

Abstract
E138K, a G→A mutation in HIV-1 reverse transcriptase (RT), is preferentially selected by etravirine (ETR) and rilpivirine over other substitutions at position E138 that offer greater drug resistance. We hypothesized that there was a mutational bias for the E138K substitution and designed an allele-specific PCR to monitor the emergence of E138A/G/K/Q/R/V during ETR selection experiments. We also performed competition experiments using mutated viruses and quantified the prevalence of E138 minority species in drug-naive patients. E138K, as well as E138G, consistently emerged first during ETR selection experiments, followed by E138A and E138Q; E138R was never selected. Surprisingly, E138K was identified as a tiny minority in 23% of drug-naive subtype B patients, a result confirmed by ultradeep sequencing (UDS). This result could reflect a low fitness cost of E138K; however, E138K was one of the least fit substitutions at codon E138, even after taking into account the deoxynucleoside triphosphate pools of the cells used in competition experiments. Further UDS analysis revealed other minority species in a pattern consistent with the mutational bias of HIV RT. There was no evidence of APOBEC3-hypermutation in these selection experiments or in patients. Our results confirm the mutational bias of HIV-1 in patients and highlight the importance of G→A mutations in HIV-1 drug resistance evolution.
INTRODUCTION
The G→A nucleotide mutation is the most common type of HIV-1 drug resistance mutation (1), and E138K in HIV-1 reverse transcriptase (RT) is such a mutation. E138K and several other substitutions at position E138 are commonly selected by newer members of the non-nucleoside RT inhibitor (NNRTI) family of drugs, rilpivirine (RPV) and etravirine (ETR) (2–4). Each of the E138K, E138A, E138G, E138Q, E138R, and E138V substitutions can be selected by these NNRTIs, while E138A is also considered to be a polymorphism (1, 5, 6). However, it is not known why E138K, a substitution that confers only modest levels of drug resistance to RPV and ETR seems to be preferentially selected over other E138 substitutions (4, 6–10), since each of the above substitutions only requires a single nucleotide mutation, with the exception of E138R, which requires two.
We believe that E138K may be selected frequently due to the HIV-1 mutational bias. First, there is a bias toward G→A mutations in both cell-free RT assays and in single cycle viral replication experiments performed in cell lines (11–13). Host factors may also modulate the mutational bias of HIV-1. For example, the APOBEC3 cytidine-deaminase family of HIV-1 host restriction factors can deaminate deoxycytidine in the viral genome to deoxyuracil, ultimately causing numerous G→A mutations, known as hypermutations (14, 15). Indeed, the frequent identification of hypermutated viruses containing E138K in the proviral reservoir of untreated patients suggests that APOBEC3 enzymes may play a major role in the appearance of E138K and related mutations (16). This suggests that it might be possible to identify small E138K minority species genetically linked to hypermutated viruses in plasma RNA.
Some studies have used bulk sequencing to estimate the mutational bias of HIV-1 and to look for hypermutation in patient plasma (17, 18). However, bulk sequencing might hide some evidence of hypermutation and mutational bias due to the effects of fitness. Analyzing small minority species that have been under negative selection for shorter periods could be a better way to evaluate HIV-1 mutational bias in patients.
We hypothesized that E138K is preferentially selected because of the mutational bias of HIV-1 and because of the activities of APOBEC3 enzymes since E138K yields lower-level drug resistance to RPV and ETR than other E138 substitutions (6). To assess this, we developed a sensitive, inexpensive, and quantitative allele-specific PCR (AS-PCR) method for detecting E138A, E138G, E138K, E138Q, E138V, and E138R (19–23). Simply sequencing the amplicon of AS-PCRs revealed mutations linked to the identified minority species (24). We used this AS-PCR technique, as well as ultradeep sequencing (UDS), to search for evidence of APOBEC3 activity in plasma RNA, as well as to gauge the mutational bias of HIV-1 in patients.
MATERIALS AND METHODS
Viral isolates, cells, drugs, and plasmid.
Viral isolates 5326, 5331, 8336, 8116, and BG-05 were obtained from drug-naive patients during acute infection (<6 months) with informed consent at our clinics in Montreal, Canada. Cord blood mononuclear cells (CBMCs) were obtained through the Department of Obstetrics, Jewish General Hospital, Montreal, Canada. Through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Disease, National Institutes of Health, the pNL4-3 vector was obtained from Malcolm Martin (25), and ETR was obtained from Janssen Pharmaceuticals, Inc. To construct standards for AS-PCR, the E138A, E138G, E138K, E138Q, E138R, and E138V substitutions were introduced into RT by site-directed mutagenesis (QuikChange XL kit; Stratagene, La Jolla, CA). Clinical isolates were used in selection experiments with ETR as previously described (26); ETR selection experiments with viral isolates 5326, 5331, and BG-05 were performed previously.
Viral RNA extraction and RT-PCR.
HIV-1 RNA was purified from EDTA-anticoagulated plasma or from cell culture supernatants (QIAamp viral RNA minikit; Qiagen Sciences, MD), according to the instructions of the manufacturer. Purified RNA (20 μl) was reverse transcribed and PCR amplified in a single step (SuperScript III one-step RT-PCR system with platinum Taq DNA polymerase; Invitrogen, Burlington, Ontario, Canada) according to the manufacturer's instructions, with cDNA synthesis performed at 53°C for 30 min and the PCR annealing temperature set at 55°C for 30 s. A total of 20 U of RNaseOUT (Invitrogen) was used in each reaction. The primers used for RT-PCR were RT_Sense and RT_Antisense (see Table S1A in the supplemental material). RT-PCR was also performed using a clinical protocol (Virco BVBA, Mechelen, Belgium). The PCR products were purified (QIAquick PCR purification kit; Qiagen) and quantified by spectrophotometry (NanoDrop 1000; Thermo Scientific).
Primers, reaction setup, and quantifying E138 minority species by AS-PCR.
The sequences of all primers were based on the 2010 Los Alamos HIV sequence compendium of HIV-1 subtype B pol (27) (see Table S1B in the supplemental material). All AS-PCR conditions were the same: 300 nM sense primer, 300 nM antisense primer, 2 μM SYTO9, 2% dimethyl sulfoxide, and 1× Platinum Quantitative PCR SuperMix-UDG (Invitrogen) in a volume of 10 μl in a transparent tube (0.1-ml Strip Tubes and Caps; Qiagen). The final DNA concentration in the AS-PCR mixture was 107 to 108 amplicons/μl. Samples were evaluated by qPCR in a Rotor-Gene 6000 real-time thermocycler (Corbett Research Pty, Ltd., Australia) using the following parameters: 50°C for 2 min (for UDG), then 94°C for 2 min, and then 50 cycles of 94°C denaturation for 15 s, 50°C annealing for 15 s, and 72°C elongation for 60 s. Assessments of the sensitivity and running of samples were performed as reported elsewhere (28). The method used to determine genetic linkage following AS-PCR is similar to a method published elsewhere (24).
Study populations.
The study included evaluation of two HIV-1 pol data sets of drug-naive populations from 2001 to 2007: the Quebec drug resistance genotyping program (ntreated = 817; ndrug naive = 406; nunknown = 167) and the Montreal Primary HIV Infection (PHI) Cohort Study (ndrug naive = 335). Patients provided informed consent for blood collection and resistance testing. From the Montreal PHI cohort study, we randomly identified drug-naive patients before January 2008 with a high viral load (>150,000) during primary infection who were assessed for minority species by AS-PCR and ultradeep sequencing (UDS). The viral load was >150,000 to achieve at least 0.05% sensitivity for AS-PCR (28).
UDS.
To remove UDS errors, and also in an attempt to increase sensitivity, we used the PrimerID method (29). After PrimerID analysis, UDS yielded 1,088 complete and unique sequences per patient from codons 105 to 194 with an average of two reads per sequence. To further compensate for potential UDS errors, we only considered mutations present when the mutational frequency was significantly greater (Fisher exact test, P < 0.05) than the frequency of this mutation in >5,000 sequence reads that should not have possessed this mutation but occasionally did due to PCR induced mutations (i.e., sequences that had the same PrimerID tag). This protocol was followed for each individual mutation and position, and this permitted the detection of mutations below 1% in proportion. These analyses were performed using Prism 5 (GraphPad Software, Inc.) and Excel (Microsoft). Alignments were performed in Clustal Omega (30, 31) and were corrected by hand in MEGA5 (32).
Competition experiments.
Competition experiments were performed with 65,000 MT-2 cells in 2 ml of RPMI medium supplemented with 10% fetal bovine serum, 1% penicillin, 1% streptomycin, and 1% l-glutamine. Virus was normalized based on the multiplicity of infection (MOI), and cells were infected with a total MOI of 0.05. Then, 1 ml of supernatant was removed, frozen at −80°C each day for future AS-PCR analysis, and replaced with 1 ml of fresh medium. Next, 1.5 mM hydroxyurea (HU; Sigma) was used in some studies because this was the highest concentration that could be used on MT-2 cells before toxic effects were noted. 0.65 mM deoxynucleosides (dN; Sigma) was used in some studies because this was the lowest concentration that partially rescued MT-2 cells subjected to excess (50 mM) HU. MT-2 cells were preincubated for 24 h with HU or dN prior to infection. The relative fitness was calculated by least-squares analysis (33).
RESULTS
Design of an AS-PCR for E138A, E138G, E138K, E138Q, E138R, and E138V.
The design of this AS-PCR offered a unique challenge in that it had to be specific for two nucleotides to adequately distinguish among six different substitutions. Therefore, the 3′ ultimate and penultimate positions of the AS primer were designed to overlap with the first two nucleotides of the E138 codon (see Table S1B in the supplemental material). Intentional mismatches were placed on two invariant adenine nucleotides of residue N137 (27, 34, 35) to increase the sensitivity. Of note, approximately half of subtype B and subtype C viruses should not have any mutations in the AS primer-binding site (27). The sensitivity of our assay was determined by diluting DNA with the indicated mutations at position E138 in wild-type DNA (Fig. 1) and in other common E138 mutants (see Fig. S1 in the supplemental material). We also confirmed that sequencing the amplicon of the E138K AS-PCR revealed linked mutations with sensitivity between 0.1 and 0.01% (see Fig. S2 in the supplemental material).
Fig 1.

Summary of empirically determined AS-PCR sensitivities (%). The final concentration of DNA was 108 amplicons per μl or 109 amplicons per reaction. The data are representative of the means ± the standard errors of the mean (SEM) of two independent trials performed in triplicate. (A to F) Mutant PCR amplicons diluted in wild-type PCR amplicons were measured by AS-PCR. The dotted line shows equivalence between actual proportion and observed proportion. NA, the PCR with the AS-primer failed to amplify. (A) E138A serially diluted in E138; (B) E138G serially diluted in E138; (C) E138K serially diluted in E138; (D) E138Q serially diluted in E138; (E) E138R serially diluted in E138; (F) E138V serially diluted in E138. (G) Numerical summary of empirically determined AS-PCR sensitivities. The intra-assay coefficient of variation ranged from 0.01 to 0.2, while the interassay coefficient of variation ranged from 0.1 to 0.4. Linearity below the proportion of 0.01% was not determined; therefore, 0.01% is the minimum sensitivity reported. Sensitivities were confirmed using RT-PCR amplicons from viruses obtained from HEK293T cell transfection.
E138K emerges first during ETR selection experiments.
We performed cell culture selection experiments with ETR using patient derived subtype B and C viruses. Four viruses had mutations in primer-binding sites, and such mutations are known to impact the observed proportion and sensitivity of AS-PCR methods (28, 36). By comparing the observed proportion of points at which E138K was measurable by bulk sequencing, we validated the assay and estimated the approximate fold decrease in the observed proportion of viruses with mutations in the primer binding sites (Fig. 2).
Fig 2.

Allele-specific PCR analysis of E138 substitutions selected in viral isolates from acute HIV-1 infections harboring wild-type and G190A viral quasispecies under etravirine pressure. Raw data from the AS-PCR analysis of E138A/G/K/Q are presented. These selections were also screened for E138R and E138V minority species by AS-PCR; however, E138R and E138V were never present at levels above the sensitivity of the assay. The shaded areas indicate time points at which E138K was measurable by bulk sequencing. Open symbols indicate proportions of mutated viruses at levels below AS-PCR sensitivity. Closed symbols show the proportions of mutants above the levels of AS-PCR sensitivity. (A) Etravirine selection experiments performed in CBMCs using viral samples taken from the plasma of HIV-1 subtype B infected, drug-naive patients. (B) Etravirine selection experiments performed in CBMCs with viral samples taken from the plasma of an HIV-1 subtype C-infected, drug-naive patient. (C) Approximate decrease in observed proportion caused by primer-binding site mismatches, determined by comparing the observed proportion of E138K at points when E138K was measurable by both AS-PCR and bulk sequencing. The sequences are arranged from 5′ to 3′.
Analysis of these ETR selections by AS-PCR also revealed the dynamic process of E138 substitution selection (Fig. 2). E138K consistently emerged first for each particular experiment. The higher limit of detection for E138G combined with primer-binding site mismatches prevented the detection of E138G above the technical limit of detection in most of these selections; nonetheless, the trend is clear that E138G also emerged very early above background measurements. In one case, an E138A minority transiently appeared shortly after E138K and E138G. In two instances, E138Q minority species increased after an accumulation of the E138K substitution. Interestingly, even though E138R conferred the highest level of resistance to ETR (6, 8, 10), this substitution did not appear above the limit of detection during the experiments. Of note, the presence of G190A, a common first-generation NNRTI resistance mutation as an initial background mutation (37), did not appear to influence the evolution of E138 minority species. The consistently early selection of E138K and E138G indicated that there is a mutational bias for these substitutions. We also analyzed the genetic linkage of E138K during these selection experiments by AS-PCR (see Table S2 in the supplemental material); there was no indication that E138K emerged on hypermutated viruses in these selection experiments.
Prevalence of substitutions at position E138 by bulk sequencing, AS-PCR, and UDS in plasma samples obtained from patients prior to the availability of ETR and RPV.
We next evaluated patients by bulk sequencing, AS-PCR, and UDS for the historic prevalence of E138 substitutions at times prior to the clinical availability of ETR and RPV. By bulk sequencing, we evaluated 1725 viral sequences obtained prior to 2008 from the plasma of different subtype B-infected patients enrolled in the Quebec Drug Resistance Genotyping Program and the Montreal PHI Cohort Study (Fig. 3A). Only E138A and E138G were identified in drug-naive patients by bulk sequencing.
Fig 3.
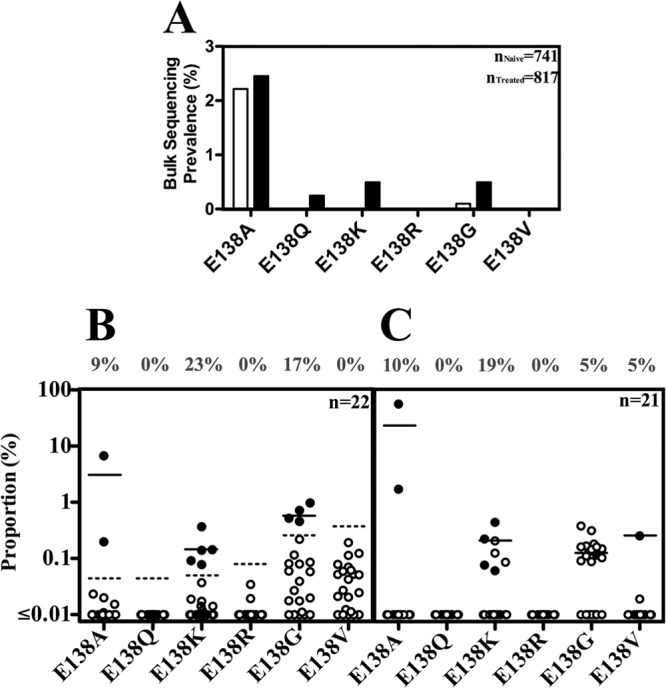
Prevalence of majority and minority E138 species in drug-naive patients prior to 2008. (A) Prevalence of E138 species identified by bulk sequencing. Closed bars represent drug-experienced patients, and open bars represent drug-naive patients. Results for patients with unknown treatment status are not shown. (B) Prevalence of minority E138 species in 22 patients measured by AS-PCR, as confirmed in two or three independent experiments. The dotted line represents the AS-PCR limit of detection for a specified substitution, taking into account a minimum limit of detection for patients based on viral load. (C) Prevalence of minority E138 species in the same patients as in panel B above, except that one patient, due to plasma availability, was also monitored by 454 UDS. Open circles indicate values below the sensitivity of the assay. Closed circles represent values above the sensitivity of the assay; for UDS, this meant a level significantly higher than that of background PCR mutations (P < 0.05, Fisher exact test). The solid line indicates the mean proportion of minority species detected at levels above the sensitivity of the assay.
We then used our AS-PCR to evaluate E138 minority species in samples obtained prior to 2008 from subtype B drug-naive persons with a high viral load (>150,000). This was done to increase the sensitivity of the assay. Due to these conditions, as well as sample availability, we were limited to analyzing only 22 patients. We found that E138K was the most prevalent minority species, found surprisingly in 23% (5/22) of these patients (Fig. 3B). E138G and E138A were also found at a high prevalence of 18% (4/22) and 9% (2/22), respectively, whereas E138Q, E138R, and E138V were never detected.
We confirmed these results by UDS, although one patient identified by AS-PCR as possessing E138K, was not analyzed due to lack of plasma availability. Identifying mutations at levels below the 1% proportion by UDS was achieved by statistically evaluating each mutation on a site and mutation-type specific basis. We were able to confirm the high prevalence of E138K at 19% (4/21), as well as E138A at 10% (2/21) (Fig. 3C). The high prevalence of E138G substitutions could not be confirmed due to a high UDS background for this specific mutation. In addition, a patient with an E138V minority was also identified.
E138K is one of the least fit substitutions at codon E138.
The early selection and high prevalence of E138K minority species could be rationalized if E138K had a low fitness cost relative to other substitutions at codon E138. Competition experiments with E138K-containing viruses competed against viruses containing E138A, E138G, or E138Q have been performed previously (38). Paradoxically, E138K was found to be more fit than E138A and E138Q, despite the fact that E138A is fit enough to be a common polymorphism, whereas E138K is not. To confirm this finding, and also to test E138R, we performed competition experiments using viruses that contained E138A, E138G, E138K, E138Q, and E138R. We capitalized on the ability of the AS-PCR method to distinguish among these substitutions by competing all viruses together in an internally controlled experiment (Fig. 4A). Consistent with the majority-species prevalence of E138A in drug-naive patients, we found that E138A and E138G were the most-fit substitutions, followed by E138R; the least-fit substitutions were E138K and E138Q.
Fig 4.

Competition experiments with mutations at codon E138 in MT-2 cells monitored by AS-PCR. The data are representative of means ± the SEM of three independent experiments. (A to D) Results of competition experiments using E138A, E138G, E138K, E138Q, and E138R mutations. (A) Competition experiments performed without altering the dNTP concentration. (B) 1.5 mM hydroxyurea (HU) was added to the medium. (C) 0.65 mM deoxynucleosides (dN) were added to the medium. (D) Fold change in the proportions of individual substitutions at the end of the competition experiment. Bars: ▧, fold change with the addition of HU; □, fold change with the addition of deoxynucleosides. (E to H) Results of competition experiments using E138K virus and wild type. Lines: solid line, wild type; dashed line, E138K. “1 + s” is the relative viral fitness with the SEM noted. (E) Competition experiment performed without altering the dNTP concentration. (F) 1.5 mM hydroxyurea (HU) was added to the medium. (G) 0.65 mM deoxynucleoside (dN) was added to the medium. (H) Both 1.5 mM HU and 0.65 mM dN were added to the medium. Note that this combination caused a minor decrease in MT-2 cell replication.
dNTP pool size affects E138 substitution relative fitness.
We have previously shown that some E138 substitutions have a deoxynucleoside triphosphate (dNTP) concentration-dependent effect on RT kinetics in vitro (10, 39, 40). We now assessed whether modified dNTP concentrations in vivo may permit E138K to become the most fit substitution at codon E138 (Fig. 4B to H). Using hydroxyurea (HU), an inhibitor of ribonucleotide reductase, to lower the dNTP concentrations of MT-2 cells resulted in a modest increase in the fitness of viruses containing E138K and E138Q relative to E138A, E138G, and E138R (Fig. 4B and D). Conversely, using deoxynucleosides (dNs) to exogenously raise the dNTP concentration of the cells resulted in E138G gaining a strong fitness advantage over the other substitutions at codon E138 (Fig. 4C and D). Under the conditions tested, E138K was consistently one of the least fit substitutions at codon E138. E138K virus was also competed against wild-type virus under similar conditions (Fig. 4E to H); E138K also had modestly improved relative fitness with the addition of HU but was nonetheless consistently less fit than wild-type virus.
Small minority species correlates to HIV-1 mutational bias.
Considerations of fitness and drug resistance cannot explain the early and preferential selection of E138K; a high forward mutation rate for this mutation may be the best rationale for this phenomenon. The high prevalence of small E138K minority species in patients might also be due to HIV-1 mutation rate and bias. Further UDS analysis revealed the pattern of small minority species was consistent with that of HIV-1 mutational bias during single cycle replication experiments (Fig. 5A) (12). The prevalence in patients of any particular G→A mutation was ca. 15%, usually as a tiny (<1%) minority. This indicates that the high prevalence of E138K as a small minority is not specific to E138K, and can be explained by the fact that it is a G→A mutation.
Fig 5.
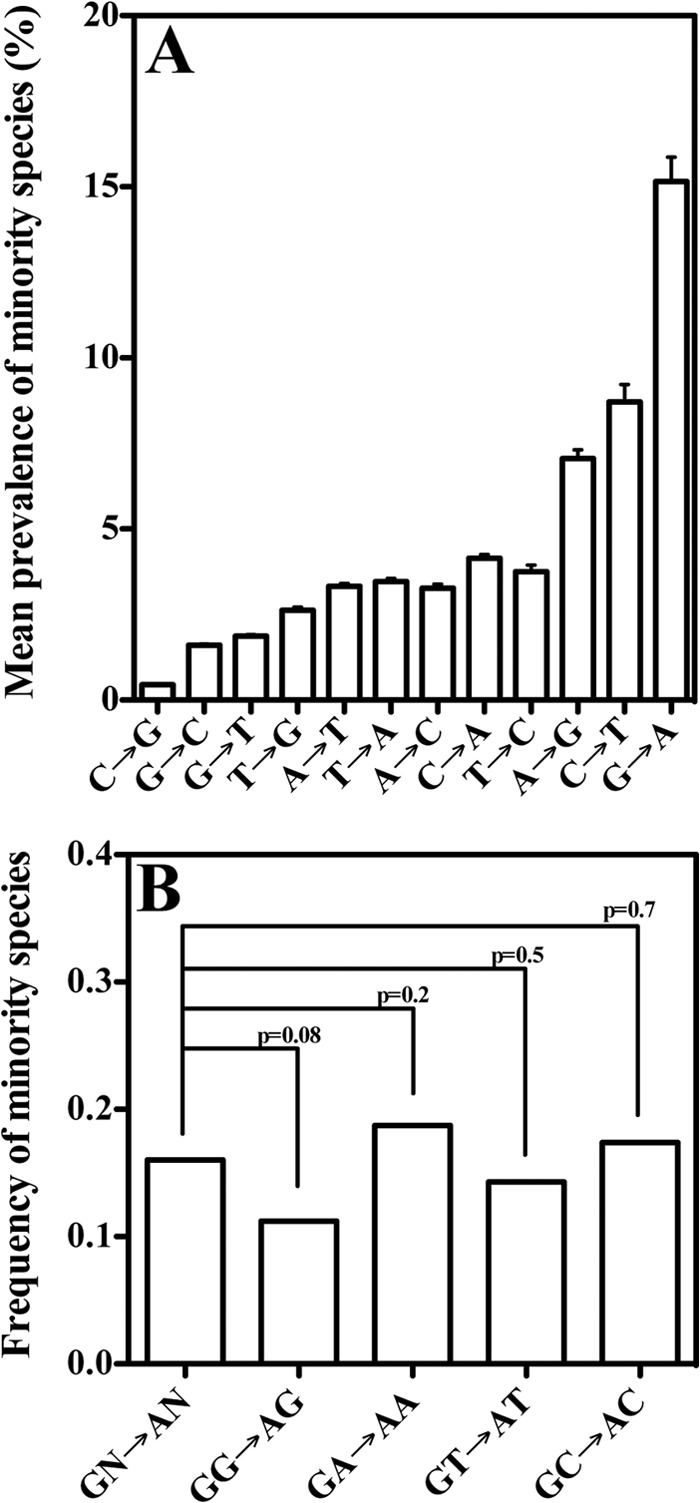
UDS revealed a prevalence of minority species per available nucleotide site in HIV pol (HXB2 2862→3131) in 21 drug-naive patients. (A) Mean prevalence of minority species (<5% proportion) stratified on the basis of mutational type. The prevalences of minority species per patient were calculated and then averaged together. Error bars represent standard deviations. (B) Frequency of A minority species at sites that were predominated by G, stratified on the basis of dinucleotide context. Frequencies were compared by Fisher exact test, with the P values noted.
Despite the fact that in vitro RT preferentially introduces G→A mutations during positive- and negative-sense DNA polymerization, positive-sense G→A mutations were significantly higher than positive-sense C→T mutations in the present study. This indicates that RT exhibits a slightly different mutational bias for RNA- and DNA-dependent DNA polymerization, as suggested previously (12), or that a host factor is responsible for additional positive-sense G→A mutations sufficient to impact the mutational bias.
Genetic linkage of E138K in drug-naive patients.
We next investigated whether these frequent G→A minority species were hypermutated, which would indicate their emergence was due to APOBEC3 activity. Sequencing the amplicons in the AS-PCR assay from drug-naive patients to reveal mutations linked to E138K yielded no indication that E138K was linked to hypermutated viruses (see Table S3 in the supplemental material). Neither did UDS analysis reveal any linkage of E138K to hypermutated viruses; indeed, there were no hypermutated viruses at all (Hypermut 2.0 Program, P <0.05). It is possible that most reactivated APOBEC3-hypermutated viruses fail to replicate sufficiently to be detectable by UDS. In this case, hypermutation, followed by early recombination, could enrich G→A minority species in specific dinucleotide contexts (41). Although this would provide indirect evidence of APOBEC3 activity, no significant enrichment of G→A mutations occurred (Fig. 5B). This questions whether APOBEC3 really does play a role in regard to the overall HIV-1 mutational bias for G→A mutations, such as E138K.
DISCUSSION
Using an AS-PCR assay and UDS to analyze ETR selection experiments and drug-naive patients has yielded novel information. To investigate substitutions at codon E138, we developed an AS-PCR method that distinguished among six substitutions at a single codon. This was accomplished by building on archetypal AS-PCR primers, which are specific for one nucleotide at the 3′ ultimate position (22, 23). In addition, we adapted a previously published technique to study linked mutations following AS-PCR (24) in order to identify E138K mutational combinations with high sensitivity. These assays have diagnostic applications, given that the E138K/M184I substitution combination was identified in the majority of virologic failures in the ECHO and THRIVE trials in which RPV was used together with two nucleoside RT inhibitors to treat previously drug-naive patients (2).
Investigating the evolution of E138 substitutions under ETR drug pressure has revealed the early selection of E138K over other E138 substitutions. Since each of the E138K, E138G, and E138A substitutions confer ETR resistance and it appeared at times that E138 still dominated the quasispecies, one might assume that their order of appearance is proportional to their mutational bias. Conversely, E138Q was selected only after E138K first became a majority species, suggesting that the E138K substitution followed by the K138Q substitution (G→A→C) may have a higher likelihood for occurrence than the E138Q (G→C) substitution, a possibility consistent with the mutational bias of HIV-1 that favors G→A or A→C mutations over G→C mutations (12). Although E138R conferred the highest levels of resistance to ETR of all the E138 substitutions, this substitution was not observed as a minority species; this is consistent with the fact that the generation of E138R requires two instead of only one nucleotide mutation. In summary, these findings are consistent with the following relative mutational bias: E138K ≤ E138G < E138A ≤ E138Q < E138R. Considering that E138K is one of the least fit E138 substitutions and also offers one of the lowest levels of NNRTI resistance, its preferential selection can best be explained by the HIV-1 mutational bias.
Although this bias is thought to be the result of both an innate G→A bias during viral replication and APOBEC3 cytosine deaminase enzymes, we could not find any evidence of APOBEC3-hypermutated viruses in viral RNA during ETR selections or in subtype B-infected drug-naive patients. That said, we were recently able to identify a subtype C patient with an obviously hypermutated E138K minority species by AS-PCR (see Table S5 in the supplemental material), showing that hypermutated viruses could be detected in plasma if they were actually present. Likewise, if APOBEC3 enzymes played a major role in the emergence of E138K, it would be expected that differences in the emergence and prevalence of E138K and the E138G minorities would be more dramatic.
The fact that E138G and E138K were both selected early and frequently identified in drug-naive patients helps to explain why both were the most common E138 substitutions in treatment failure during clinical trials with RPV, despite the fact that E138G only confers low-level RPV resistance (2, 8). A different study indicated that E138K was selected over E138G by TSAO derivatives—a group of NNRTIs that have not been approved for clinical use—primarily because of the mutational bias of HIV-1. Using exogenously provided deoxycytidine (dC) and tetrahydrouridine (THU) to artificially increase the concentration of dCTPs in cells lead to the selection of E138G instead of E138K by TSAO derivatives (42). This was interpreted to be the result of an augmented dCTP/dTTP ratio that skewed the RT mutational bias. However, we show here that E138G often emerges alongside E138K, and that high dNTP concentrations provide E138G with a strong fitness advantage. We hope to test whether dC and THU augments the fitness of E138G over E138K as opposed to altering the mutational bias of HIV-1.
The prevalence of minority species identified here is unexpectedly high. However, this finding does not likely represent an artifact of PCR, since we specifically controlled for this during the AS-PCR optimization and during UDS analysis. Likewise, the prevalence of minority species revealed by UDS was consistent with the HIV-1 mutational bias and was very distinct from that of the Taq polymerase mutational bias (43). Furthermore, this is not the first study to note unfit very small minority species in patients who are not under drug pressure (28, 44, 45). Nonetheless, our findings serve as a warning to interpret with caution the discovery of minority drug resistance mutations. The presence of E138A minority species at the start of some of the ETR selection experiments shows that not every detectable minority species will be positively selected under drug pressure, since most circulating viruses are not infectious (46, 47).
On the other hand, the frequent identification of minority species appears to reflect a mutational bias that could favor the selection of a particular mutation. For example, we previously identified a genetic template bias for the selection of K65R in subtype C compared to subtype B (48). Likewise, 9% of drug-naive subtype C-infected patients harbor tiny K65R minority species, while this is true for only 2% of subtype B-infected patients (44). Consequently, a relatively high number of subtype C patients have failed first-line therapy with the K65R mutation (49, 50). The fact that G→A mutations were the most common minority species detected here, combined with the fact that G→A mutations are the most common type of HIV-1 drug resistance mutation, further underlines the critical role that mutational bias plays in the evolution of clinically relevant HIV-1 drug resistance.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Sloan for reviewing the manuscript, D. A. Donahue for helpful discussions, S. Germinario and C. Collazos for technical assistance, and B. Spira for managerial aid.
This study was supported by the Canadian Institutes of Health Research. M.M. is the recipient of a 2012-2013 Fonds de la Recherche en Santé du Québec Master's Fellowship and a McGill University Max Stern Recruitment Fellowship.
Footnotes
Published ahead of print 15 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01029-13.
REFERENCES
- 1.Johnson VA, Calvez V, Günthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. 2011. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 19:156–164 [PMC free article] [PubMed] [Google Scholar]
- 2.Rimsky L, Vingerhoets J, Van Eygen V, Eron J, Clotet B, Hoogstoel A, Boven K, Picchio G. 2012. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J. Acquir. Immune Defic. Syndr. 59:39–46 [DOI] [PubMed] [Google Scholar]
- 3.Tambuyzer L, Vingerhoets J, Azijn H, Daems B, Nijs S, de Béthune M-P, Picchio G. 2010. Characterization of genotypic and phenotypic changes in HIV-1-infected patients with virologic failure on an etravirine-containing regimen in the DUET-1 and DUET-2 clinical studies. AIDS Res. Hum. Retrovir. 26:1197–1205 [DOI] [PubMed] [Google Scholar]
- 4.Schader SM, Oliveira M, Ibanescu R-I, Moisi D, Colby-Germinario SP, Wainberg MA. 2012. In vitro resistance profile of the candidate HIV-1 microbicide drug dapivirine. Antimicrob. Agents Chemother. 56:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherrer AU, Hasse B, von Wyl V, Yerly S, Boni J, Burgisser P, Klimkait T, Bucher HC, Ledergerber B, Gunthard HF. 2009. Prevalence of etravirine mutations and impact on response to treatment in routine clinical care: the Swiss HIV Cohort Study (SHCS). HIV Med. 10:647–656 [DOI] [PubMed] [Google Scholar]
- 6.Tambuyzer L, Nijs S, Daems B, Picchio G, Vingerhoets J. 2011. Effect of mutations at position E138 in HIV-1 reverse transcriptase on phenotypic susceptibility and virologic response to etravirine. J. Acquir. Immune Defic. Syndr. 58:18–22 [DOI] [PubMed] [Google Scholar]
- 7.Asahchop EL, Oliveira M, Wainberg MA, Brenner BG, Moisi D, Toni TD, Tremblay CL. 2011. Characterization of the E138K resistance mutation in HIV-1 reverse transcriptase conferring susceptibility to etravirine in B and non-B HIV-1 subtypes. Antimicrob. Agents Chemother. 55:600–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azijn H, Tirry I, Vingerhoets J, de Bethune MP, Kraus G, Boven K, Jochmans D, Van Craenenbroeck E, Picchio G, Rimsky LT. 2010. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob. Agents Chemother. 54:718–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asahchop EL, Wainberg MA, Oliveira M, Xu H, Brenner BG, Moisi D, Ibanescu IR, Tremblay C. 2013. Distinct resistance patterns to etravirine and rilpivirine in viruses containing nonnucleoside reverse transcriptase inhibitor mutations at baseline. AIDS 27:879–887 [DOI] [PubMed] [Google Scholar]
- 10.Xu HT, Colby-Germinario SP, Asahchop EL, Oliveira M, McCallum M, Schader SM, Han Y, Quan Y, Sarafianos SG, Wainberg MA. 2013. Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob. Agents Chemother. 57:3100–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. 2010. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 84:9864–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez MA, Vartanian JP, Wain-Hobson S. 1994. Hypermutagenesis of RNA using human immunodeficiency virus type 1 reverse transcriptase and biased dNTP concentrations. Proc. Natl. Acad. Sci. U. S. A. 91:11787–11791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulder LC, Harari A, Simon V. 2008. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. U. S. A. 105:5501–5506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 1:e6. 10.1371/journal.ppat.0010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fourati S, Malet I, Lambert S, Soulie C, Wirden M, Flandre P, Fofana DB, Sayon S, Simon A, Katlama C, Calvez V, Marcelin AG. 2012. E138K and M184I mutations in HIV-1 reverse transcriptase coemerge as a result of APOBEC3 editing in the absence of drug exposure. AIDS 26:1619–1624 [DOI] [PubMed] [Google Scholar]
- 17.Turner D, Brenner B, Mosis D, Liang C, Wainberg MA. 2005. Substitutions in the reverse transcriptase and protease genes of HIV-1 subtype B in untreated individuals and patients treated with antiretroviral drugs. MedGenMed 7:69. [PMC free article] [PubMed] [Google Scholar]
- 18.Deforche K, Camacho R, Laethem KV, Shapiro B, Moreau Y, Rambaut A, Vandamme AM, Lemey P. 2007. Estimating the relative contribution of dNTP pool imbalance and APOBEC3G/3F editing to HIV evolution in vivo. J. Comput. Biol. 14:1105–1114 [DOI] [PubMed] [Google Scholar]
- 19.Buckton AJ, Harris RJ, Pillay D, Cane PA. 2011. HIV type-1 drug resistance in treatment-naive patients monitored using minority species assays: a systematic review and meta-analysis. Antivir. Ther. 16:9–16 [DOI] [PubMed] [Google Scholar]
- 20.Halvas EK, Aldrovandi GM, Balfe P, Beck IA, Boltz VF, Coffin JM, Frenkel LM, Hazelwood JD, Johnson VA, Kearney M, Kovacs A, Kuritzkes DR, Metzner KJ, Nissley DV, Nowicki M, Palmer S, Ziermann R, Zhao RY, Jennings CL, Bremer J, Brambilla D, Mellors JW. 2006. Blinded, multicenter comparison of methods to detect a drug-resistant mutant of human immunodeficiency virus type 1 at low frequency. J. Clin. Microbiol. 44:2612–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paredes R, Clotet B. 2008. HIV drug resistance testing. Eur. Infect. Dis. 2:52–62 [Google Scholar]
- 22.Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham AF. 1989. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17:2503–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cha RS, Zarbl H, Keohavong P, Thilly WG. 1992. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. Genome Res. 2:14–20 [DOI] [PubMed] [Google Scholar]
- 24.Johnson JA, Li JF, Wei X, Lipscomb J, Bennett D, Brant A, Cong ME, Spira T, Shafer RW, Heneine W. 2007. Simple PCR assays improve the sensitivity of HIV-1 subtype B drug resistance testing and allow linking of resistance mutations. PLoS One 2:e638. 10.1371/journal.pone.0000638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliveira M, Moisi D, Spira B, Cox S, Brenner BG, Wainberg MA. 2009. Apricitabine does not select additional drug resistance mutations in tissue culture in human immunodeficiency virus type 1 variants containing K65R, M184V, or M184V plus thymidine analogue mutations. Antimicrob. Agents Chemother. 53:1683–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuiken C, Foley B, Leitner T, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B. 2010. HIV sequence compendium 2010 Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM [Google Scholar]
- 28.Paredes R, Marconi VC, Campbell TB, Kuritzkes DR. 2007. Systematic evaluation of allele-specific real-time PCR for the detection of minor HIV-1 variants with pol and env resistance mutations. J. Virol. Methods 146:136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. 2011. Accurate sampling and deep sequencing of the HIV-1 protease gene using PrimerID. Proc. Natl. Acad. Sci. U. S. A. 108:20166–20171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38:W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum-parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma J, Dykes C, Wu T, Huang Y, Demeter L, Wu H. 2010. vFitness: a web-based computing tool for improving estimation of in vitro HIV-1 fitness experiments. BMC Bioinformatics 11:261. 10.1186/1471-2105-11-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auwerx J, Van Nieuwenhove J, Rodríguez-Barrios F, de Castro S, Velázquez S, Ceccherini-Silberstein F, De Clercq E, Ma Camarasa J, Perno C-F, Gago F, Balzarini J. 2005. The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes of anti-HIV-1 drugs. FEBS Lett. 579:2294–2300 [DOI] [PubMed] [Google Scholar]
- 35.Mulky A, Vu BC, Conway JA, Hughes SH, Kappes JC. 2007. Analysis of amino acids in the beta7-beta8 loop of human immunodeficiency virus type 1 reverse transcriptase for their role in virus replication. J. Mol. Biol. 365:1368–1378 [DOI] [PubMed] [Google Scholar]
- 36.Palmer S, Boltz V, Maldarelli F, McIntyre J, Morris L, Hopley M, Mayers D, Robinson P, Mellors J, Coffin J. 2006. Optimization of allele-specific PCR for drug-resistant HIV variants using patient-specific consensus sequences for primer design. 13th Conference on Retroviruses and Opportunistic Infections, Denver, CO [Google Scholar]
- 37.Llibre JM, Santos JR, Puig T, Molto J, Ruiz L, Paredes R, Clotet B. 2008. Prevalence of etravirine-associated mutations in clinical samples with resistance to nevirapine and efavirenz. J. Antimicrob. Chemother. 62:909–913 [DOI] [PubMed] [Google Scholar]
- 38.Pelemans H, Aertsen A, Van Laethem K, Vandamme AM, De Clercq E, Pérez-Pérez MJ, San-Félix A, Velázquez S, Camarasa MJ, Balzarini J. 2001. Site-directed mutagenesis of human immunodeficiency virus type 1 reverse transcriptase at amino acid position 138. Virology 280:97–106 [DOI] [PubMed] [Google Scholar]
- 39.Xu HT, Oliveira M, Quashie PK, McCallum M, Han Y, Quan Y, Brenner BG, Wainberg MA. 2012. Subunit-selective mutational analysis and tissue culture evaluations of the interactions of the E138K and M184I mutations in HIV-1 reverse transcriptase. J. Virol. 86:8422–8431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol. 85:11300–11308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armitage AE, Katzourakis A, de Oliveira T, Welch JJ, Belshaw R, Bishop KN, Kramer B, McMichael AJ, Rambaut A, Iversen AK. 2008. Conserved footprints of APOBEC3G on hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J. Virol. 82:8743–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balzarini J, Camarasa MJ, Perez-Perez MJ, San-Felix A, Velazquez S, Perno CF, De Clercq E, Anderson JN, Karlsson A. 2001. Exploitation of the low fidelity of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase and the nucleotide composition bias in the HIV-1 genome to alter the drug resistance development of HIV. J. Virol. 75:5772–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bracho MA, Moya A, Barrio E. 1998. Contribution of Taq polymerase-induced errors to the estimation of RNA virus diversity. J. Gen. Virol. 79(Pt 12):2921–2928 [DOI] [PubMed] [Google Scholar]
- 44.Kozal MJ, Chiarella J, St John EP, Moreno EA, Simen BB, Arnold TE, Lataillade M. 2011. Prevalence of low-level HIV-1 variants with reverse transcriptase mutation K65R and the effect of antiretroviral drug exposure on variant levels. Antivir. Ther. 16:925–929 [DOI] [PubMed] [Google Scholar]
- 45.Hedskog C, Mild M, Jernberg J, Sherwood E, Bratt G, Leitner T, Lundeberg J, Andersson B, Albert J. 2010. Dynamics of HIV-1 quasispecies during antiviral treatment dissected using ultra-deep pyrosequencing. PLoS One 5:e11345. 10.1371/journal.pone.0011345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bourinbaiar AS. 1994. The ratio of defective HIV-1 particles to replication-competent infectious virions. Acta Virol. 38:59–61 [PubMed] [Google Scholar]
- 47.Kwon YJ, Hung G, Anderson WF, Peng CA, Yu H. 2003. Determination of infectious retrovirus concentration from colony-forming assay with quantitative analysis. J. Virol. 77:5712–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coutsinos D, Invernizzi CF, Xu H, Moisi D, Oliveira M, Brenner BG, Wainberg MA. 2009. Template usage is responsible for the preferential acquisition of the K65R reverse transcriptase mutation in subtype C variants of human immunodeficiency virus type 1. J. Virol. 83:2029–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sunpath H, Wu B, Gordon M, Hampton J, Johnson B, Moosa MY, Ordonez C, Kuritzkes DR, Marconi VC. 2012. High rate of K65R for antiretroviral therapy-naive patients with subtype C HIV infection failing a tenofovir-containing first-line regimen. AIDS 26:1679–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Theys K, Vercauteren J, Snoeck J, Zazzi M, Camacho RJ, Torti C, Schulter E, Clotet B, Sonnerborg A, De Luca A, Grossman Z, Struck D, Vandamme AM, Abecasis AB. 2013. HIV-1 subtype is an independent predictor of reverse transcriptase mutation K65R in HIV-1 patients treated with combination antiretroviral therapy including tenofovir. Antimicrob. Agents Chemother. 57:1053–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.