relative light units. Results represent the mean and standard deviation from 3 experiments per construct. All numbering of the nucleotide sequences is relative to the ATG start codon.
Characterization of the P. carinii msg promoter.
For P. carinii msg, multiple transcription sites were previously identified, with the most prominent one 17 bp upstream of the ATG start codon (17). The initial sequence (1,414 bp) used in our promoter assay contained 1,397 bp upstream of this transcription start site. To define the minimal sequence required for promoter function, plasmids with progressive deletions from the 5′ end were constructed. Figure 3A shows the fold increase in Renilla luciferase activity for these deletion constructs compared to the activity of the no-msg-insert control. There was a gradual increase in activity with increasing deletions, with the highest activity, ∼24-fold, in a construct starting at −196 (Del4). Deletion of another 80 bp (Del5; bp −196 to −117) resulted in a decrease in the activity by ∼50%, to ∼11-fold, and a further deletion of 13 bp (Del6) abolished promoter activity. Thus, the 13-bp sequence (CATATTAAGGGGA, bp −116 to −104) appeared to be important for P. carinii msg promoter activity.
Fig 3.

Promoter activity of 5′-deletion constructs of the P. carinii msg gene. (A) A series of deletions were made from the 5′ end of the promoter region. The schematic diagram of the deletion constructs is shown in the left panel, with the full-length sequence shown at the top. The fold increase in Renilla luciferase activity of each construct over the activity of the control vector is shown in the right panel. All data are normalized to firefly luciferase activity. (B and C) Promoter activity of additional constructs with deletions or mutations targeting the two conserved 13-bp regions (bp −135 to −123 and −116 to −104). Panel B shows a schematic of the constructs on the left and the fold change in Renilla luciferase activity on the right, while panel C shows aligned sequences of the same constructs, in which deletions are marked by a dash and mutations are boxed. Deletion or mutation of the second 13-bp region alone led to an increase rather than decrease in activity, while any combination of deletion and mutation of both regions markedly diminished activity. The numbering on the left in panel B is shown only for the constructs with no internal deletions. In panels A and B, the segments that were deleted are shown in different shades of gray to highlight their locations in the different constructs. In panel B, the thick vertical bars represent the region that was deleted (white, Del8b and Del9a) or mutated (black, Del8c and Del9a-c). (D) Electrophoretic mobility shift assay. A biotinylated PCR product corresponding to bp −196 to −90 of the P. carinii promoter was incubated with yeast nuclear extracts in the presence or absence of 400-fold excess of unlabeled probes. DNA-protein complexes were electrophoretically separated on a polyacrylamide gel, transferred to a nylon membrane, and detected by chemiluminescence using peroxidase-conjugated streptavidin. Lane 1, biotinylated probe alone. Lane 2, biotinylated probe plus nuclear extracts. DNA-protein complex formation is observed. Lane 3, biotinylated probe plus nuclear extracts plus unlabeled PCR product that is otherwise identical to the labeled probe. Lane 4, biotinylated probe plus nuclear extracts plus unlabeled PCR product corresponding to bp −144 to −49 of the P. murina promoter. Both unlabeled PCR products largely eliminated a specific DNA-protein complex band (shown by a box across the lanes). The numbers above the box represent the relative density of the boxed band compared to its density in lane 2. Relative density was determined using ImageJ software (28).
To explore this further, we utilized additional longer constructs with internal deletions or mutations of this region. Del8, which started at −171, showed similar activity to Del4. Deletion of 27 bp internally, from −116 to −90, which included the 13-bp region, did not decrease activity but rather resulted in increased activity (Del8a) (Fig. 3B and C). We identified a near duplication of the 13-bp region (CAATATTTAAGGG, bp −135 to −123) that could potentially account for this promoter activity. Deletion of both of these 13-bp regions (Del8b) eliminated promoter activity. In addition, mutation of 4 bases (AGGG to TTTT, bp −126 to −123 [Del8c]) rather than deleting this region also markedly reduced activity. Furthermore, utilizing Del4, a longer construct, we found that either mutation of the homologous 4 bp (AGGG to TTTT) in both 13-bp regions (Del9b) or deletion of the first and mutation of the second (Del9a) also markedly reduced activity, while mutation of only the second region (Del9) did not (Fig. 3B and C).
We performed an electrophoretic mobility shift assay (EMSA) using S. cerevisiae nuclear extract to further study the role of these regions (Fig. 3D). A biotinylated 107-bp PCR product containing these regions was used as the probe. DNA-protein binding was observed when this probe was incubated with nuclear extract (lane 2 versus lane 1). Competition with 400-fold excess of unlabeled PCR product decreased the intensity of a specific DNA-protein complex band (lane 2 versus lane 3, shown by box). Competition with an unlabeled PCR product corresponding to bp −144 to −49 of the P. murina promoter provided a similar level of inhibition (lane 4), suggesting that the same protein was able to bind to both promoter regions. Attempts to identify this protein by affinity chromatography followed by peptide sequencing were unsuccessful.
Characterization of the P. jirovecii msg promoter.
We next characterized the promoter region of P. jirovecii msg in a similar manner. For P. jirovecii msg, only one transcription start site has been identified, and it is located 16 bp upstream of the initiation ATG (19). The P. jirovecii msg upstream sequence (1,308 bp) used in our construct contained 1,292 bp upstream of this transcription start site, and it showed an ∼11-fold increase in promoter activity compared to that of the control (Fig. 4). Progressive deletions resulted in small changes in activity, until Del 5, a deletion of 14 bp (bp −110 to −97), which largely abolished activity. Thus, for P. jirovecii msg, a 14-bp region (TTTCTATAACTGGT, bp −110 to −97) is important for promoter activity.
Fig 4.
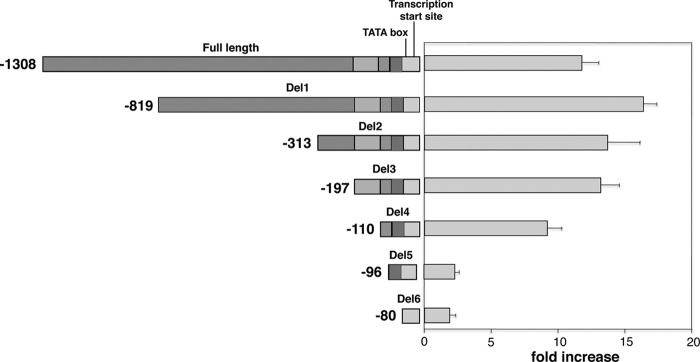
Promoter activity of 5′-deletion constructs of the P. jirovecii msg gene. A series of deletions were made from the 5′ end of the 1,308-bp promoter construct. The schematic diagram of the deletion constructs is shown in the left panel. The fold increase in Renilla luciferase activity of each construct over the activity of the control vector is shown in the right panel. All data are normalized to firefly luciferase activity. Results represent the mean and standard deviation of 3 experiments per construct. All numbering of the nucleotide sequences is relative to the ATG start codon. The segments that were deleted are shown in different shades of gray to highlight their locations in the different constructs.
Characterization of the P. murina msg promoter.
For P. murina msg, the transcription start site has not been identified. To characterize the promoter, the 144-bp region upstream of the initiation ATG was subjected to deletions. Deletion of 32 bp from the 5′ end (Del1, bp −112) reduced the activity by half, to ∼19-fold (Fig. 5), while a further deletion of 14 bp (Del2, bp −112 to −99) resulted in the loss of promoter activity. Thus, for P. murina msg, the 14-bp sequence (CGTATTAAAGGGGT) spanning bp −112 to −99 is important for promoter activity.
Fig 5.
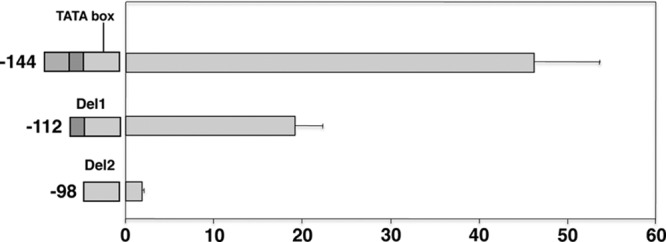
Promoter activity of 5′-deletion constructs of the P. murina msg gene. Deletions were made from the 5′ end of the 144-bp promoter construct. The schematic diagram of the deletion constructs is shown in the left panel. The fold increase in Renilla luciferase activity of each construct over the activity of the vector alone is shown in the right panel. All data are normalized to firefly luciferase activity. Results represent the mean and standard deviation of 3 experiments per construct. All numbering of the nucleotide sequences is relative to the ATG start codon. The segments that were deleted are shown in different shades of gray to highlight their locations in the different constructs.
Alignment of promoter sequences of msg from three different Pneumocystis species.
Sequence alignments of the proximal regions of the respective upstream sequences and of the 13- to 14-bp regions of the P. carinii, P. murina, and P. jirovecii msg promoters that are important for activity are shown in Fig. 6A and B, respectively. In Fig. 6A, the 13- to 14-bp sequences important for promoter activity (shown in boxes) and potential TATA boxes (underlined) are highlighted. The 144-bp 5′-flanking region of P. murina msg showed 72% identity to that of P. carinii. The 5′-flanking region of P. jirovecii msg showed 58 and 61% identities to those of P. murina and P. carinii, respectively, in the overlapping regions. Figure 6B shows that the 13- to 14-bp region is well conserved in the two P. carinii sequences and the single P. murina sequence but not in the P. jirovecii sequence.
Fig 6.

(A) Alignment of 5′-flanking sequence of msg from P. carinii (196 bp), P. jirovecii (197 bp), and P. murina (144 bp). The alignment was performed by ClustalW in MacVector (MacVector, Inc.), using default conditions. The 13- to 14-bp sequences important for promoter activity in S. cerevisiae are boxed. The transcription start sites for P. carinii and P. jirovecii are indicated by arrows, and a presumptive TATA box is underlined. (B) Alignment of the 13- to 14-bp sequences important for promoter activity in S. cerevisiae. Conserved nucleotides are shaded in both panels A and B.
DISCUSSION
In this study, we have characterized the promoter activity of the region upstream of the msg genes from three different species of Pneumocystis, using a heterologous yeast system, and have identified a 13- to 14-bp region in each that is important for promoter activity in this system. While the upstream region among all 3 species is reasonably well conserved, the 13- to 14-bp region that appears critical for promoter function is conserved between the more closely related species P. carinii and P. murina but not the more distant species P. jirovecii. EMSAs utilizing P. carinii-specific sequences suggest that an S. cerevisiae nuclear protein binds specifically to this region; attempts to identify this protein utilizing affinity chromatography and mass spectroscopy were, however, unsuccessful. Furthermore, because this region in P. carinii and P. murina included the stress response element (STRE; AGGGG), which is the binding site of MSN2 and MSN4 in S. cerevisiae (27), we examined promoter activity in yeast strains in which MSN2, MSN4, or both had been knocked out. However, we found no diminution in promoter activity, and we did not identify MSN2 or MSN4 in the mass spectroscopy analysis.
We undertook these studies to better understand the regulation of Msg expression and to identify potential promoter sequences that could be utilized in vectors for transfection of Pneumocystis. We selected the msg promoter because Msg is the most abundant protein of Pneumocystis, based on Coomassie blue-stained gels of Pneumocystis protein extracts, suggesting that its promoter is likely to be highly active. Because Pneumocystis cannot be cultured or genetically manipulated at present, we needed to utilize a heterologous system and chose S. cerevisiae because multiple Pneumocystis proteins have been previously expressed in this related fungus. A previous study utilizing ∼1.1 kb upstream of the P. carinii msg expression site also found high levels of promoter activity in S. cerevisiae, especially compared to those of the promoter of Pneumocystis p55, although no detailed characterization of the promoter region of msg was performed (22).
Our studies suggest that heterologous systems can provide biologically relevant information about gene regulation in Pneumocystis, although these results will need to be validated directly in Pneumocystis when such methods are available. The promoter vector that we constructed can be used for characterization of promoter activity of other Pneumocystis genes in S. cerevisiae, although it is not clear that it would provide an advantage over available yeast promoters. Ultimately, it can potentially be used to facilitate development of methods for genetic manipulation of Pneumocystis species to better explore the biology and pathogenicity of this opportunistic organism.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health Clinical Center and the National Cancer Institute.
We thank Rene Costello and Howard Mostowski for providing animal care.
The authors have no conflicting financial interests.
Footnotes
Published ahead of print 26 July 2013
REFERENCES
- 1.Kovacs JA, Masur H. 2009. Evolving health effects of Pneumocystis: one hundred years of progress in diagnosis and treatment. JAMA 301:2578–2585 [DOI] [PubMed] [Google Scholar]
- 2.Thomas CF, Jr, Limper AH. 2004. Pneumocystis pneumonia. N. Engl. J. Med. 350:2487–2498 [DOI] [PubMed] [Google Scholar]
- 3.Dei-Cas E, Mazars E, Ferragut CO, Durand I, Aliouat EM, Dridba M, Palluault F, Cailliez JC, Seguy N, Tibayrenc M, et al. 1994. Ultrastructural, genomic, isoenzymatic and biological features make it possible to distinguish rabbit Pnenumocystis from other mammal Pneumocystis strains. J. Eukaryot. Microbiol. 41:84S. [PubMed] [Google Scholar]
- 4.Sinclair K, Wakefield AE, Banerji S, Hopkin JM. 1991. Pneumocystis carinii organisms derived from rat and human hosts are genetically distinct. Mol. Biochem. Parasitol. 45:183–184 [DOI] [PubMed] [Google Scholar]
- 5.Stringer JR, Stringer SL, Zhang J, Baughman R, Smulian AG, Cushion MT. 1993. Molecular genetic distinction of Pneumocystis carinii from rats and humans. J. Eukaryot. Microbiol. 40:733–741 [DOI] [PubMed] [Google Scholar]
- 6.Ma L, Imamichi H, Sukura A, Kovacs JA. 2001. Genetic divergence of the dihydrofolate reductase and dihydropteroate synthase genes in Pneumocystis carinii from 7 different host species. J. Infect. Dis. 184:1358–1362 [DOI] [PubMed] [Google Scholar]
- 7.Cushion MT. 2004. Molecular and phenotypic description of Pneumocystis wakefieldiae sp. nov., a new species in rats. Mycologia 96:429–438 [PubMed] [Google Scholar]
- 8.Keely SP, Fischer JM, Cushion MT, Stringer JR. 2004. Phylogenetic identification of Pneumocystis murina sp. nov., a new species in laboratory mice. Microbiology 150:1153–1165 [DOI] [PubMed] [Google Scholar]
- 9.Stringer JR, Beard CB, Miller RF, Wakefield AE. 2002. A new name (Pneumocystis jiroveci) for Pneumocystis from humans. Emerg. Infect. Dis. 8:891–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garbe TR, Stringer JR. 1994. Molecular characterization of clustered variants of genes encoding major surface antigens of human Pneumocystis carinii. Infect. Immun. 62:3092–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haidaris PJ, Wright TW, Gigliotti F, Haidaris CG. 1992. Expression and characterization of a cDNA clone encoding an immunodominant surface glycoprotein of Pneumocystis carinii. J. Infect. Dis. 166:1113–1123 [DOI] [PubMed] [Google Scholar]
- 12.Kovacs JA, Powell F, Edman JC, Lundgren B, Martinez A, Drew B, Angus CW. 1993. Multiple genes encode the major surface glycoprotein of Pneumocystis carinii. J. Biol. Chem. 268:6034–6040 [PubMed] [Google Scholar]
- 13.Radding JA, Armstrong MY, Ullu E, Richards FF. 1989. Identification and isolation of a major cell surface glycoprotein of Pneumocystis carinii. Infect. Immun. 57:2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright TW, Bissoondial TY, Haidaris CG, Gigliotti F, Haidaris PJ. 1995. Isoform diversity and tandem duplication of the glycoprotein A gene in ferret Pneumocystis carinii. DNA Res. 2:77–88 [DOI] [PubMed] [Google Scholar]
- 15.Keely SP, Stringer JR. 2009. Complexity of the MSG gene family of Pneumocystis carinii. BMC Genomics 10:367. 10.1186/1471-2164-10-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keely SP, Linke MJ, Cushion MT, Stringer JR. 2007. Pneumocystis murina MSG gene family and the structure of the locus associated with its transcription. Fungal Genet. Biol. 44:905–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edman JC, Hatton TW, Nam M, Turner R, Mei Q, Angus CW, Kovacs JA. 1996. A single expression site with a conserved leader sequence regulates variation of expression of the Pneumocystis carinii family of major surface glycoprotein genes. DNA Cell Biol. 15:989–999 [DOI] [PubMed] [Google Scholar]
- 18.Wada M, Sunkin SM, Stringer JR, Nakamura Y. 1995. Antigenic variation by positional control of major surface glycoprotein gene expression in Pneumocystis carinii. J. Infect. Dis. 171:1563–1568 [DOI] [PubMed] [Google Scholar]
- 19.Kutty G, Ma L, Kovacs JA. 2001. Characterization of the expression site of the major surface glycoprotein of human-derived Pneumocystis carinii. Mol. Microbiol. 42:183–193 [DOI] [PubMed] [Google Scholar]
- 20.Sunkin SM, Stringer JR. 1997. Residence at the expression site is necessary and sufficient for the transcription of surface antigen genes of Pneumocystis carinii. Mol. Microbiol. 25:147–160 [DOI] [PubMed] [Google Scholar]
- 21.Haidaris CG, Medzihradsky OF, Gigliotti F, Simpson-Haidaris PJ. 1998. Molecular characterization of mouse Pneumocystis carinii surface glycoprotein A. DNA Res. 5:77–85 [DOI] [PubMed] [Google Scholar]
- 22.Broomall K, Collins M, Smulian AG. 1997. Pneumocystis carinii promoter analysis in a heterologous Saccharomyces cerevisiae assay system. J. Eukaryot. Microbiol. 44:10S–11S [DOI] [PubMed] [Google Scholar]
- 23.Lundgren B, Lipschik GY, Kovacs JA. 1991. Purification and characterization of a major human Pneumocystis carinii surface antigen. J. Clin. Invest. 87:163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovacs JA, Halpern JL, Swan JC, Moss J, Parrillo JE, Masur H. 1988. Identification of antigens and antibodies specific for Pneumocystis carinii. J. Immunol. 140:2023–2031 [PubMed] [Google Scholar]
- 25.Zupancic ML, Frieman M, Smith D, Alvarez RA, Cummings RD, Cormack BP. 2008. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68:547–559 [DOI] [PubMed] [Google Scholar]
- 26.Ma L, Borio L, Masur H, Kovacs JA. 1999. Pneumocystis carinii dihydropteroate synthase but not dihydrofolate reductase gene mutations correlate with prior trimethoprim-sulfamethoxazole or dapsone use. J. Infect. Dis. 180:1969–1978 [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Pastor MT, Marchler G, Schuller C, Marchler-Bauer A, Ruis H, Estruch F. 1996. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 15:2227–2235 [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]