

Abstract
The identification of novel antiretroviral agents is required to provide alternative treatment options for HIV-1-infected patients. The screening of a phenotypic cell-based viral replication assay led to the identification of a novel class of 4,5-dihydro-1H-pyrrolo[3,4-c]pyrazol-6-one (pyrrolopyrazolone) HIV-1 inhibitors, exemplified by two compounds: BI-1 and BI-2. These compounds inhibited early postentry stages of viral replication at a step(s) following reverse transcription but prior to 2 long terminal repeat (2-LTR) circle formation, suggesting that they may block nuclear targeting of the preintegration complex. Selection of viruses resistant to BI-2 revealed that substitutions at residues A105 and T107 within the capsid (CA) amino-terminal domain (CANTD) conferred high-level resistance to both compounds, implicating CA as the antiviral target. Direct binding of BI-1 and/or BI-2 to CANTD was demonstrated using isothermal titration calorimetry and nuclear magnetic resonance (NMR) chemical shift titration analyses. A high-resolution crystal structure of the BI-1:CANTD complex revealed that the inhibitor bound within a recently identified inhibitor binding pocket (CANTD site 2) between CA helices 4, 5, and 7, on the surface of the CANTD, that also corresponds to the binding site for the host factor CPSF-6. The functional consequences of BI-1 and BI-2 binding differ from previously characterized inhibitors that bind the same site since the BI compounds did not inhibit reverse transcription but stabilized preassembled CA complexes. Hence, this new class of antiviral compounds binds CA and may inhibit viral replication by stabilizing the viral capsid.
INTRODUCTION
The advent of highly active antiretroviral therapy has led to significant reductions in morbidity and mortality associated with HIV/AIDS. There are currently 26 FDA-approved drugs for the treatment of HIV-1 (1). These drugs fall into six distinct classes that target different sites on 4 of the 15 viral proteins, in addition to one host protein. Although these drugs are generally effective, poor adherence, toxicity associated with long-term treatment, and multidrug resistance can ultimately limit their efficacy. The identification of novel inhibitors of HIV-1 replication that exhibit novel mechanisms of action and favorable resistance and safety profiles will expand potential treatment options.
The viral Gag polyprotein mediates the assembly and budding of immature virions (2–4). As the virus buds, Gag is cleaved by the viral protease to create a series of smaller proteins (MA, CA, and NC) and peptides (SP1, SP2, and p6). The newly processed proteins then rearrange in a process called maturation. Mature virions contain a conical core particle that has an outer shell (the “capsid”) composed of CA subunits. The capsid surrounds a ribonucleoprotein complex comprising the viral RNA genome, the NC protein, and the viral enzymes reverse transcriptase (RT) and integrase (IN) (2, 3). The conical capsid lattice follows the geometry of a fullerene cone, with ∼200 CA hexamers comprising the body of the cone and the required declination provided by 12 CA pentamers: 7 at the wide end and 5 at the narrow end (5, 6). The amino-terminal domain of CA (CANTD, amino acid residues 1 to 146) forms the hexameric (or pentameric) rings, whereas the carboxyl-terminal domain of CA (CACTD, amino acid residues 151 to 231) forms a “belt” around the rings and makes dimeric interactions that connect adjacent rings (7–9).
Amino acid substitutions within HIV-1 CA can impair either the late-stage event of virion assembly or early postentry events such as reverse transcription, capsid uncoating, and/or nuclear entry (2, 10–12). Two observations of particular relevance to the current study are that (i) CA amino acid substitutions such as E128A/R132A that appear to stabilize the viral capsid also reduce the efficiency of reverse transcription (12), and (ii) other detrimental CA amino acid substitutions, such as Q63A/Q67A, can increase the levels of CA associated with the preintegration complex (PIC), suggesting that they may impair capsid uncoating (13).
There is growing interest in HIV-1 CA as a target of antiviral inhibitors, and several peptides and small molecules that bind CA and inhibit viral replication have been identified (reviewed in reference 14). A phage display approach led to the identification of a peptide that binds the CACTD and inhibits the in vitro assembly of both immature and mature particles (15, 16). A small molecule, CAP-1, was shown to target a pocket (site 1) at the base of the CANTD formed by helices 1 to 4 (17, 18), and more potent inhibitors that bind this pocket have subsequently been reported (19–21). All of these compounds inhibit CA assembly in vitro but can have distinct effects in inhibiting either virion production or capsid assembly (20). A distinct family of small molecules was recently reported to bind to a separate site on CANTD, site 2, formed by helices 3, 4, 5, and 7 (22). These compounds perturb viral capsid assembly and appear to both enhance the rate of CA multimerization in vitro and accelerate capsid dissociation in cells (22, 23).
Here we describe a new family of 4,5-dihydro-1H-pyrrolo[3,4-c]pyrazol-6-one (pyrrolopyrazolone) small molecules that bind within CANTD site 2 and inhibit HIV-1 replication. These compounds differ from previously reported site 2 inhibitors (22, 23) since they stabilize HIV-1 CA assemblies and prevent uncoating of viral capsids in vitro. The pyrrolopyrazolones also reduce the accumulation of 2 long terminal repeat (2-LTR) circles without affecting reverse transcription in infected cells. Thus, these compounds may exert their primary effect at the level of nuclear import of the PIC and therefore represent a new class of anti-HIV inhibitors.
MATERIALS AND METHODS
Plasmids.
Plasmids used to produce vesicular stomatitis virus envelope glycoprotein (VSV-G) pseudotyped replication-incompetent HIV-1 (HIV-1 helper plasmid, pTV-Luc transfer plasmid, and pIADL-VSV-G envelope plasmid) and to express HIV-1NL4-3 CA-NC were described previously (20). BI-2 drug resistance mutations were introduced into the HIV-1 helper plasmid and CA-NC expression plasmids using the QuikChange II site-directed mutagenesis kit (Agilent Technologies), according to the manufacturer's instructions.
Cells.
C8166, SupT1, and 293FT cells were obtained from John Sullivan (University of Massachusetts Medical Center), ATCC (CRL-1942), and Life Technologies (R700-07), respectively, and maintained at 37°C, 5% CO2. The establishment of the C8166-LTR-Luc cells was described previously (20). C8166, C8166-LTR-Luc, and SupT1 cells were maintained in RPMI 1640 medium (Wisent Inc.) supplemented with 10% fetal bovine serum (FBS; HyClone) and 1% penicillin-streptomycin (Life Technologies). C8166-LTR-Luc cells were cultured in the presence of 5 μg/ml blasticidin S-HCl (Life Technologies), but blasticidin S-HCl was omitted from all antiviral assays. 293FT cells were maintained in Dulbecco's modified Eagle medium (DMEM; Wisent Inc.) supplemented with 10% FBS and 1% penicillin-streptomycin.
Preparation of test compounds for antiviral assays.
Ten-point serial dilutions of test compounds were prepared in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin. All samples, including the negative and positive controls, contained the same concentration of dimethyl sulfoxide (DMSO) in assay medium (≤0.5%). Diluted compound (30 or 100 μl) was added to quadruplicate or triplicate wells of 384- or 96-well assay plates, prior to single-cycle (384-well) or multicycle (96-well) viral replication assays, respectively.
Single-cycle viral replication assays.
The activity of the BI-1 and BI-2 inhibitors in the postentry phase of the HIV-1 replication cycle was evaluated by infecting SupT1 cells with VSV-G pseudotyped viral vectors in the presence of the compounds. VSV-G pseudotyped HIV-1 was prepared as described previously (20). Cells and virus were mixed and added to 384-well assay plates (30 μl of medium containing 15,000 SupT1 cells and virus equivalent to 1 ng of CA). Uninfected cells were included as negative controls. Seventy-two hours postinfection, firefly luciferase activity was evaluated by adding 10 μl/well of Steady-Glo (Promega). Luminescence was measured using a TopCount-HTS plate reader (PerkinElmer Life Sciences). The activity of the BI-1 and BI-2 inhibitors in the postintegration (or viral assembly) phase of the HIV-1 replication cycle was evaluated by adding test compounds to 293FT cells during the production of VSV-G pseudotyped HIV-1, exactly as described previously (20). The 50% effective concentration (EC50) is the concentration of compound that produced half-maximal inhibition of HIV-1 viral replication in both assays. EC50 values were calculated by nonlinear regression analysis using SAS software (SAS Institute).
Multiple-cycle viral replication assays.
Antiviral activity in a 72-h multicycle viral replication assay was determined using C8166-LTR-Luc cells and HIV-1 2.12, exactly as described in reference 20.
MTT cytotoxicity assays.
The antiviral and cytotoxicity activities of compounds were measured in parallel. Fifty thousand SupT1 or C8166-LTR-Luc cells in 100 μl of media were added to 96-well plates that contained 100 μl of diluted compound. After 72 h, mitochondrial function and cell viability were assessed by evaluating the reduction of the tetrazolium salt MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide; Sigma-Aldrich) with the addition of 40 μl of a 5-mg/ml MTT solution in RPMI 1640 medium lacking FBS. Assay plates were incubated 3 to 5 h, 150 μl of medium was removed, and the formazan product was solubilized following the addition of 50 μl 10% Triton X-100 in 0.01N HCl. Formazan concentrations were quantified by measuring the optical density at 570 nm using a SpectraMax plate reader (Molecular Devices). The 50% cytotoxic concentration (CC50) represents the concentration of test compound that reduced cell viability by 50%. CC50 values were calculated by nonlinear regression analysis using SAS software (SAS Institute).
Quantitative PCR assays.
Twenty-four hours prior to infection, 40,000 C8166 cells/well were seeded in 96-well plates (Corning) precoated with 25 μg/ml poly-d-lysine (Sigma-Aldrich). Immediately prior to infection, VSV-G pseudotyped HIV-1 virus was treated with DNase I (Life Technologies) for 60 min at 37°C. Cells were infected for 2 h at 37°C with virions (equivalent to 25 ng of CA per well) in the presence of DMSO, 1.1 μM nevirapine, 70 μM BI-1, or 20 nM of the integrase strand transfer inhibitor L-870,810 (24) (in a volume of 200 μl). Cells were then washed once with phosphate-buffered saline (PBS) to remove unbound virus, and fresh inhibitor was applied. At 10 or 24 h postinfection, the cells were washed once with PBS and total DNA was extracted using the DNA blood minikit (Qiagen) according to the manufacturer's instructions. DNA was eluted in 200 μl AE buffer (supplied with the kit), and 5-μl samples were used in quantitative PCRs. Primers and TaqMan probes specific for late reverse transcription (RT) products or 2-LTR circles were multiplexed with primers and TaqMan probes specific for mitochondrial DNA, as described previously (25). Absolute copy numbers were quantified from standard curves prepared using cloned PCR products. Late RT products and 2-LTR levels were normalized against mitochondrial levels in each sample.
Selection of HIV-1 variants resistant to BI-2.
C8166 cells were infected with HIV-1 NL4.3, and resistance to BI-2 was selected starting at an inhibitor concentration of approximately twice its EC50 (5 μM), in accordance with the protocol described previously (20). Resistance mutations were identified by clonal sequencing.
Capsid assembly assays.
Purification of CA-NC (wild type and A42D) and the immobilized CA-NC assembly assay were performed as described previously (20).
EM analyses of CA-NC tubes.
CA-NC tubes were assembled in solution by incubating 9 μM CA-NC and 1 μM oligonucleotide [d(TG)50] in 100 μl of assay buffer containing 50 mM Tris-Cl (pH 8.0), 400 mM NaCl, and 5 mM β-mercaptoethanol, in the presence of either 20 or 200 μM BI-2 or DMSO alone (vehicle control), for 16 h at 4°C. Five-microliter suspensions of assembled CA-NC tubes were processed and analyzed by electron microscopy (EM) as described previously (26).
Capsid stabilization assay.
Reacti-Bind NeutrAvidin-coated black 384-well microplates (Pierce) were washed once with 80 μl/well of buffer B (50 mM Tris-Cl [pH 8.0], 350 mM NaCl, 10 μM ZnSO4, 0.0025% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfate [CHAPS] (wt/vol), 50 μg/ml bovine serum albumin [BSA], 1 mM dithiothreitol [DTT]). To each well was added a 25 nM solution (50 μl/well) of 5′-biotin-labeled d(TG)25 oligonucleotide (Integrated DNA Technologies) in buffer C (same composition as buffer B, except containing 5 mg/ml BSA). Following an overnight incubation (∼12 to 16 h), the wells were washed twice with buffer B (80 μl/well). CA-NC complexes were assembled by incubation of 2 μM CA-NC protein in buffer B supplemented with 100 nM 5′-fluorescein-labeled d(TG)25 oligonucleotide (Integrated DNA Technologies) in a total volume of 60 μl/well for 2 h at room temperature. Unassembled material was removed by washing once with 80 μl/well of buffer B. Test compounds (60 μl/well), serially diluted in buffer D (same composition as buffer B, except containing 250 mM NaCl) to a final DMSO concentration of 1%, were added to these preassembled CA-NC complexes. Following a 2-h incubation at room temperature, disassembled material was removed by two washes with buffer B (80 μl/well). During this disassembly incubation, complexes spontaneously disassembled at a given rate, and the amount of fluorescein-labeled oligonucleotide remaining in each well, which is proportional to the stability of the complexes, was determined following the addition of 80 μl/well of buffer B supplemented with 0.1% SDS (wt/vol) and quantification of fluorescence on a Victor2 plate reader (PerkinElmer Life Sciences). The negative-control wells contained no compound but were incubated for 2 h to assess the extent of disassembly during that period. The positive-control wells had no compound, and the disassembly incubation was omitted. The fluorescence associated with these wells was determined following the addition of buffer B supplemented with 0.1% SDS (wt/vol) and represented 100% of the assembled CA-NC complexes present prior to the disassembly incubation. The capacity of a test compound to stabilize capsid complexes (i.e., inhibit the dissociation of the preassembled CA-NC complexes) was considered to be proportional to the captured fluorescence remaining following the disassembly incubation. For each well, the percent stabilization was calculated using the following equation: % stabilization = {1 − [(Fpositive control − Ftest well)/(Fpositive control − Fnegative control)]} ∗ 100, where Fpositive control and Fnegative control are the averages of each fluorescence (F) value from a series of wells that were incubated under the relevant conditions, as described above. In the absence of compound, the percent stabilization is 0%, and in the absence of the disassembly incubation, the percent stabilization is 100%. The 50% inhibitory concentration (IC50) is the concentration of compound that produced half-maximal stabilization of CA complexes. IC50 values were calculated by nonlinear regression analysis using SAS software (SAS Institute).
ITC.
Isothermal titration calorimetry (ITC) was performed using 200 μM CANTD and CACTD-NC in a buffer containing 50 mM Tris-Cl (pH 8.0), 350 mM NaCl, and 1% DMSO using a VP-ITC microcalorimeter (GE Healthcare Life Sciences). Titrations were performed at 25°C, as described previously (20).
NMR spectroscopy.
To prepare 15N- and 13C/15N-labeled CANTD, the proteins were expressed, as described previously (20), in Spectra 9 media (15N, 98% and 13C, 98%; 15N, 98%) (Cambridge Isotope Laboratories Inc.). At the last step of purification, the proteins (at 720 μM) were exchanged into 25 mM sodium phosphate buffer (pH 5.5) and 2 mM deuterated-DTT using a NAP-5 column (GE Healthcare Life Sciences). For 1H-15N chemical shift perturbation studies, 12.5 μl of 15N CANTD was added to 527 μl of 25 mM sodium phosphate (pH 5.5) in H2O, followed by 60 μl of D2O. To this sample, increasing amounts of a 10 mM stock solution of BI-1 in DMSO-d6 were added. The CANTD–BI-1 final concentrations were as follows: 15 μM CANTD and 0 μM BI-1; 15 μM CANTD and 15 μM BI-1; 15 μM CANTD and 30 μM BI-1; 15 μM CANTD and 60 μM BI-1; and 15 μM CANTD and 120 μM BI-1.1H-15N nuclear magnetic resonance (NMR) experiments were acquired at 35°C using a 600-MHz Avance II spectrometer (Bruker BioSpin) equipped with a 5-mm z-gradient triple-resonance cryoprobe. Two-dimensional 1H-15N heteronuclear single quantum coherence (HSQC) and 1H-15N transverse relaxation optimized spectroscopy (TROSY) spectra were acquired using standard pulse sequences and parameters. Backbone resonances were assigned using a 1 mM 13C/15N-labeled HIV-1 CANTD (residues 1 to 146) sample in 25 mM sodium phosphate (pH 5.5) via standard triple-resonance assignment strategies as previously reported (27). All NMR data were processed and analyzed using Topspin (Bruker BioSpin). Binding isotherms from 1H-15N NMR titration experiments were calculated using Prism 4 software (GraphPad Software, Inc.).
X-ray crystallography.
Cocrystallization with BI-1 was carried out in a similar manner as described previously (20, 28, 29). A small molecule, BI-3, which is structurally distinct from BI-1 and BI-2 and binds to a different site on CANTD, was used to facilitate crystallization of the CANTD (29). A protein solution comprised of 15 mg/ml CANTD (residues 1 to 146), 15 mM morpholineethanesulfonic acid (MES) (pH 6.5), 1.5 mM BI-1, 1.5 mM BI-3 (PDB ligand 0OE), and 2% DMSO was mixed in a 1:1 ratio with a solution containing 22.5% PEG8000, 0.1 M TAPS [N-Tris(hydroxymethyl)methyl-3-aminopropanesulfonic acid] (pH 9.0), and 0.1 M sodium acetate, microseeded, and suspended over a reservoir containing the same solution. Large, block-like crystals appeared within a day and continued to grow for 1 to 2 weeks. Immediately prior to flash-freezing in liquid nitrogen, individual crystals were briefly transferred to a solution containing 22.5% PEG8000, 0.1 M TAPS (pH 9.0), 0.1 M sodium acetate, 1.5 mM BI-1, and 12% DMSO for cryoprotection. Data were collected at 100 K on an FR-E X-ray generator equipped with Osmic HiRes2 optics and an MA345dtb image plate detector. Data reduction and scaling were performed using HKL2000 (30). Preliminary models were obtained via rigid body refinement in Phenix using a model ultimately derived from PDB ID 1GWP (31). Further iterations of refinement using Phenix and manual model building using Coot (32) yielded the final model. The data processing and final model refinement statistics are provided in Table S1 in the supplemental material. The stereochemical quality of the model was assessed with the program Molprobity (33); there were no Ramachandran outliers.
Protein structure accession number.
The coordinates for the CANTD-BI-1 structure have been deposited in the PDB under accession code 4J93.
RESULTS
Identification of a small-molecule inhibitor of HIV-1 replication.
A novel class of HIV-1 inhibitors was identified using a cell-based screen that employed a single-cycle HIV-1 reporter vector pseudotyped with the vesicular stomatitis viral envelope glycoprotein (VSV-G). Compounds were added at the time of infection and could therefore target only the early stages of replication (postentry events). A preselected subset of ∼60,000 compounds that reflected the diversity and complexity of our entire corporate compound collection were screened. One of the hits identified in this screen, BI-1, a representative of the pyrrolopyrazolones, was active in both single and multicycle viral replication assays with EC50 values of 8.2 and 7.5 μM, respectively (Table 1). A more potent analogue, BI-2, was identified from our full compound collection using similarity searches. BI-2 displayed EC50s of 1.8 and 1.4 μM, in single and multicycle viral replication assays, respectively (Table 1). Neither BI-1 nor BI-2 altered the production of infectious virions when added to producer cells (Table 1), implying that these compounds were not active during the late phase of the HIV-1 replication cycle (postintegration to virus maturation). Thus, the pyrrolopyrazolone family of compounds targets the early stage of the HIV-1 replication cycle.
Table 1.
Antiviral profiles of BI compounds 1 and 2a
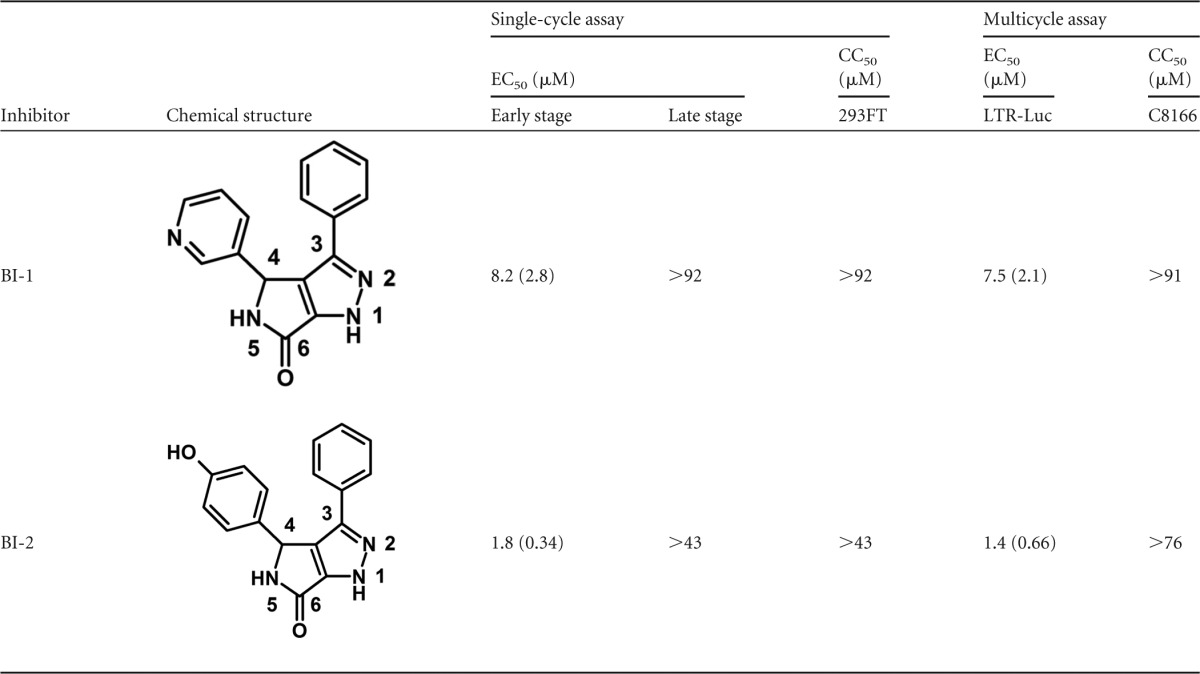
Values represent the averages of at least three independent experiments; standard deviation of the mean is indicated in parentheses.
Mechanistic studies.
Given that the pyrrolopyrazolones are active only during the early phase of the replication cycle, BI-1 and BI-2 were tested for activity against viruses with known nonnucleoside reverse transcriptase inhibitor (NNRTI) resistance mutations. Both compounds maintained activity against all mutant viruses tested (see Table S2 in the supplemental material). To begin to define the mode of action of the pyrrolopyrazolone compounds, quantitative PCR (qPCR) assays were performed to monitor the accumulation of late reverse transcription products and 2-LTR circles within cells infected with VSV-G pseudotyped HIV-1 and treated with BI-1. Positive controls for this experiment included treatment with the NNRTI nevirapine (NVP) and the integrase strand transfer inhibitor L-870,810 (24). Late reverse transcript levels and 2-LTR circles were normalized against mitochondrial DNA levels (mtDNA) present in each sample and are reported relative to a DMSO control sample (Fig. 1). Both positive controls behaved as expected: NVP inhibited reverse transcription (Fig. 1A) and consequently, also reduced the accumulation of 2-LTR circles (Fig. 1B), whereas L-870,810 had no effect on reverse transcription (Fig. 1A) but enhanced the levels of 2-LTR circles (Fig. 1B). As shown in Fig. 1A, BI-1 did not affect the production of late reverse transcripts but did reduce the levels of 2-LTR circles by nearly 1 order of magnitude at both 10 and 24 h postinfection (Fig. 1B). BI-1 did not inhibit either RT or IN enzyme activity in a series of biochemical assays or PIC activity in assays performed using lysates from infected cells treated with inhibitors (data not shown). Instead, the reduction in 2-LTR circles and the lack of an effect on either reverse transcription or integration suggested that BI-1 might inhibit nuclear import of the PIC.
Fig 1.
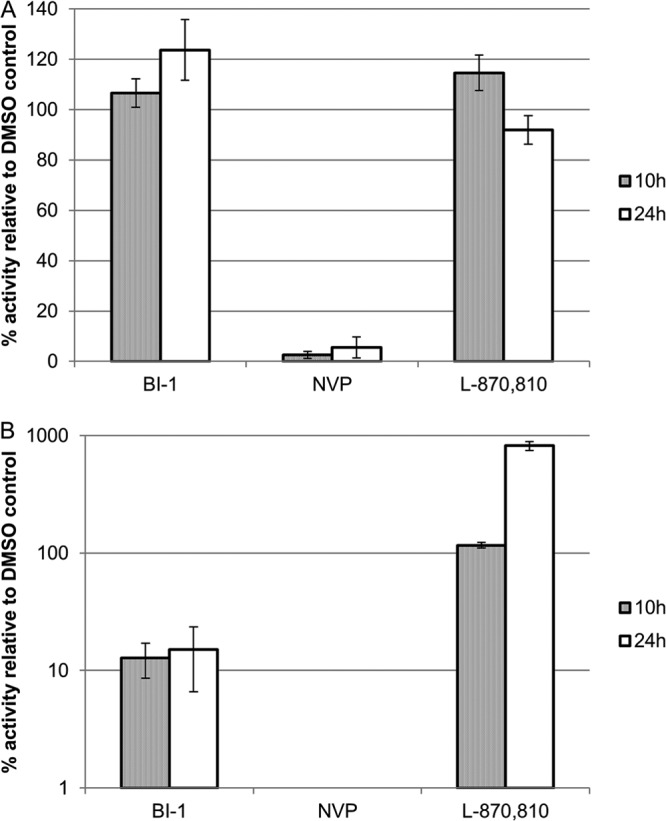
Effects of BI-1 on late reverse transcription and 2-LTR circle formation. C8166 cells were infected in the presence of DMSO, 70 μM BI-1, 1.1 μM nevirapine (NVP), or 20 nM L-870,810 for 2 h at 37°C. Cells were washed with PBS, and fresh inhibitor or DMSO was added to the cells. The cells were harvested 10 or 24 h postinfection, and total DNA was isolated and used in quantitative PCRs specific for late RT (A) or 2-LTR circle (B) products. DNA levels were normalized to mitochondrial DNA levels and then to HIV-1 DNA levels in control, DMSO-treated cells (defined as 100%). Shown are data from a representative experiment (of two independent experiments) with the averages and standard deviations of the means for duplicate samples.
CA substitution mutations confer BI-2 resistance.
Resistance selection studies were performed to identify the target of the BI compounds. To select for resistance, HIV-1 NL4-3 was cultured in C8166 cells in the presence of increasing concentrations of the more potent BI-2 inhibitor. As the cytopathic effect escalated (implying viral replication), the drug concentration was increased 2-fold. Proviral DNA was isolated, amplified, and sequenced at passages 4, 6, 8, 10, and 12. These analyses revealed the accumulation of substitution mutations at positions A105 and T107 within the N-terminal domain of CA. The initial mutation to emerge was an A105T substitution. This mutation was still maintained during passages 8 to 12 but was found at lower frequency as additional mutations emerged at position 107. Viruses with T107 substitutions, either T107A or T107N, emerged during these later passages, and ultimately these became the most prevalent mutations in the population.
To confirm that the A105T, T107A, and T107N substitutions were responsible for conferring resistance to the BI compounds, each mutation was introduced separately into an otherwise wild-type HIV-1 vector. None of the individual resistance mutations affected viral replication significantly in the absence of inhibitor treatment (data not shown). However, both BI-1 and BI-2 were less effective against all three mutants than against the wild-type virus, thereby confirming that these substitutions were responsible for conferring inhibitor resistance (Table 2). Specifically, the EC50 for BI-1 increased 6-fold against the T107A mutant virus and more than 8-fold against the A105T and T107N mutants, remaining inactive at 70 μM, the highest concentration of BI-1 tested. Similarly, the EC50 for BI-2 increased 5- to 10-fold against all three mutant viruses (Table 2). In contrast, each of the mutant viruses maintained susceptibility to NVP (Table 2), demonstrating that resistance was specific for the BI compounds. The identification of drug resistance mutations within the CANTD implies that CA is the functional target of the BI compounds.
Table 2.
Susceptibility of viruses containing CA resistance mutations to antiviral inhibition by BI-1 and BI-2
| Virus inhibitor | Avg EC50 fold change (SD)a |
||
|---|---|---|---|
| CA A105T | CA T107A | CA T107N | |
| BI-1 | >8 | >6 | >8 |
| BI-2 | 5 (1) | 7 (0) | 10 (4) |
| Nevirapine | 0.8 (0.2) | 1 (0.5) | 0.8 (0.4) |
The average EC50 fold change values represent the ratio of the EC50 against the mutant virus to the EC50 against the wild-type virus. Values represent the averages of at least 3 independent experiments, except for T107A, for which the average of 2 independent experiments is shown.
BI compounds bind directly to CANTD.
Isothermal titration calorimetry and NMR chemical shift experiments were used to test whether the BI compounds bound directly to pure recombinant CANTD. As summarized in Table 3, ITC measurements demonstrated that BI-1 and BI-2 bound to CANTD in a 1:1 stoichiometry, with dissociation constants (Kd) of 20 and 3.0 μM, respectively. Control experiments with a CACTD-NC fusion protein showed no compound binding (not shown), indicating that the interactions observed for CANTD were specific.
Table 3.
Inhibitor binding to CANTD measured by ITCa
| Inhibitor | Kd(μM) | ΔG(kcal/mol) | ΔH(kcal/mol) | −TΔS(kcal/mol) |
|---|---|---|---|---|
| BI-1 | 20 | −6.3 | −15.3 | 9.0 |
| BI-2 | 3.0 | −7.5 | −14.9 | 7.4 |
Values represent the averages of two and three independent experiments for BI-1 and BI-2, respectively. ΔG, Gibbs free energy change; ΔH, enthalpy change; T, temperature; ΔS, entropy change.
Binding of BI-1 to CANTD was also evident in NMR chemical shift perturbation studies (Fig. 2). Figure 2A shows overlays of two-dimensional (2D) 1H-15N HSQC TROSY spectra of 15N-labeled CANTD in the presence of increasing concentrations of BI-1. Although most resonances were unaffected by the addition of BI-1, a significant subset were shifted or broadened beyond detection, indicating site-specific inhibitor binding. Residues that were maximally affected by inhibitor binding are labeled in Fig. 2A, and their positions are mapped onto the structure of the free CANTD protein in Fig. 2B. The shifted residues cluster on one face of CANTD defined by helices 3, 4, 5, and 7. 1H and 15N chemical shift changes (Δδ) for residues D103, G106, T108, and L138 were plotted as a function of inhibitor concentration and fit to a 1:1 binding model, yielding an average Kd of 20 ± 2.2 μM (Fig. 2C). Thus, our NMR and ITC measurements of BI-1 compound binding to CANTD are in excellent agreement and together reveal that both BI-1 and BI-2 bind directly and specifically to the amino-terminal domain of HIV-1 CA.
Fig 2.

Identification of the inhibitor binding site within CANTD. (A) Overlay of 2D 1H-15N HSQC TROSY spectra obtained upon titration of BI-1 into CANTD (15 μM 15N CANTD, residues 1 to 146). Color coding and CANTD to BI-1 ratios were as follows: black, 1:0; red, 1:1; cyan, 1:4; blue, 1:8. Backbone NH resonances that shift upon inhibitor addition are labeled with the corresponding residue number. (B) Ribbon diagram showing the CANTD structure and highlighting the residues most affected by BI-1 binding. Significantly shifted resonances were F32, A47, Q50, L52, T54, L56, N57, T58, G60, G61, H62, Q63, K70, E71, T72, N74, A77, D81, D103, A105, G106, T107, T108, I134, and L138. (C) 1H and 15N NMR chemical shifts for CA residues D103, G106, T108, and L138. Fits show 1:1 binding models corresponding to the designated dissociation constants (average Kd, 20 ± 2.2 μM).
Crystal structure of the BI-1:CANTD complex.
Cocrystals of CANTD and BI-1 produced high-quality diffraction data to 1.74-Å resolution, and the structure was refined to 19.7% Rcryst and 23.3% Rfree (see Table S1 in the supplemental material for full details). Density for BI-1 was clear and revealed that the inhibitor bound in a relatively shallow binding pocket formed by residues from helices 3, 4, and 5 (Fig. 3), in good agreement with the 1H-15N HSQC TROSY data (Fig. 2). The pocket appears to be preformed because BI-1 binding did not induce significant protein backbone movements. The pyrrolopyrazolone core stacks against the amide side chain of CA residue N53, parallel to the surface of the protein, and buttressed on both sides by residues N57 and T107. The N57 side chain becomes more ordered upon inhibitor binding, forming one direct and one water-mediated hydrogen bond with the N2 and N1 nitrogens of the inhibitor, respectively. The T107 side chain rotates to form a hydrogen bond between the T107 Oγ and the inhibitor N5 atom. A water-mediated hydrogen bond is also formed between the carbonyl oxygens of BI-1 and the G106 backbone. The C3 phenyl substituent of BI-1 is rotated 12° relative to the pyrrolopyrazolone core and sits in a hydrophobic environment created by the side chains of L56, M66, L69, K70, and I73. It is clear that only the S enantiomer at position C5 of BI-1 can bind CANTD because inversion of the chiral center would produce a highly unfavorable steric clash between the pyridine ring and the protein. The C5 pyridine is oriented perpendicular to the plane of the inhibitor, and the nitrogen likely faces the solvent, although carbon and nitrogen cannot be discriminated at this resolution. The pyridine binding pocket is more open than the C3 phenyl binding pocket and is formed primarily by residues K70, I73, N74, and Y130.
Fig 3.
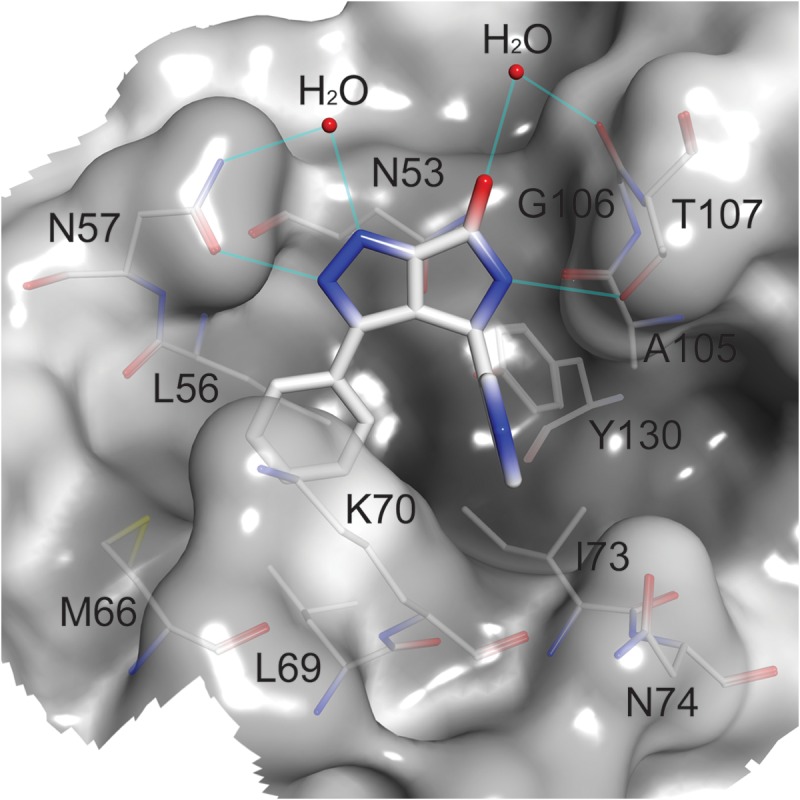
Interactions of BI-1 within the CANTD site 2 pocket. Semitransparent surface representation of the BI-1 binding pocket, shaded from dark to light by increasing solvent exposure. BI-1 (thick sticks) and residues forming the pocket (thin sticks) are colored by atom type. Water molecules are indicated by red spheres and hydrogen bonds by cyan lines.
BI-1 promotes HIV-1 CA-NC tube assembly.
Pure recombinant CA and CA-NC proteins can assemble in vitro, forming helical tubes that are composed of hexagonal arrays of CA rings that mimic the surface lattice of the viral capsid (6, 34). The influence of BI-1 binding on CA-NC assembly was analyzed using a fluorescence assay that measured the degree of CA-NC assembly on the surface of NeutrAvidin-coated microplates (20). CA-NC assembly was nucleated by immobilized, biotin-conjugated single-stranded d(TG)25 oligonucleotides, and the extent of assembly was quantified by measuring the levels of soluble, fluorescein-labeled d(TG)25 oligonucleotides that remained incorporated following a wash step. Remarkably, BI-1 stimulated CA-NC assembly in a concentration-dependent manner in this assay, resulting in a 2-fold increase in the fluorescence signal at a BI-1 concentration of 25 μM and an NaCl concentration of 350 mM (Fig. 4A). CA-NC assembly is salt dependent, and BI-1 also stimulated CA-NC assembly at NaCl concentrations of 250 mM and 200 mM NaCl, with maximal increases of 9- and 5-fold, respectively (Fig. 4B).
Fig 4.

BI-1 and BI-2 enhance assembly and stability of CA-NC tubes. (A) Assembly of wild-type and A42D CA-NC was assayed in the presence of increasing concentrations of BI-1 (2-fold dilutions, 1.6 to 25 μM) in a buffer containing 350 mM NaCl. Assembly assays were performed in NeutrAvidin-coated microplates, precoupled with d(TG)25 5′ biotin-labeled oligonucleotide, by incubating 2 μM CA-NC proteins and 100 nM d(TG)25 5′ fluorescein-labeled oligonucleotide for 2 h at 23°C. Assembled CA-NC tubes were quantified by measuring the fluorescein signal remaining following a wash step. (B) Wild-type CA-NC assembly levels at different NaCl concentrations (150, 200, and 250 mM) in the presence of increasing BI-1 concentrations. (C) Assembly of CA-NC tubes in solution [9 μM CA-NC, 1 μM d(TG)50, 400 mM NaCl] during 16 h of incubation at 4°C in the presence of DMSO (control) or 20 or 200 μM BI-2. Suspended CA-NC tubes were visualized by negative-staining electron microscopy. (D) Effects of BI-1 and BI-2 on CA-NC tube stability. CA-NC tubes were preassembled in microplates and washed, the buffer salt concentration was reduced from 350 to 250 mM in serial dilutions of BI-1 or BI-2, and the CA-NC tubes were allowed to disassemble for 2 h at 23°C. The remaining fluorescein signal was measured following a wash step and used to quantify the extent of CA stabilization (0% represents no stabilization). Shown are data from a representative experiment (of at least two independent experiments) with the averages and standard deviations of the means for duplicate samples.
Several lines of evidence indicate that the BI compounds specifically stimulated the formation of authentic CA-NC tubes in this assay. First, the fluorescent signal did not increase when BI-1 was added to a control CA-NC protein that carried the CA A42D amino acid substitution, which has previously been shown to inhibit assembly of the hexagonal CA lattice (Fig. 4A) (10, 11). Second, BI-2 also stimulated the formation of CA-NC tubes in a homogeneous solution assay (Fig. 4C; see Fig. S1 in the supplemental material). In this assay, 9 μM CA-NC and 1 μM d(TG)50 were incubated together in a buffer that contained 300, 400, or 500 mM NaCl and different concentrations of BI-2. CA-NC tube formation was analyzed both by quantifying the amount of pelletable CA-NC tubes (see Fig. S1 in the supplemental material) and by electron microscopy with negative staining (Fig. 4C). Consistent with the results from the heterogeneous plate-based fluorescence assembly assay, the number of CA-NC tubes increased with increasing BI-2 concentrations, and the CA-NC tubes had normal sizes and morphologies (Fig. 4C and data not shown). We therefore conclude that the BI compounds bind HIV-1 CA in a conformation that stimulates assembly of a hexagonal lattice.
BI compounds stabilize HIV-1 CA-NC complexes.
Given that the BI compounds stimulated CA-NC assembly, we hypothesized that they might also stabilize assembled CA-NC tubes. To test this hypothesis, we established an in vitro CA stabilization assay. Briefly, CA-NC tubes were preassembled on NeutrAvidin-coated microplates in 350 mM NaCl and then allowed to disassemble in the presence or absence of compound, in a buffer that contained only 250 mM NaCl. The degree of CA-NC tube stabilization was evaluated by measuring the fluorescence remaining following a final wash step (see Materials and Methods for details). In the absence of compound, ∼70% of the CA-NC tubes disassembled during the low-salt incubation (defined as 0% stabilization). As shown in Fig. 4D, both BI-1 and BI-2 stabilized CA-NC tubes in a concentration-dependent manner. The IC50 values for BI-1 and BI-2 were 19 and 3.3 μM, respectively, in good agreement with their binding affinities for free CANTD (Table 3 and Fig. 2C). The compounds also modestly stabilized purified HIV-1 cores (data not shown) against dissociation in vitro (35), consistent with the CA assembly experiments. Taken together, our data demonstrate that BI compound binding must increase the thermodynamic stability of the hexagonal CA lattice because the compounds enhance assembly and inhibit disassembly of CA complexes.
Resistance mutations prevent BI-2 stabilization of CA-NC tubes in vitro.
To test whether the activity of BI-2 in the CA stabilization assay correlated with CANTD pocket binding and antiviral activity, we examined the effects of the A105T and T107A resistance mutations on CA-NC stability. The intrinsic activities of the mutant CA-NC proteins were similar to that of the wild-type protein in both the CA assembly and stabilization assays (data not shown). However, BI-2 failed to stabilize either of the mutant CA-NC proteins in the CA stabilization assay (IC50, >30 μM compared to 3.3 μM for WT CA-NC; fold change [FC], >10), consistent with the loss of susceptibility of each of these mutants to BI-2 in viral replication assays (Table 2).
CA residue Asn-57 contributes to BI compound binding by forming a hydrogen bond with the pyrazole ring (Fig. 3). To test whether this residue also contributes to BI-2 activity, we analyzed the CA-NC N57S mutant protein in CA stabilization and viral replication assays. This amino acid substitution reduced CA-NC assembly but did not reduce the intrinsic rate of CA-NC dissociation in the CA stabilization assay (data not shown). Importantly, BI-2 failed to stabilize the CA-NC N57S mutant in the CA stabilization assay (IC50 > 30 μM; FC > 10). Similarly, a CA N57S mutant virus was resistant to BI-2 (EC50 > 65 μM, FC > 36). Thus, it was possible to use the BI-1:CANTD structure to introduce an amino acid substitution that conferred resistance to BI-2 stabilization and antiviral activity. The excellent correspondence between the requirements for BI inhibitor binding, activity in the CA assembly and stabilization assays, and antiviral activity implies that inhibitor binding within CANTD pocket 2 is responsible for antiviral activity.
DISCUSSION
Cell-based antiviral phenotypic screens can identify novel inhibitors of viral replication that target either viral or cellular targets. Such screens have led to the discovery of anti-HIV-1 inhibitors with novel mechanisms of action (22, 36–40) that probably could not have been identified in more specifically targeted assays. Although it can be challenging to identify their functional targets and mechanisms of action, such compounds can also provide valuable tools for identifying and analyzing key steps in viral replication.
In the present case, we screened for inhibitors of postentry steps in HIV-1 infection. Our screen identified a novel series of pyrrolopyrazolone small-molecule inhibitors that are mechanistically distinct from other classes of HIV-1 antiviral compounds. Quantitative PCR analysis of reverse transcription products and 2-LTR circles indicated that the BI compounds target a step following reverse transcription, but prior to (or concomitant with) nuclear import of the preintegration complex (PIC). Amino acid substitutions that confer inhibitor resistance map to a series of residues that surround a conserved pocket within the N-terminal domain of CA (site 2), and BI-1 was shown to bind in this pocket by ITC, NMR, and X-ray crystallographic analyses. Thus, CANTD site 2 is the functional target of the BI inhibitors.
To replicate efficiently, HIV-1 must form capsids of the appropriate stability (3, 10–12), and we speculate that the BI compounds may function, at least in part, by stabilizing the viral capsid. We have shown that the BI compounds accelerate CA-NC tube assembly and stabilize preassembled CA-NC tubes against dissociation. However, the mechanism by which the BI pyrrolopyrazolones stabilize capsid complexes is uncertain because the inhibitor binding site does not reside directly within any inter- or intrahexamer CA interfaces that stabilize the capsid lattice (see Fig. S2 in the supplemental material). It is possible, however, that the compounds act by reducing CANTD flexibility and/or stabilizing CA interfaces allosterically. The latter possibility is consistent with the observation that BI-1 binding alters the backbone amide chemical shift environment of several residues that lie outside the binding pocket. Distal shifted residues include Phe32 and His62, which reside within the flexible loops between α1/α2 and α3/α4, respectively, both of which are positioned near intrahexamer CA interfaces. In other contexts, amino acid substitutions that stabilize the HIV-1 capsid can inhibit viral replication at a step between reverse transcription and integration, as was recently shown for the E45A mutant (10–12, 41). Similarly, the Q63A/Q67A mutant exhibits delayed uncoating in target cells and is impaired for PIC nuclear entry and integration (13). Thus, it seems possible that the BI compounds act in a similar manner to prevent nuclear localization of the PIC.
The previously reported PF-3450074 inhibitor also binds in the CANTD site 2 pocket (22), and PF-3450074 and BI-1 make similar interactions with many of the same residues (Fig. 5A). Nevertheless, the activities of these two chemically distinct inhibitors appear to differ significantly. Key differences include the following: (i) PF-3450074 is active during both the early and late phases of the replication cycle (22, 23), whereas the BI pyrrolopyrazolones are active only during the early phase; (ii) reverse transcription is inhibited in infected cells treated with PF-3450074 (22) but not in infected cells treated with the BI compounds; and (iii) PF-3450074 stimulates capsid dissociation (23), whereas the BI compounds stabilize CA assemblies. Thus, the two different chemical classes of inhibitors appear to exert distinct effects on early steps of viral replication, despite binding in the same pocket on CANTD. Although the structural basis for their differing activities remains to be determined, we note that the indole moiety of PF-3450074 extends beyond the region of site 2 that is occupied by the BI pyrrolopyrazolones (Fig. 5A), resulting in additional contacts with CA residues Met66, Gln67, Lys70, and Gln63 (22). We speculate that these additional contacts beyond the core of site 2 may result in capsid destabilization.
Fig 5.

BI-1, PF-3450074, and CPSF6313–327 all bind in CANTD pocket 2. (A) Superposition of the BI-1:CANTD (PDB 4J93) and PF-3450074:CANTD (PDB 2XDE) structures. The CANTD surface is shown with atomic color coding. The bound BI-1 and PF-3450074 inhibitors (sticks) are shown in green and silver, respectively. To create this figure, the CANTD complexes were superimposed, and the figure shows the superimposed inhibitors, mapped onto the CANTD structure from the BI-1:CANTD complex. Note that the PF-3450074 inhibitor appears to clash sterically with Lys-70 because this side chain occupies a different position in the PF-3450074:CANTD structure. (B) Superposition of BI-1:CANTD (PDB 4J93) and CPSF6313–327:CANTD (PDB 4B4N) structures. The orientations and color coding of the CANTD and BI-1 inhibitors are the same as in Fig. 5A, and the CPSF6313–327 backbone is shown in tan, with three key binding side chains shown explicitly and with residue labels.
HIV-1 CA binds several host factors that influence HIV-1 activity, including cyclophilin A, TRIM5α, and TNPO3 (reviewed in references 42–46). A role for CA in the nuclear import of the PIC is well established, in part through the interactions of CA with nucleoporins (47–52). We speculate that BI pyrrolopyrazolones binding to CA site 2 may disrupt the interaction of some host factors with capsid cores. TNPO3 binds HIV-1 CA proteins (53, 54) and CA-NC tubes (54) and stimulates core dissociation in vitro (55). TNPO3 depletion reduces viral infectivity (49, 53–62), and CA amino acid substitutions, both within CANTD site 2 (T54A, N57A, K70R, N74D, A105T, and T107N) and elsewhere, can render the virus resistant to this effect (49, 54, 60–62). The interaction between TNPO3 and CA has not yet been characterized in molecular detail, however, and it is not yet clear how the BI inhibitors would affect TNPO3 binding.
A C-terminally truncated mutant of CPSF6 (CSPF61–358) restricts the infectivity of wild-type HIV-1 (47). Like the BI pyrrolopyrazolones, CSPF61–358 has no effect on reverse transcription but prevents subsequent accumulation of 2-LTR circles (47, 61, 63). CANTD amino acid substitutions within site 2, like N74D, that inhibit CPSF61–358 binding, abrogate viral restriction (47, 63, 64), and this amino acid substitution renders HIV-1 noninfectious in macrophages, suggesting that host factor binding to site 2 may be important in some contexts (47). The region of CSPF61–358 required for HIV-1 restriction maps to residues 301 to 358 (63), and a peptide encompassing CPSF6 residues 313 to 327 has been cocrystallized in complex with CANTD (64). Moreover, CA complexes are stabilized in infected cells expressing CSPF61–358 (61, 62). As illustrated in Fig. 5B, CPSF6313–327 binds in CANTD pocket 2, overlapping the BI-1 and PF-3450074 binding sites. PF-3450074 competitively inhibits CPSF6313–327 binding (64), and it is clear that BI compound binding is also likely to sterically occlude CPSF6313–327 binding. We therefore suggest that the BI compounds may mimic the action of CSPF61–358 (47, 61, 62, 64) and/or TNPO3 (61, 62) and anticipate that the BI compounds will be useful tools for helping to define precisely how CPSF6 functions in viral replication.
In summary, we used a cell-based antiviral screen to identify novel inhibitors of HIV-1 replication that bind and stabilize HIV-1 capsids. Our discovery of a new class of antiviral compounds that bind CANTD site 2 and inhibit distinct steps in HIV replication indicates that this pocket warrants continued investigation as an antiretroviral target. Further optimization of BI-1 and BI-2 or the discovery of a new chemical series with increased potency may enable the identification of compounds suitable for the treatment of HIV-1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Richard Bethell, Pierre Bonneau, and Michael Cordingley for leadership and guidance in biology and structural research. Our gratitude is extended to Chantal Grand-Maître, Olivier Lepage, and Anne-Marie Faucher for the resynthesis of BI-1 and BI-2 and to Steve Alam for assistance in creating Fig. 5.
V. Shav and C. Aiken were supported by NIH grant R01 AI089401. C. Langelier and W. Sundquist were supported by Boehringer Ingelheim (Canada) Ltd. and NIH grant P50 GM082545 . C. Langelier was also supported by a National Institute of Allergy and Immunology Microbial Pathogenesis training grant.
Footnotes
Published ahead of print 1 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00985-13.
REFERENCES
- 1.Arts EJ, Hazuda DJ. 2012. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2:a007161. 10.1101/cshperspect.a007161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell NM, Lever AM. 2013. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 21:136–144 [DOI] [PubMed] [Google Scholar]
- 3.Sundquist WI, Krausslich HG. 2012. HIV-1 assembly, budding, and maturation. Cold Spring Harb, Perspect, Med. 2:a006924. 10.1101/cshperspect.a006924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bharat TA, Davey NE, Ulbrich P, Riches JD, de Marco A, Rumlova M, Sachse C, Ruml T, Briggs JA. 2012. Structure of the immature retroviral capsid at 8 A resolution by cryo-electron microscopy. Nature 487:385–389 [DOI] [PubMed] [Google Scholar]
- 5.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83 [DOI] [PubMed] [Google Scholar]
- 6.Li S, Hill CP, Sundquist WI, Finch JT. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409–413 [DOI] [PubMed] [Google Scholar]
- 7.Ganser-Pornillos BK, Cheng A, Yeager M. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70–79 [DOI] [PubMed] [Google Scholar]
- 8.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dismuke DJ, Aiken C. 2006. Evidence for a functional link between uncoating of the human immunodeficiency virus type 1 core and nuclear import of the viral preintegration complex. J. Virol. 80:3712–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bocanegra R, Rodriguez-Huete A, Fuertes MA, Del Alamo M, Mateu MG. 2012. Molecular recognition in the human immunodeficiency virus capsid and antiviral design. Virus Res. 169:388–410 [DOI] [PubMed] [Google Scholar]
- 15.Sticht J, Humbert M, Findlow S, Bodem J, Muller B, Dietrich U, Werner J, Krausslich HG. 2005. A peptide inhibitor of HIV-1 assembly in vitro. Nat. Struct. Mol. Biol. 12:671–677 [DOI] [PubMed] [Google Scholar]
- 16.Ternois F, Sticht J, Duquerroy S, Krausslich HG, Rey FA. 2005. The HIV-1 capsid protein C-terminal domain in complex with a virus assembly inhibitor. Nat. Struct. Mol. Biol. 12:678–682 [DOI] [PubMed] [Google Scholar]
- 17.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013–1020 [DOI] [PubMed] [Google Scholar]
- 18.Kelly BN, Kyere S, Kinde I, Tang C, Howard BR, Robinson H, Sundquist WI, Summers MF, Hill CP. 2007. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 373:355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fader LD, Bethell R, Bonneau P, Bos M, Bousquet Y, Cordingley MG, Coulombe R, Deroy P, Faucher AM, Gagnon A, Goudreau N, Grand-Maitre C, Guse I, Hucke O, Kawai SH, Lacoste JE, Landry S, Lemke CT, Malenfant E, Mason S, Morin S, O'Meara J, Simoneau B, Titolo S, Yoakim C. 2011. Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg. Med. Chem. Lett. 21:398–404 [DOI] [PubMed] [Google Scholar]
- 20.Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier JF, Wardrop E, Faucher AM, Coulombe R, Banik SS, Fader L, Gagnon A, Kawai SH, Rancourt J, Tremblay M, Yoakim C, Simoneau B, Archambault J, Sundquist WI, Mason SW. 2012. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 86:6643–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tremblay M, Bonneau P, Bousquet Y, DeRoy P, Duan J, Duplessis M, Gagnon A, Garneau M, Goudreau N, Guse I, Hucke O, Kawai SH, Lemke CT, Mason SW, Simoneau B, Surprenant S, Titolo S, Yoakim C. 2012. Inhibition of HIV-1 capsid assembly: optimization of the antiviral potency by site selective modifications at N1, C2 and C16 of a 5-(5-furan-2-yl-pyrazol-1-yl)-1H-benzimidazole scaffold. Bioorg. Med. Chem. Lett. 22:7512–7517 [DOI] [PubMed] [Google Scholar]
- 22.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 6:e1001220. 10.1371/journal.ppat.1001220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J. Virol. 85:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr, Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen IW, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD. 2004. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl. Acad. Sci. U. S. A. 101:11233–11238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631–634 [DOI] [PubMed] [Google Scholar]
- 26.Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI. 2008. Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 82:11682–11694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gitti RK, Lee BM, Walker J, Summers MF, Yoo S, Sundquist WI. 1996. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 273:231–235 [DOI] [PubMed] [Google Scholar]
- 28.Goudreau N, Lemke CT, Faucher AM, Grand-Maitre C, Goulet S, Lacoste JE, Rancourt J, Malenfant E, Mercier JF, Titolo S, Mason SW. 2013. Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem. Biol. 8:1074–1082 [DOI] [PubMed] [Google Scholar]
- 29.Lemke CT, Titolo S, Goudreau N, Faucher A-M, Mason SW, Bonneau P. 2013. A novel inhibitor-binding site on the HIV-1 capsid N-terminal domain leads to improved crystallization via compound-mediated dimerization. Acta Crystallogr. D 69:1115–1123 [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 31.Tang C, Ndassa Y, Summers MF. 2002. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 9:537–543 [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 33.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah VB, Aiken C. 2011. In vitro uncoating of HIV-1 cores. J. Vis. Exp. 10.3791/3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. 1994. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 57:243–247 [DOI] [PubMed] [Google Scholar]
- 37.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 100:11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jochmans D, Deval J, Kesteleyn B, Van Marck H, Bettens E, De Baere I, Dehertogh P, Ivens T, Van Ginderen M, Van Schoubroeck B, Ehteshami M, Wigerinck P, Gotte M, Hertogs K. 2006. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J. Virol. 80:12283–12292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blair WS, Cao J, Jackson L, Jimenez J, Peng Q, Wu H, Isaacson J, Butler SL, Chu A, Graham J, Malfait AM, Tortorella M, Patick AK. 2007. Identification and characterization of UK-201844, a novel inhibitor that interferes with human immunodeficiency virus type 1 gp160 processing. Antimicrob. Agents Chemother. 51:3554–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blair WS, Cao J, Fok-Seang J, Griffin P, Isaacson J, Jackson RL, Murray E, Patick AK, Peng Q, Perros M, Pickford C, Wu H, Butler SL. 2009. New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob. Agents Chemother. 53:5080–5087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang R, Shi J, Byeon IJ, Ahn J, Sheehan JH, Meiler J, Gronenborn AM, Aiken C. 2012. Second-site suppressors of HIV-1 capsid mutations: restoration of intracellular activities without correction of intrinsic capsid stability defects. Retrovirology 9:30. 10.1186/1742-4690-9-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sokolskaja E, Luban J. 2006. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr. Opin. Microbiol. 9:404–408 [DOI] [PubMed] [Google Scholar]
- 43.Towers GJ. 2007. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 4:40. 10.1186/1742-4690-4-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arhel N. 2010. Revisiting HIV-1 uncoating. Retrovirology 7:96. 10.1186/1742-4690-7-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grutter MG, Luban J. 2012. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2:142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz-Griffero F. 2012. The Role of TNPO3 in HIV-1 Replication. Mol. Biol. Int. 2012:868597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matreyek KA, Engelman A. 2011. The requirement for nucleoporin NUP153 during human immunodeficiency virus type 1 infection is determined by the viral capsid. J. Virol. 85:7818–7827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 7:e1002439. 10.1371/journal.ppat.1002439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Nunzio F, Danckaert A, Fricke T, Perez P, Fernandez J, Perret E, Roux P, Shorte S, Charneau P, Diaz-Griffero F, Arhel NJ. 2012. Human nucleoporins promote HIV-1 docking at the nuclear pore, nuclear import and integration. PLoS One 7:e46037. 10.1371/journal.pone.0046037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koh Y, Wu X, Ferris AL, Matreyek KA, Smith SJ, Lee K, KewalRamani VN, Hughes SH, Engelman A. 2013. Differential effects of human immunodeficiency virus type 1 capsid and cellular factors nucleoporin 153 and LEDGF/p75 on the efficiency and specificity of viral DNA integration. J. Virol. 87:648–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Nunzio F, Fricke T, Miccio A, Valle-Casuso JC, Perez P, Souque P, Rizzi E, Severgnini M, Mavilio F, Charneau P, Diaz-Griffero F. 2013. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 440:8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou L, Sokolskaja E, Jolly C, James W, Cowley SA, Fassati A. 2011. Transportin 3 promotes a nuclear maturation step required for efficient HIV-1 integration. PLoS Pathog. 7:e1002194. 10.1371/journal.ppat.1002194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valle-Casuso JC, Di Nunzio F, Yang Y, Reszka N, Lienlaf M, Arhel N, Perez P, Brass AL, Diaz-Griffero F. 2012. TNPO3 is required for HIV-1 replication after nuclear import but prior to integration and binds the HIV-1 core. J. Virol. 86:5931–5936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. 2013. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J. Virol. 87:422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926 [DOI] [PubMed] [Google Scholar]
- 57.Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, Debyser Z. 2008. Transportin-SR2 imports HIV into the nucleus. Curr. Biol. 18:1192–1202 [DOI] [PubMed] [Google Scholar]
- 58.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krishnan L, Matreyek KA, Oztop I, Lee K, Tipper CH, Li X, Dar MJ, Kewalramani VN, Engelman A. 2010. The requirement for cellular transportin 3 (TNPO3 or TRN-SR2) during infection maps to human immunodeficiency virus type 1 capsid and not integrase. J. Virol. 84:397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Iaco A, Luban J. 2011. Inhibition of HIV-1 infection by TNPO3 depletion is determined by capsid and detectable after viral cDNA enters the nucleus. Retrovirology 8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. 2013. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10:20. 10.1186/1742-4690-10-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fricke T, Valle-Casuso JC, White TE, Brandariz-Nunez A, Bosche WJ, Reszka N, Gorelick R, Diaz-Griffero F. 2013. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 10:46. 10.1186/1742-4690-10-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee K, Mulky A, Yuen W, Martin TD, Meyerson NR, Choi L, Yu H, Sawyer SL, Kewalramani VN. 2012. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J. Virol. 86:3851–3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 8:e1002896. 10.1371/journal.ppat.1002896 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.