

Abstract
Faldaprevir (BI 201335) is a selective NS3/4A protease inhibitor under development for the treatment of chronic hepatitis C virus (HCV) infection. NS3/4A genotyping and NS3 protease phenotyping analyses were performed to monitor the emergence of resistance in patients with HCV genotype 1 infection receiving faldaprevir alone or combined with pegylated interferon alfa 2a and ribavirin (PegIFN-RBV) during a phase 1b study. Among all baseline variants, a maximum 7-fold reduction in in vitro sensitivity to faldaprevir was observed for a rare NS3 (V/I)170T polymorphism. During faldaprevir monotherapy in treatment-naive patients, virologic breakthrough was common (77%, 20/26) and was associated with the emergence of resistance mutations predominantly carrying NS3 substitutions R155K in GT1a and D168V in GT1b. D168V conferred a greater reduction in faldaprevir sensitivity (1,800-fold) than R155K (330-fold); however, D168V was generally less fit than R155K in the absence of selective drug pressure. Treatment-experienced patients treated with faldaprevir-PegIFN-RBV triple therapy showed higher viral load reductions, lower rates of breakthrough (8%, 5/62), and less frequent emergence of resistance-associated variants compared with faldaprevir monotherapy. (This study has been registered at ClinicalTrials.gov under registration no. NCT00793793.)
INTRODUCTION
Worldwide prevalence of hepatitis C virus (HCV) infection is approximately 170 million, and since 2007, HCV has surpassed HIV as a cause of death in the United States (1). Chronically infected individuals with liver disease require effective, well-tolerated therapies that offer an improvement over the previous standard of care of pegylated interferon alfa (PegIFN) and ribavirin (RBV). The HCV-carried nonstructural NS3/4A protease is essential for viral replication and was one of the first clinically validated drug targets (2–4). The current NS3/4A inhibitors used in clinical practice are telaprevir and boceprevir, which form a reversible covalent bond with NS3 (5–8). Combined with PegIFN-RBV, telaprevir or boceprevir significantly increase sustained virologic response rates compared with those of PegIFN-RBV therapy alone in patients with chronic HCV genotype 1 (GT1) infection. These triple-drug combination regimens are now considered the current standard of care (9, 10).
HCV has a high replication rate and a high mutation frequency during viral RNA replication (11), leading to the evolution of multiple subpopulations, some of which have amino acid substitutions in the NS3/4A protein that may confer resistance to protease inhibitors. During antiviral therapy with NS3/4A protease inhibitors, treatment failure has been associated with the emergence of resistant variants (12–14). Compounds that are distinct from telaprevir and boceprevir and that inhibit NS3/4A solely through noncovalent interactions with the catalytic site are in advanced clinical trials. These include faldaprevir (BI 201335), a selective linear tripeptide (15) that inhibits HCV RNA replication in vitro with 50% effective concentrations (EC50s) of 6.5 and 3.1 nM against HCV GT1a and GT1b, respectively (16). In vitro studies with faldaprevir showed that NS3 R155K was the predominant resistant variant selected in GT1a, whereas substitutions at D168 were observed mostly in GT1b (17). The NS3 R155K variant is common to all classes of NS3/4A protease inhibitors (12–14, 17, 18). By contrast, changes at V36 and T54, which confer resistance to telaprevir and boceprevir, were not associated with resistance to faldaprevir in vitro (17). The NS3 D168V variant confers resistance to the macrocyclic peptidomimetic class of NS3/4A protease inhibitors, such as simeprevir, and to faldaprevir (12, 13, 17, 18). Faldaprevir antiviral activity was first evaluated in a phase 1b clinical trial (trial identifier NCT00793793) (19), and we report here the results of NS3/4A genotyping and NS3 phenotyping analyses to monitor the emergence of resistance during treatment with faldaprevir. Furthermore, we also describe the on-treatment virologic response to faldaprevir in additional cohorts of the phase 1b study that have not previously been reported.
MATERIALS AND METHODS
Patients.
The 1220.2 phase 1b study (trial identifier NCT00793793) was a randomized, multicenter, multiple-rising-dose trial of faldaprevir in treatment-naive (TN) or treatment-experienced (TE) patients chronically infected with HCV GT1. Patient inclusion and exclusion criteria have been described previously (19), and following a protocol amendment, additional cohorts of TE patients with or without compensated liver cirrhosis (Child-Pugh A) were included in the study.
The trial protocol and supporting documentation were submitted to the independent ethics committee responsible for the trial center of the coordinating investigator. The trial was performed in compliance with the protocol, the Declaration of Helsinki (1996 version), the International Committee on Harmonization (ICH) Harmonized Tripartite Guideline for Good Clinical Practice (GCP), and applicable regulatory requirements. Prior to participation in the trial, written informed consent was obtained from each patient according to the ICH-GCP.
Study treatments.
TN patients without cirrhosis were randomized to receive, in successive cohorts, faldaprevir monotherapy (20, 48, 120, and 240 mg once daily [QD]) for 14 days as a powder-in-bottle (PiB) oral solution or placebo (Fig. 1). In patients with an HCV RNA decrease of ≥1 log10 from baseline (on day 10), faldaprevir was combined with PegIFN-α-2a (180 μg/week) and weight-based RBV (1,000 or 1,200 mg/day) from days 14 to 28. TE patients without cirrhosis received faldaprevir PiB oral solution, at 48, 120, or 240 mg QD, and PegIFN-RBV for 28 days (Fig. 1). The additional cohorts of TE patients with or without cirrhosis that were added after a protocol amendment received 240 mg faldaprevir QD or twice daily (BID) as a soft gel capsule (SGC) and PegIFN-RBV for 28 days. All patients were invited to extend PegIFN-RBV to week 48, with an additional 24 weeks of follow-up at the discretion of the investigator. Initial efficacy, tolerability, and pharmacokinetic results for faldaprevir PiB oral solution with and without PegIFN-RBV have been reported previously (19).
Fig 1.
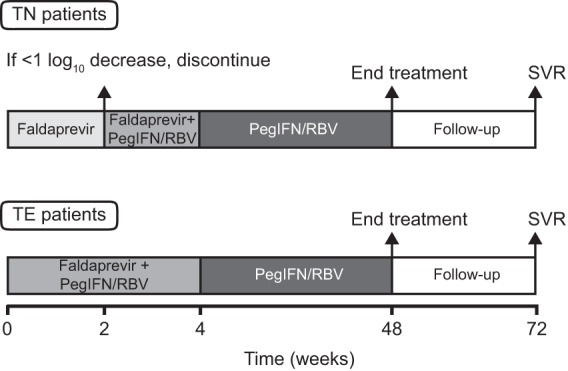
Trial design. PegIFN-RBV, pegylated interferon α-2a and ribavirin; SVR, sustained virologic response; TE, treatment experienced; TN, treatment naive.
Viral RNA extraction and PCR amplification.
Viral RNA was isolated from patient plasma samples at baseline, at indicated time points during treatment, and during follow-up using the QIAamp viral RNA extraction kit (Qiagen). A DNA amplicon of 2.4-kbp containing the complete NS3/4A region was synthesized using the Superscript III one-step reverse transcription-PCR (RT-PCR) system (Invitrogen). After purification of the first RT-PCR product, a 2.3-kbp product spanning the entire NS3/4A domain or a 0.7-kbp product containing only the NS3 protease domain was generated using KOD Hot Start DNA polymerase (Novagen). The analysis was restricted to samples with viral loads (VLs) of >1,000 IU/ml, which was the lower limit of amplification (LLOA) for the RT-PCR amplification method (data not shown) and is similar to the limit established by other methods (20).
Sequence and phylogenetic analyses.
Population and clonal sequencing of virus isolates was performed at baseline (day 1) for all patients and on days 14 and 28 for TN and TE patients, respectively, if VL was above the LLOA. If VL was below the LLOA at day 14 or 28, sequence analysis was performed if a VL increase was detected posttreatment. Clonal sequencing was performed at additional on-treatment and follow-up time points, to monitor the longitudinal evolution of resistant variants.
The NS3/4A amplicon (2.3 kbp) was used for population-based sequencing of the NS3/4A region using BigDye Terminator version 3.1 (Applied Biosystems) and an ABI Prism 3130XL Genetic Analyzer (Applied Biosystems). The resulting nucleotide sequences were analyzed with SeqScape version 2.5 (Applied Biosystems). The 0.7-kbp DNA fragment was used to generate the clone-based (ZeroBlunt TOPO cloning kit; Invitrogen) sequences of the 181 amino acids of the NS3 protease domain. For each sample, 96 clones were picked and sequenced using universal primers with ABI Prism BigDye Terminator cycle PCR sequencing (Applied Biosystems). Two single-pass sequences were generated for each NS3/4A or NS3 nucleotide region, resulting in 90 to 100% double-strand coverage analyzed with Mutation Surveyor version 3.0 (SoftGenetics).
HCV subtypes were determined by alignment of baseline NS3/4A sequences to reference sequences (NCBI GenBank accession number AF009606 for GT1a and AJ39799 for GT1b). All amino acid changes in baseline and postbaseline sequences were determined with respect to the reference. Postbaseline amino acid changes were compared with the respective baseline to identify the emergence of amino acid substitutions.
Drug sensitivity assay.
A bicistronic HCV replicon shuttle vector (pIT2) comprising a luciferase reporter gene and an adapted Con-1 NS3/NS5B region was modified to create two unique restriction sites (MluI and SpeI) at NS3 codons 11 and 225, which enabled the insertion of compatible NS3 amplicons from patient plasma samples. The first-step RT-PCR product (2.4 kbp of NS3/4A) was synthesized as described above and was used to amplify a 0.65-kbp DNA fragment with primer pairs encoding the MluI and SpeI restriction sites to facilitate unidirectional insertion into the shuttle vector. Amplicons were ligated in the shuttle vector, and reconstituted plasmid DNA was used to generate HCV subgenomic replicon RNA transcripts (T7 Ribomax kit; Promega, Mannheim, Germany). The in vitro-transcribed RNA was electroporated into Huh-7.5 cells which were cultured in 96-well plates, and after 24 h, serial dilutions of faldaprevir or IFN-α were added. HCV RNA replication was quantified by luciferase assays at 4 and 96 h postelectroporation. The EC50 for inhibition of HCV RNA replication was determined for all baseline samples and from faldaprevir on-treatment and posttreatment samples, including follow-up. For individual patient samples, EC50 values were determined from three or more independent experiments.
Statistical analyses.
The relationship between NS3 baseline polymorphisms and baseline susceptibility to faldaprevir was investigated using a Wilcoxon rank-sum test comparing EC50 values of baselines carrying a specific NS3 variant with those lacking the NS3 variant.
RESULTS
Viral load response.
Virologic breakthrough, plateau, and continuous VL decline responses during 14-day faldaprevir monotherapy in TN patients and during 28-day triple faldaprevir-PegIFN-RBV therapy were evaluated in a post hoc analysis (Table 1). The median reduction in VL during 14-day faldaprevir monotherapy ranged from 3.0 to 4.4 log10 depending on the dose of faldaprevir, and the rates of breakthrough, plateau, and continuous decline were 77% (20/26), 15% (4/26), and 7.7% (2/26), respectively. All TN patients given placebo experienced a negligible reduction in VL.
Table 1.
Virologic response during faldaprevir treatment by dose group and HCV subtyped
| Patient type | No. of patients | Dose | Dosage method | Median VL change from baseline (log10 IU/ml)a | No. of patients with a faldaprevir on-treatment virologic responsea |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Breakthrough |
Plateau |
Decline |
||||||||
| GT1a | GT1b | GT1a | GT1b | GT1a | GT1b | |||||
| TN | 8 | Placebo | PiB | −0.06b | 4/4 | 4/4 | ||||
| 6 | 20 mg QD | PiB | −3.0b | 3/3 | 2/3 | 1/3 | ||||
| 7 | 48 mg QD | PiB | −3.6b | 1/2 | 4/5 | 1/2 | 1/5 | |||
| 7 | 120 mg QD | PiB | −3.7b | 1/3 | 4/4 | 2/3 | ||||
| 6 | 240 mg QD | PiB | −4.4b | 3/3 | 2/3 | 1/3 | ||||
| TE | 6 | 48 mg QD | PiB | −5.0b | 1/3 | 1/3 | 2/3 | 2/3 | ||
| 7 | 120 mg QD | PiB | −5.2b | 1/4 | 1/4 | 2/4 | 3/3 | |||
| 6 | 240 mg QD | PiB | −5.3b | 4/4 | 2/2 | |||||
| 15 | 240 mg BID | SGC | −5.3 | 2/10 | 8/10 | 5/5 | ||||
| 15 | 240 mg QD | SGC | −5.2 | 2/9 | 7/9 | 6/6 | ||||
| TE-Cirr | 6 | 240 mg QD | SGC | −4.8 | 1/2 | 1/2 | 4/4 | |||
| 7 | 240 mg BID | SGC | −5.5 | 5/5c | 2/2 | |||||
Median VL change from baseline and virologic responses were evaluated during 14-day faldaprevir monotherapy for TN patients and 28-day faldaprevir-PegIFN-RBV therapy for TE patients. Virologic responses include breakthrough (VL increase of >0.8 log10 above nadir), plateau (≤0.8 log10 VL increase above nadir), and continuous decline (profiles not consistent with breakthrough or plateau).
Previously published results (19).
One GT1a patient terminated treatment early at day 4 and showed virologic decline.
BID, twice daily; Cirr, patients with cirrhosis; GT, genotype; PiB; powder-in-bottle oral solution; QD, once daily; SGC, soft gel capsule; TE, treatment experienced; TN, treatment naive; VL, viral load.
In TE patients without liver cirrhosis who were treated with faldaprevir plus PegIFN-RBV, the median reduction in VL ranged from 5.0 to 5.3 log10, with no clear relationship to the dose of faldaprevir. The rates of continuous decline, breakthrough, and plateau were 83.7% (41/49), 10.2% (5/49), and 6.1% (3/49), respectively. All 13 TE patients with cirrhosis received 240 mg faldaprevir and PegIFN-RBV. The median reduction in VL was 4.8 log10 in the QD group and 5.5 log10 in the BID group. The overall rates of continuous decline, breakthrough, and plateau in patients with cirrhosis were 92.3% (12/13), 0%, and 7.7% (1/13), respectively. Given the small sample sizes in each dose group, differences in breakthrough frequency between GT1a and GT1b during faldaprevir treatment were not evaluated statistically.
Baseline genotyping.
Phylogenetic analysis based on the NS3/4A nucleotide sequence showed that 52 patients were infected with GT1a and 44 with GT1b. Among amino acids within the NS3 protease domain that have been reported to be associated with resistance to any HCV protease inhibitors (21), the greatest baseline sequence diversity was observed at amino acid 80 in GT1a and amino acids 132 and 170 in GT1b (Fig. 2A). NS3 Q80K was detected by population sequencing in 29% (15/52) of GT1a baselines and in none of the GT1b isolates. (I/V)132 and (I/V)170 were common polymorphisms, with isoleucine as the predominant amino acid for GT1a and valine for GT1b. A rare V170T polymorphism appeared in only 1 of 44 GT1b baselines (Fig. 2B).
Fig 2.

Frequency of baseline polymorphisms detected among key amino acids in the NS3 protease domain. (A) Population sequence analysis of 96 baseline samples at clinically relevant NS3 amino acid positions (21). Prevalence of population-based baseline sequences that do not carry the wild type with respect to the subtype reference sequence are shown (GT1a, AF009606; GT1b, AJ238799). (B) Prevalence of NS3 amino acids at three polymorphic residues from panel A, detected by population sequencing. (C) Baseline samples in which clonal sequencing detected an amino acid change at NS3 R155 or NS3 D168 (average of 83 clones sequenced per baseline).
Clonal sequence analysis (Fig. 2C) detected R155 polymorphism G, K, W, or S in 33% (17/52) of GT1a baseline samples (23%, 6%, 2%, and 2%, respectively) and Q in 2.2% (1/44) of GT1b baseline samples. D168 polymorphism G, N, or E was observed in 27% (14/52) of GT1a baseline samples (23%, 2%, and 2%, respectively) and G and N in 32% (14/44) of GT1b baselines (27% and 5%, respectively). The theoretical lower limit of NS3 variant detection is 4% (95% confidence interval) for sequencing of 80 clones (22). Only two baseline samples contained clones that carried R155 or D168 substitutions above this limit of detection: one baseline sample with 5% of clones carrying GT1b D168G (TE, 240-mg QD dose group) and one GT1a baseline sample with 53% of clones carrying D168E (TN, placebo).
In vitro phenotyping of the baseline NS3 protease domain.
The susceptibilities of baseline NS3 protease to faldaprevir were similar between GT1a and GT1b. Mean EC50 values for GT1a (12 ± 8 nM; range, 3 to 42 nM) and GT1b (10 ± 9 nM; range, 2 to 63 nM) baseline-derived samples were within the range of the values obtained with nonchimeric control replicons (12 ± 4 nM, n = 96) (Fig. 3A). The mean EC50 values for IFN-α, used as a control for intrinsic assay variability among GT1a and GT1b NS3 protease chimeric replicons, were the same: 0.19 ± 0.06 IU/ml (Fig. 3B).
Fig 3.

Baseline susceptibility of NS3 protease domains isolated from patients infected with GT1a or GT1b. EC50 values for faldaprevir (A) and for IFN-α (B) for each baseline sample are shown. Each point represents ≥3 independent in vitro phenotyping experiments. (C) Fold changes of individual EC50 values were calculated relative to an EC50 reference value for wild-type replicons treated with faldaprevir (12 ± 4 nM). Wilcoxon P values are shown. Horizontal lines represent the means. One of 96 patient-derived NS3 chimeric replicons did not replicate in vitro and could not be phenotyped.
In a comprehensive analysis of all NS3 protease baseline polymorphisms, the only notable changes in faldaprevir in vitro susceptibility were associated with Q80K or V170T variants. Clinical isolates with GT1a Q80K (n = 15) were associated with a mean 2.4 ± 1.6-fold decrease (P < 0.0001) in faldaprevir susceptibility relative to isolates without Q80K (Fig. 3C), which is a fold change consistent with Q80K substitutions generated in standard laboratory strains (17). Notably, the baseline Q80K polymorphism did not have a noticeable impact on the initial VL decline during faldaprevir therapy (see Fig. S1 in the supplemental material).
The largest fold change in baseline sensitivity to faldaprevir in GT1b was observed for a TN patient in the 20-mg QD group. Genotypic information for this sample revealed a V170T amino acid change, a rare polymorphism, which conferred a 7-fold reduction (P = 0.09) in faldaprevir susceptibility relative to that of isolates without V170T (Fig. 3C). At day 6, the VL decline achieved by this patient was 1.2 log10 IU/ml, compared to the 3.7-log10 IU/ml decline for patients lacking V170T within the same dose group (see Fig. S2 in the supplemental material); however, the patient with V170T still achieved a maximum VL decline from baseline of 1.5 log10 IU/ml at day 14 (data not shown) and, per protocol, was eligible for combination therapy of faldaprevir and PegIFN-RBV from day 15 through day 28.
Emergence of NS3/4A variants.
The frequency of emergent NS3/4A variants detected during faldaprevir monotherapy in TN patients was higher than the frequency observed in TE patients who received the combination regimen, and the predominant amino acid substitutions were NS3 R155K in GT1a and NS3 D168V in GT1b (Table 2). GT1b D168E emerged in two TN patients treated with a 20-mg or 120-mg dose of faldaprevir.
Table 2.
Emergence of resistance-associated NS3 variants identified during FDV therapye
| Predominant NS3 resistance varianta | No. of samplesf |
|||||
|---|---|---|---|---|---|---|
| Placebo (TN) |
FDV monotherapy (TN) |
FDV-PegIFN-RBV (TE) |
||||
| GT1a | GT1b | GT1a | GT1b | GT1a | GT1b | |
| Breakthrough | ||||||
| Total | NA | NA | 7 | 12 | 4 | 1 |
| R155K | 6 | 0 | 2 | 0 | ||
| D168V | 1 | 10 | 1 | 1 | ||
| D168E | 0 | 2 | 0 | 0 | ||
| R155(K/R) + D168(D/V/Y/F)b | 0 | 0 | 1 | 0 | ||
| Plateau | ||||||
| Total | 4 | 4 | 2 | <LLOA | 2 | <LLOA |
| R155K | 0 | 0 | 2 | 2 | ||
| No RAV detected | 4 | 4 | 0 | 0 | ||
| Decline | ||||||
| Total | NA | NA | NA | 1 | 1 | <LLOA |
| R155K | 0 | 1 | ||||
| No RAV detected | 1 | 0 | ||||
| RAVs detected in ≥4% of clonesc | ||||||
| Total | 4 | 4 | 9 | 13 | 5 | 1 |
| R155K only | 0 | 0 | 6 | 0 | 5 | 0 |
| R155K > R155S | 0 | 0 | 1 | 0 | 0 | 0 |
| R155K > R155W > R155G | 0 | 0 | 1 | 0 | 0 | 0 |
| D168V only | 0 | 0 | 0 | 4 | 0 | 0 |
| D168V > R155K | 0 | 0 | 0 | 1 | 0 | 0 |
| D168V > D168A | 0 | 0 | 1 | 2 | 0 | 0 |
| D168V > D168T | 0 | 0 | 0 | 1 | 0 | 1 |
| D168V > D168H > D168Y | 0 | 0 | 0 | 1 | 0 | 0 |
| D168V > A156Vd | 0 | 0 | 0 | 1 | 0 | 0 |
| D168E > D168G | 0 | 0 | 0 | 1 | 0 | 0 |
| D168E > D168V > R155W > D168A | 0 | 0 | 0 | 1 | 0 | 0 |
| No RAV detected | 4 | 4 | 0 | 1 | 0 | 0 |
Predominant emerging NS3 variants were detected by population sequencing and confirmed by clonal sequencing during 14-day faldaprevir monotherapy in TN patients and 28-day faldaprevir-PegIFN-RBV therapy in TE patients.
Clonal sequencing not available for confirmation of predominant RAV.
Threshold was based on theoretical limit of 4% for 80 clonal sequences per sample (95% confidence interval) (22). > indicates relative frequency.
A156V was detected in 6% of clones; A156T was detected in 3% of clones; A156(T/A) was detected by population sequencing.
<LLOA, VL was below the lower limit of sequence amplification (<1,000 IU/ml); LLOA, lower limit of amplification; NA, not applicable; TE, treatment experienced; TN, treatment naive; RAV, resistance-associated variant; VL, viral load.
All TE patients with cirrhosis received FDV-PegIFN-RBV and were <LLOA during treatment; therefore, no sequence data were available.
Samples from patients with cirrhosis were below the lower limit of sequence amplification at the end of faldaprevir therapy, and therefore no sequence data were available (Table 2). However, after faldaprevir treatment, six of the 13 patients with cirrhosis experienced virologic rebound and had available sequences; the results were as follows. One GT1a patient in the 240-mg QD, SGC group, who had experienced an on-treatment virologic plateau, carried NS3 R155K variants in posttreatment sequences. Three GT1a patients with virologic decline during treatment carried R155K variants posttreatment. One GT1b and one GT1a patient (both with on-treatment VL decline) had available posttreatment sequences but lacked detectable resistance-associated variants.
Emerging variants detected at low frequency during faldaprevir treatment at NS3 positions other than 155 and 168 included GT1b A156V detected in 6% of clones from one sample that also carried the predominant D168V substitution (Table 2).
No emerging NS3/4A-resistant variants were detected in the placebo group. Amino acids spanning the NS3/4A cleavage site were also monitored; however, no emerging variants were detected.
In vitro phenotyping of treatment-emergent NS3 variants.
Phenotypic analyses of patient-derived NS3 protease domains evaluated in transient replicon assays (Table 3) showed a reduction in sensitivity to faldaprevir inhibition that ranged from 130- to 520-fold for the NS3 R155K variant. The D168 variants conferred a broader range of reduced susceptibility to faldaprevir. NS3 proteases that predominantly carried the D168V substitution conferred larger decreases in faldaprevir susceptibility (1,100- to 2,800-fold changes) than D168E and D168T (respectively, 180- and 690-fold changes). Amino acid changes at 155 and 168 did not substantially impact sensitivity to IFN-α. The lack of emergent resistance-associated variants in TN patients from the placebo group was reflected by an unchanged susceptibility to faldaprevir.
Table 3.
Susceptibility to faldaprevir and IFN-α of plasma sample-derived NS3 protease with treatment-emergent variants in chimeric replicons
| Genotype | Placebo or NS3 varianta | No. of samples | Faldaprevir EC50 fold change relative to baseline |
IFN-α EC50 fold change relative to baseline |
||
|---|---|---|---|---|---|---|
| Mean | Range | Mean | Range | |||
| GT1a | Placebo | 4 | 1.8 | 0.8–3.8 | 1.1 | 0.8–1.7 |
| R155K | 27 | 330 | 130–520 | 1.3 | 0.5–3.1 | |
| D168Vb | 1 | 1800 | 1.3 | |||
| GT1b | Placebo | 4 | 1.2 | 1.0–1.3 | 1.0 | 0.6–1.9 |
| R155K | 1 | 130 | 1.3 | |||
| D168V | 11 | 1700 | 1,100–2,800 | 1.6 | 0.7–3.6 | |
| D168E | 2 | 180 | 140–210 | 1.3 | 0.5–2.1 | |
| D168T | 1 | 690 | 3.9 | |||
NS3 proteases from on-treatment or post-faldaprevir treatment samples. Changes in susceptibility for the placebo group were determined at day 14 relative to the baseline.
GT1a D168V emerged in a TN patient who had an R155 codon nucleotide sequence, CGG, similar to subtype 1b.
NS3/4A-resistant variant dynamics and persistence.
Clonal sequence analysis revealed that NS3 D168 substitutions that emerged during faldaprevir therapy were more diverse than R155 substitutions (Table 2 and Fig. 4). In a GT1b-infected TN patient (20-mg dose group), a mixture of D168 substitutions (V, H, Y, and A) emerged by day 14 (Fig. 4), but only D168V was detected at the following time point, day 56. Although D168V was the major variant that emerged during faldaprevir monotherapy in this patient, the incidence of R155K increased to 14% in post-faldaprevir treatment clonal sequence analysis. In certain cases, specific D168 and R155 substitutions were linked at a low frequency, such as the GT1b NS3 R155Q-D168N combination (see Fig. S3B in the supplemental material).
Fig 4.
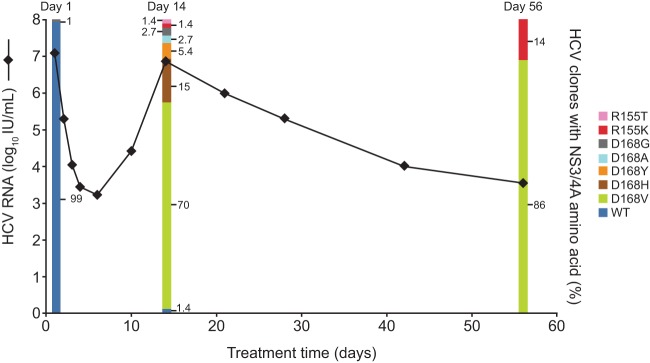
NS3/4A-resistant variant dynamics. Viral load profile of virologic breakthrough and percentage of clones carrying NS3 R155 and/or D168 substitutions over time for a TN GT1b-infected patient from the 20-mg dose group. WT, wild type.
The persistence of NS3 R155K and D168V resistance variants in patients who experienced virologic breakthrough and had clonal sequence data available during the post-faldaprevir treatment phase was evaluated. Four GT1a-infected patients had breakthrough virus containing 72 to 100% R155K substitutions by day 28, and in another GT1a patient, virus with 99% R155K substitution was first detected at day 57 (Fig. 5A). R155K was the predominant variant detected in all four of the patients with available sequences during the dual PegIFN-RBV treatment phase (93 to 100% of clones). Furthermore, in three patients with available sequences during posttreatment follow-up (in one case up to day 506), R155K persisted in 72 to 100% of clones. Outgrowth of virus predominantly carrying wild-type R155 was not observed in these five patients up to day 506. In a representative GT1a-infected patient, viral breakthrough at day 14 was associated with emergence of NS3 R155K, which persisted to day 338 in 72% of clones (Fig. 5A; see also Fig. S3A in the supplemental material).
Fig 5.

Longitudinal clonal sequence analysis. Detection of NS3 R155K (A) or NS3 D168V (B) by clonal sequencing in patients with virologic breakthrough and post-faldaprevir treatment sequences. Each row represents one patient. Wild-type (WT) sequence represents a lack of detectable R155K or D168V substitutions. FDV, faldaprevir; P/R, PegIFN-ribavirin; TE, treatment experienced; TN, treatment naive.
Five GT1b patients with breakthrough viruses that had 85 to 100% D168V variants by day 28 were evaluated (Fig. 5B). Complete outgrowth of wild-type D168 virus and a lack of detectable D168V variants occurred in 2/3 and 4/4 patients with available sequences, respectively, during the PegIFN-RBV dual-therapy phase and during follow-up. In a representative GT1b-infected patient, D168V was detected in 100% of clones during viral breakthrough with faldaprevir-PegIFN-RBV therapy, but D168V was no longer detected after day 85 due to outgrowth of wild-type D168 (Fig. 5B; see also Fig. S3B in the supplemental material). Thus, D168V variants did not persist as long as R155K variants.
DISCUSSION
The first evaluation of faldaprevir in a phase 1b clinical trial in HCV-infected patients was conducted to evaluate antiviral potency and safety (19). Virology samples from this early trial were used in a post hoc analysis to comprehensively assess the baseline NS3/4A genotype and NS3 phenotype, as well as to monitor the emergence of resistance-associated variants during treatment with faldaprevir.
All patient baseline samples were genotyped, and among all NS3 protease polymorphisms detected by population sequencing from the 96 samples, only Q80K and (V/I)170T conferred notable changes in in vitro sensitivity to faldaprevir in replicon phenotyping experiments. However, Q80K was associated with only a small reduction (mean change, 2.4 ± 1.6-fold) in sensitivity to faldaprevir and did not affect VL decline at any dose. Q80K has been shown to alter sensitivity to the macrocyclic protease inhibitor simeprevir (TMC-435) by 7.7- or 14-fold in transient GT1b replicon assays (17, 23). The greatest fold shift in sensitivity to faldaprevir of a baseline NS3 isolate—a 7-fold shift associated with (V/I)170T detected in one patient relative to isolates without this substitution—did not preclude a continuous VL decline during faldaprevir monotherapy, even with the suboptimal 20-mg dose. This rare NS3 polymorphism occurs at a residue that has been shown to alter sensitivity to boceprevir (24). The current observations are based on a small sample size and a limited duration of faldaprevir treatment but may indicate that the presence of Q80 polymorphisms will not prevent reduction of VL by faldaprevir at the doses currently being investigated in larger clinical trials. The significance of (V/I)170T and Q80K to treatment response is being explored in ongoing studies.
One of three predominant NS3 substitutions—R155K, D168V, or D168E—was associated with all virologic breakthrough during faldaprevir therapy observed in this study. The fold shifts associated with these NS3 substitutions (330-, 1,700-, or 180-fold, respectively) are consistent with single-amino-acid site-directed mutant replicon resistance studies (17). The divergence in nucleotide sequence at codon 155 between GT1a and GT1b may explain the higher prevalence of the R155K variant in GT1a. As noted previously (13), this substitution requires a single-nucleotide transition in GT1a but two in GT1b. Notably, this study enrolled a rare GT1a patient with a 155 codon that would have required a two-nucleotide transition to yield R155K and thus favored the emergence of D168V in this GT1a patient.
NS3 protease single-amino-acid substitutions that reduced sensitivity to faldaprevir, in addition to R155 and D168, emerged at position A156 in cell culture during in vitro selection experiments (17). A GT1b NS3 protease variant carrying A156V was detected at low prevalence by clonal sequencing [and A156(T/A) was detected by population sequencing] in only one patient (TN, 120 mg faldaprevir). The predominant NS3 variant that emerged in this patient was D168V, and this suggests that emergence of NS3 A156 variants is rare in patients treated with faldaprevir.
Virologic breakthrough and the emergence of resistance variants in NS3/4A were more common with faldaprevir monotherapy than when faldaprevir was combined from day 1 with PegIFN-RBV. Virologic response during the PegIFN-RBV add-on period suggests that viruses carrying key NS3 resistance-associated variants remained sensitive to PegIFN-RBV therapy and could be cleared since IFN acts on host immune responses rather than the HCV replication cycle (25–27). Faldaprevir with PegIFN-RBV reduced VL and suppressed breakthrough more potently than faldaprevir monotherapy, notably in TE patients despite their previous failed treatment with PegIFN-based regimens lacking an HCV protease inhibitor.
Longitudinal analysis during the follow-up period revealed the gradual outgrowth of the wild-type virus with concomitant loss of NS3 D168V in GT1b-infected patients and suggests this variant may have a low replicative fitness. Other studies have also shown that NS3/4A protease inhibitor-resistant variants, particularly those associated with GT1b, are generally less fit than the wild type (12, 13, 18, 28). The NS3 R155K variants persisted during follow-up, indicating high replicative fitness of this variant. The implications of this observation are unclear in view of the small size of this study and the limited number of NS3 variants available for follow-up but suggest that NS3 R155K variants can persist longer in the absence of drug pressure than the D168V variants.
In conclusion, the major HCV NS3 variants that emerged with faldaprevir treatment were R155K in GT1a and D168V in GT1b. These substitutions do not alter the sensitivity to PegIFN-RBV, and the majority of treatment-naive patients with resistant virus subsequently displayed antiviral responses during combination therapy. Combination of faldaprevir with PegIFN-RBV from day 1 of treatment suppresses the emergence of resistance-associated variants, reduces breakthrough, and increases VL reduction. The significance of baseline polymorphisms such as Q80K and (V/I)170T to treatment response, and of the persistence of R155K and D168V in posttreatment follow-up, is being examined in larger phase 2 and phase 3 studies with faldaprevir-PegIFN-RBV.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mithun Ranga (Boehringer Ingelheim Pharmaceuticals Inc., Ridgefield, CT) for statistical programming and analysis of baseline variants.
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Richard Murphy of Adelphi Communications Ltd. during the preparation of the manuscript.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00822-13.
REFERENCES
- 1.Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. 2012. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156:271–278 [DOI] [PubMed] [Google Scholar]
- 2.Kolykhalov AA, Mihalik K, Feinstone SM, Rice CM. 2000. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J. Virol. 74:2046–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St. George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186–189 [DOI] [PubMed] [Google Scholar]
- 4.Hinrichsen H, Benhamou Y, Wedemeyer H, Reiser M, Sentjens RE, Calleja JL, Forns X, Erhardt A, Cronlein J, Chaves RL, Yong CL, Nehmiz G, Steinmann GG. 2004. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology 127:1347–1355 [DOI] [PubMed] [Google Scholar]
- 5.Chatel-Chaix L, Baril M, Lamarre D. 2010. Hepatitis C virus NS3/4A protease inhibitors: a light at the end of the tunnel. Viruses 2:1752–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin C, Kwong AD, Perni RB. 2006. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 6:3–16 [DOI] [PubMed] [Google Scholar]
- 7.Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C. 2006. Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease. Antimicrob. Agents Chemother. 50:899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prongay AJ, Guo Z, Yao N, Pichardo J, Fischmann T, Strickland C, Myers J, Jr, Weber PC, Beyer BM, Ingram R, Hong Z, Prosise WW, Ramanathan L, Taremi SS, Yarosh-Tomaine T, Zhang R, Senior M, Yang RS, Malcolm B, Arasappan A, Bennett F, Bogen SL, Chen K, Jao E, Liu YT, Lovey RG, Saksena AK, Venkatraman S, Girijavallabhan V, Njoroge FG, Madison V. 2007. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 50:2310–2318 [DOI] [PubMed] [Google Scholar]
- 9.European Association for the Study of the Liver 2011. EASL clinical practice guidelines: management of hepatitis C virus infection. J. Hepatol. 55:245–264 [DOI] [PubMed] [Google Scholar]
- 10.Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. 2011. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54:1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107 [DOI] [PubMed] [Google Scholar]
- 12.Halfon P, Sarrazin C. 2012. Future treatment of chronic hepatitis C with direct acting antivirals: is resistance important? Liver Int. 32(Suppl 1):79–87 [DOI] [PubMed] [Google Scholar]
- 13.Sarrazin C, Zeuzem S. 2010. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138:447–462 [DOI] [PubMed] [Google Scholar]
- 14.Susser S, Welsch C, Wang Y, Zettler M, Domingues FS, Karey U, Hughes E, Ralston R, Tong X, Herrmann E, Zeuzem S, Sarrazin C. 2009. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology 50:1709–1718 [DOI] [PubMed] [Google Scholar]
- 15.Llinas-Brunet M, Bailey MD, Goudreau N, Bhardwaj PK, Bordeleau J, Bos M, Bousquet Y, Cordingley MG, Duan J, Forgione P, Garneau M, Ghiro E, Gorys V, Goulet S, Halmos T, Kawai SH, Naud J, Poupart MA, White PW. 2010. Discovery of a potent and selective noncovalent linear inhibitor of the hepatitis C virus NS3 protease (BI 201335). J. Med. Chem. 53:6466–6476 [DOI] [PubMed] [Google Scholar]
- 16.White PW, Llinas-Brunet M, Amad M, Bethell RC, Bolger G, Cordingley MG, Duan J, Garneau M, Lagace L, Thibeault D, Kukolj G. 2010. Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob. Agents Chemother. 54:4611–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagacé L, White PW, Bousquet C, Dansereau N, Do F, Llinas-Brunet M, Marquis M, Massariol MJ, Maurice R, Spickler C, Thibeault D, Triki I, Zhao S, Kukolj G. 2012. In vitro resistance profile of the hepatitis C virus NS3 protease inhibitor BI 201335. Antimicrob. Agents Chemother. 56:569–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cento V, Mirabelli C, Salpini R, Dimonte S, Artese A, Costa G, Mercurio F, Svicher V, Parrotta L, Bertoli A, Ciotti M, Di PD, Sarrecchia C, Andreoni M, Alcaro S, Angelico M, Perno CF, Ceccherini-Silberstein F. 2012. HCV genotypes are differently prone to the development of resistance to linear and macrocyclic protease inhibitors. PLoS One 7:e39652. 10.1371/journal.pone.0039652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manns MP, Bourliere M, Benhamou Y, Pol S, Bonacini M, Trepo C, Wright D, Berg T, Calleja JL, White PW, Stern JO, Steinmann G, Yong CL, Kukolj G, Scherer J, Boecher WO. 2011. Potency, safety, and pharmacokinetics of the NS3/4A protease inhibitor BI201335 in patients with chronic HCV genotype-1 infection. J. Hepatol. 54:1114–1122 [DOI] [PubMed] [Google Scholar]
- 20.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, Zhou Y, Chu HM, Lin C, Weegink C, Reesink H, Zeuzem S, Kwong AD. 2007. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology 132:1767–1777 [DOI] [PubMed] [Google Scholar]
- 21.Phenotype Working Group HCV, Drug Development Advisory Group HCV 2012. Clinically relevant HCV drug resistance mutations. Ann. Forum Collab. HIV Res. 14:1–10 [Google Scholar]
- 22.Kwong AD, Najera I, Bechtel J, Bowden S, Fitzgibbon J, Harrington P, Kempf D, Kieffer TL, Koletzki D, Kukolj G, Lim S, Pilot-Matias T, Lin K, Mani N, Mo H, O'Rear J, Otto M, Parkin N, Pawlotsky JM, Petropoulos C, Picchio G, Ralston R, Reeves JD, Schooley RT, Seiwert S, Standring D, Stuyver L, Sullivan J, Miller V. 2011. Sequence and phenotypic analysis for resistance monitoring in hepatitis C virus drug development: recommendations from the HCV DRAG. Gastroenterology 140:755–760 [DOI] [PubMed] [Google Scholar]
- 23.Lenz O, Verbinnen T, Lin TI, Vijgen L, Cummings MD, Lindberg J, Berke JM, Dehertogh P, Fransen E, Scholliers A, Vermeiren K, Ivens T, Raboisson P, Edlund M, Storm S, Vrang L, de Koch H, Fanning GC, Simmen KA. 2010. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob. Agents Chemother. 54:1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tong X, Bogen S, Chase R, Girijavallabhan V, Guo Z, Njoroge FG, Prongay A, Saksena A, Skelton A, Xia E, Ralston R. 2008. Characterization of resistance mutations against HCV ketoamide protease inhibitors. Antiviral Res. 77:177–185 [DOI] [PubMed] [Google Scholar]
- 25.Asselah T, Bieche I, Sabbagh A, Bedossa P, Moreau R, Valla D, Vidaud M, Marcellin P. 2009. Gene expression and hepatitis C virus infection. Gut 58:846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Touzot M, Soumelis V, Asselah T. 2012. A dive into the complexity of type I interferon antiviral functions. J. Hepatol. 56:726–728 [DOI] [PubMed] [Google Scholar]
- 28.Kieffer TL, De Meyer S, Bartels DJ, Sullivan JC, Zhang EZ, Tigges A, Dierynck I, Spanks J, Dorrian J, Jiang M, Adiwijaya B, Ghys A, Beumont M, Kauffman RS, Adda N, Jacobson IM, Sherman KE, Zeuzem S, Kwong AD, Picchio G. 2012. Hepatitis C viral evolution in genotype 1 treatment-naive and treatment-experienced patients receiving telaprevir-based therapy in clinical trials. PLoS One 7:e34372. 10.1371/journal.pone.0034372 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.