

Abstract
Hepatitis C virus (HCV) envelope protein (E1E2) is essential for virus binding to host cells. Aptamers have been demonstrated to have strong promising applications in drug development. In the current study, a cDNA fragment encoding the entire E1E2 gene of HCV was cloned. E1E2 protein was expressed and purified. Aptamers for E1E2 were selected by the method of selective evolution of ligands by exponential enrichment (SELEX), and the antiviral actions of the aptamers were examined. The mechanism of their antiviral activity was investigated. The data show that selected aptamers for E1E2 specifically recognize the recombinant E1E2 protein and E1E2 protein from HCV-infected cells. CD81 protein blocks the binding of aptamer E1E2-6 to E1E2 protein. Aptamers against E1E2 inhibit HCV infection in an infectious cell culture system although they have no effect on HCV replication in a replicon cell line. Beta interferon (IFN-β) and IFN-stimulated genes (ISGs) are not induced in virus-infected hepatocytes with aptamer treatment, suggesting that E1E2-specific aptamers do not induce innate immunity. E2 protein is essential for the inhibition of HCV infection by aptamer E1E2-6, and the aptamer binding sites are located in E2. Q412R within E1E2 is the major resistance substitution identified. The data indicate that an aptamer against E1E2 exerts its antiviral effects through inhibition of virus binding to host cells. Aptamers against E1E2 can be used with envelope protein to understand the mechanisms of HCV entry and fusion. The aptamers may hold promise for development as therapeutic drugs for hepatitis C patients.
INTRODUCTION
Hepatitis C virus (HCV) infects 3% of the world's population, and persistent virus infection causes chronic hepatitis, liver cirrhosis, and even hepatocellular carcinoma (1). There is no vaccine available, and alpha interferon (IFN-α)-based therapy is the current treatment for patients with chronic hepatitis C (2). Many patients do not response to the therapy. There is an urgent need to develop well-tolerated and effective therapeutic drugs against HCV infection (3).
HCV is an enveloped, single positive-strand RNA virus of the Flaviviridae family. The 9.6-kb viral genome encodes one polyprotein that is processed by viral and cellular proteases to produce structural proteins including core protein and envelope proteins E1 and E2 as well as the nonstructural proteins consisting of the p7 ion channel, NS2-3 protease, NS3 serine protease, RNA helicase, NS4A polypeptide, NS4B, NS5A proteins, and NS5B RNA-dependent RNA polymerase. E1 and E2 are type I membrane glycoproteins and form a noncovalent complex, which is believed to be the building block for the viral envelope (4). E2 is thought to be primarily responsible for receptor binding (5). Infection of the host cells by HCV is initiated through the interactions between the E1E2 protein and several previously identified HCV entry receptors, including CD81, scavenger receptor class B type I (SR-BI), claudin-1 (CLDN1), and occludin (OCLN) (6–9). The essential role of E1E2 in HCV entry makes this protein an attractive target for the development of specific antiviral drugs. The recent development of the infectious HCV clone JFH1 provides a powerful tool for the study of the HCV replication cycle and discovery of inhibitors of viral infection (10–12).
The development of an approach using selective evolution of ligands by exponential enrichment (SELEX) allows the isolation of nucleic acid ligands, termed aptamers, that display a high degree of affinity and specificity for many targets (13, 14). SELEX involves a series of enrichment cycles and counterselection based on repetitive binding that ultimately selects for a group of aptamers binding to the targets. Aptamers can specifically recognize their targets or regulate the functions of the targets. Aptamers possess many advantages over antibodies as therapeutic drugs, including easy synthesis and modification with high batch fidelity, low cost, and long-term stability (15, 16). The list of inhibitory aptamers for therapeutic use is growing, and the vascular endothelial growth factor-specific aptamer pegaptanib sodium (Macugen) has been used for the treatment of age-related macular degeneration (17). Aptamer AS1411 is in a phase II clinical trial for renal and pancreatic cancer (18).
In this study, we obtained aptamers for HCV E1E2 using in vitro SELEX and developed inhibitors of HCV infection. Our data show that these aptamers exert antiviral effects through blocking virus binding to the host cells and that their binding sites are located in E2. The aptamers against E1E2 can be used with E1E2 to understand the mechanisms of HCV entry. The data indicate that the aptamer against E1E2 protein may hold promise for the development of a novel approach for hepatitis C patients.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents.
FL-Neo, an HCV full-length replicon cell line, and Huh7.5 cells were kindly provided by Charles Rice (Rockefeller University, New York, NY) (19). The plasmids pH 77-S and pH 77-S/ΔE1P7 were from Stanley M. Lemon (University of North Carolina, Chapel Hill, NC) (20). pJFH1 and pJFH1/GND plasmids were generously provided by Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) (12). The replicon cells were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum, l-glutamine, nonessential amino acids, and 500 μg/ml of G418 (Sigma, St. Louis, MO). For each experiment, the cells were seeded in a six-well plate in the absence of G418. Mouse monoclonal anti-NS5A antibody was a gift from Chen Liu (University of Florida, Gainesville, FL) (21).
Expression and purification of E1E2 protein.
The protein expression vector of full-length E1E2 was constructed by PCR amplification using pJFH1 plasmid as the template. The full-length E1E2 sequence tagged with six His molecules at the N terminus was PCR amplified, digested with EcoRI and HindIII, and inserted into pET-32a(+) (Novagen, Madison, MI) to produce pET32a-E1E2. E1E2 protein was expressed in Escherichia coli BL21(DE3) cells (Invitrogen, Carlsbad, CA). Cells were grown in LB medium and induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). E1E2 protein was purified using Ni-nitrilotriacetic acid (NTA) affinity chromatography columns (Invitrogen, Carlsbad, CA). The protein was eluted with phosphate-buffered saline (PBS) and identified using anti-His antibody (Sigma, St. Louis, MO) via Western blotting, as described below.
In vitro selection of aptamers against E1E2.
A synthesized DNA library pool (referred to as the control library) with an overall complexity of ∼1014 was used for in vitro selection. The sequence of the random DNA is 5′-ACGCTCGGATGCCACTACAG-(N40)-CTCATGGACGTGCTGGTGAC-3′, where N40 represents 40 nucleotides with equimolar incorporation of A, G, C, and T at each position. The selection and amplification procedure were performed as previously described (15). After eight rounds of selection, the amplified DNA was cloned, and several clones were sequenced.
Enzyme-linked oligonucleotide assay (ELONA).
Streptavidin-precoated microtiter plates were coated with biotin-labeled aptamer against E1E2. The plates were washed with PBS containing 1 mM MgCl2, 0.1% bovine serum albumin (BSA), and 0.05% Tween 20. Serial dilutions of His-tagged E1E2 protein was added into the plates and incubated at 37°C for 30 min. After a washing step to remove the unbound target protein, mouse anti-His monoclonal antibody was added into the plates. After incubation at 37°C for 1 h, peroxidase-labeled goat anti-mouse IgG was added and incubated at 37°C for 30 min. Color development was performed by addition of freshly prepared substrate solution for 10 min at room temperature. After the reaction was stopped with stopping buffer, the plates were read with an enzyme-linked immunosorbent assay (ELISA) reader, and absorbance of each sample was measured at 450 nm.
MTS assay.
One day before aptamer treatment, 1.0 × 103 cells were seeded in triplicate in a 96-well plate. The cells were cultured with or without aptamer for the indicated time periods at 37°C. Twenty microliters of CellTiter AQ Solution, which contains a tetrazolium compound [3,4-(5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt] (MTS) and an electron coupling reagent (Promega), was added to each well. After 2 h of incubation at 37°C, the absorbance at 490 nm was measured. Cell viability was calculated with respect to the control samples. Three independent experiments were performed.
Selection of resistance-conferring substitutions.
HCV-infected Huh7.5 cells were treated with 100 nM E1E2-6 for 3 weeks. Control cells were maintained with a 100 nM concentration of the library. Total RNA was isolated from the cells. E1E2 cDNA was recovered from cells by reverse transcription-PCR (RT-PCR). The oligonucleotide primers used to amplify JFH1 E1E2 cDNA were GCCCAGGTGAAGAATACCAG (forward) and TGCTTCGGCCTGGCCCAACAAG (reverse). E1E2 amplicons were used for direct population sequencing and to generate cDNA clones with a TOPO-TA cloning kit (Invitrogen). The substitution Q412R was introduced into the pJFH1 plasmid by using a QuikChange site-directed mutagenesis kit (Stratagene). In vitro transcripts of wild-type JFH1 and JFH1 with the Q412R substitution were generated and transfected into Huh7.5 cells.
Real-time PCR assays.
Total cellular RNA was isolated using TRIzol. The primers targeting HCV, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IFN-β, G1P3, and 1-8U have been reported previously, and real-time PCR was performed as described previously (21).
Immunofluorescence.
The immunofluorescence protocol was published previously (22).
Western blot analysis.
The procedure for Western blotting was reported previously (22).
Titration of infectious HCV.
The protocol for titration of infectious HCV was performed as published previously (11).
Statistical analysis.
Differences between mean values were compared using Student's t test. A P value of< 0.05 was considered statistically significant.
RESULTS
Purification of HCV E1E2 and selection of aptamers against E1E2 protein.
The cDNA fragment encoding the entire E1E2 gene of HCV was amplified by PCR. The amplified product was cloned into the expression vector pET-32a(+) and confirmed by DNA sequencing analysis. E1E2 protein was expressed and purified by its N-terminal His tag. The purified E1E2 protein was confirmed by immunoblot analysis (Fig. 1A).
Fig 1.
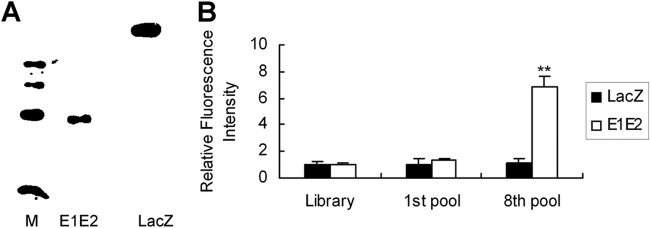
Purification of HCV E1E2 and selection of aptamers against E1E2 protein. (A) His-tagged E1E2 was expressed by IPTG induction in E. coli BL21(DE3) and confirmed using mouse anti-His monoclonal antibody via Western blot analysis. (B) Fluorescein isothiocyanate-labeled DNA pools from the control library, round 1, or round 8 were incubated with E1E2 or control LacZ protein in binding buffer. The density of the fluorescence was measured and normalized to that of the library. Error bars represent means ± standard deviations. **, P < 0.01 versus the library.
A nucleotide library was obtained from a pool of ∼1014 single-stranded DNA molecules. The DNA library was mixed with His-tagged E1E2 protein. E1E2-DNA complexes were precipitated with nickel beads, and pellets were washed. DNAs were recovered, amplified by PCR, and used for subsequent rounds of selection. To remove the nonspecifically bound DNA, we applied a counterselection step using the control protein LacZ, followed by selection using the E1E2 protein in each cycle. Eight subsequent rounds of selection were performed. After eight rounds of selection, there was a marked increase in the binding of round 8 DNA pools to E1E2 protein compared to that of the pools of round 1 (Fig. 1B). The selected aptamers were cloned and sequenced. The topmost sequences were used as representative members for this study. We selected some aptamers and named them E1E2-1, E1E2-2, E1E2-3, E1E2-4, E1E2-5, and E1E2-6. Their sequences are listed in Table 1.
Table 1.
Sequences of E1E2 aptamers
| Aptamer name | Sequence |
|---|---|
| E1E2-1 | GGTATGTGGAATAATCTAGCACCTACC |
| E1E2-2 | GCAGTGTGGAATAATCTAGCACCCTGC |
| E1E2-3 | CAGGAGGAATAATCTAGCTCCTG |
| E1E2-4 | CAGGTCGATTAATCTAGGACCTG |
| E1E2-5 | CACGTCGATTAAGATTGGACGTG |
| E1E2-6 | CACGTCTATTAAGATTGGGACGTG |
Analysis of binding affinity of aptamers for HCV E1E2 protein.
An ELONA was performed to select the aptamers with high affinity to the E1E2 protein. As shown in Fig. 2A, aptamers E1E2-1, E1E2-2, E1E2-3, E1E2-4, E1E2-5, and E1E2-6 showed a higher affinity for E1E2 protein than the control library. Various aptamers bound to E1E2 protein, and this interaction was retained in the presence of excess yeast tRNA in the binding buffer, suggesting that their binding to E1E2 was specific. To test whether E1E2-6 and human CD81 share similar binding sites in the HCV E1E2 protein, we carried out competition experiments. An ELONA was performed to determine the binding affinity between aptamer E1E2-6 and E1E2 protein in the presence of human CD81 protein. As shown in Fig. 2B, CD81 protein could block the binding of aptamer E1E2-6 to E1E2 protein. The data suggest that aptamer E1E2-6 and human CD81 may share similar binding sites on E1E2 protein and that the E1E2-6 aptamer may block E1E2 protein binding to CD81 protein.
Fig 2.
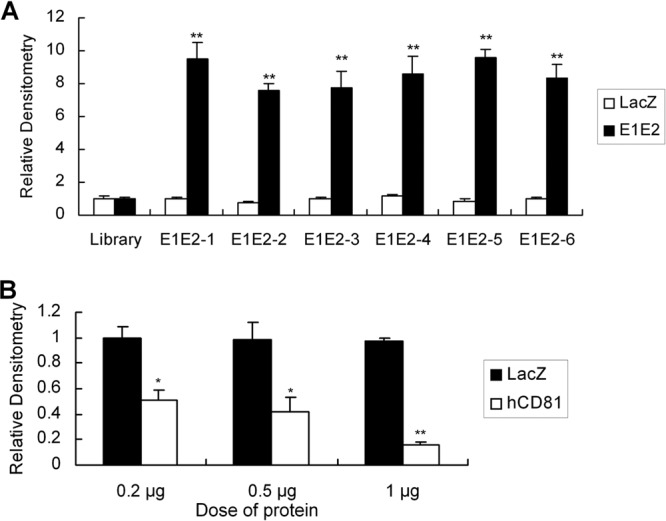
Binding affinity of selected aptamers for E1E2. (A) The binding affinity of aptamers. Each biotin-labeled aptamer was added to a microtiter plate previously coated with streptavidin, and an ELONA was performed. Purified E1E2 or control LacZ protein was added to the plates. After a washing step, mouse anti-His monoclonal antibody was added and incubated at 37°C for 1 h. Horseradish peroxidase-conjugated goat anti-mouse IgG was added to the plates. Color development was performed, and the plates were read with an ELISA reader. The absorbance of each sample was measured at 450 nm and normalized to the level of the library. The data shown are means ± standard deviations from three independent experiments performed in duplicate. **, P < 0.01 versus the library. (B) Aptamer E1E2-6 and human CD81 protein share similar binding sites on the HCV E1E2 protein. A fixed concentration of aptamer E1E2-6 was coated on the plates. Increasing concentrations of human CD81 (hCD81) protein or control LacZ protein were mixed with a fixed concentration of E1E2 protein, and the mixture was added to the plates. An ELONA was performed. The data were obtained as described for panel A. The data represent the means of three different experiments. Error bars represent means ± standard deviations. *, P < 0.05; **, P < 0.01, versus the LacZ control.
Inhibition of HCV infection by aptamers against E1E2.
To test whether aptamers for E1E2 inhibit HCV replication in full-length replicon cells, different E1E2 aptamers were used to treat FL-Neo cells. The HCV RNA replication level in the cells was not affected by E1E2 aptamers (Fig. 3A).
Fig 3.

Inhibition of HCV infection by aptamers against E1E2. (A) The effect of E1E2 aptamers on HCV RNA replication in full-length replicon cells. FL-Neo cells were incubated with a 100 nM concentration of each E1E2 aptamer for 48 h. Intracellular viral RNA was detected with real-time PCR. The data were normalized with GAPDH and represent three different experiments. (B) E1E2 aptamers inhibit HCV infection in an infectious cell culture system. JFH1 virus suspension at a multiplicity of infection of 0.1 (focus-forming units [FFU] per cell) was mixed with 100 nM each E1E2 aptamer at room temperature for 2 h and used to infect Huh7.5 cells. The supernatant was removed at 2 h postinfection. Total RNA was isolated from the cells at 48 h postinfection, and viral RNA was quantified by real-time PCR. The data were normalized with GAPDH and represent the means of three different experiments. (C) The dose-response curves of aptamers. Huh7.5 cells were treated as described for panel B. Viral RNA was detected with real-time PCR and normalized with GAPDH. The data represent the means of three different experiments. (D) The effect of E1E2 aptamer on the viability of Huh7.5 cells. Huh7.5 cells were treated as described for panel B. The effect of aptamer on the viability of the cells was measured by MTS assay. The data were normalized to the level of the control and represent the means of three independent experiments. (E) Aptamers for E1E2 inhibit the production of infectious virus particles. Huh7.5 cells were treated as described for panel B. The supernatants were harvested at 48 h postinfection, and titers were determined by FFU assay. The infectivity titers in the supernatant from aptamer-treated cells were normalized to the titer of the library treatment group and are presented as the means and standard errors of three independent infections. (F) Aptamers inhibit H77-S viral infection. H77-S virus was mixed with 100 nM E1E2 aptamer at room temperature for 2 h, and the mixtures were used to infect Huh7.5 cells. The supernatant was removed at 2 h postinfection. Total cellular RNA was isolated at 48 h postinfection. Viral RNA was quantified by real-time PCR and normalized to GAPDH. The data shown are means ± standard deviations from three independent experiments. *, P < 0.05; **, P < 0.01, versus the library.
JFH1 virus, a genotype 2 virus, was cloned from a Japanese patient with fulminate hepatitis C. The infection of human hepatoma Huh7.5 cells by JFH1 virus provides a useful tool to develop novel drugs for HCV infection. We decided to test whether E1E2 aptamer inhibits HCV infection in an infectious cell culture system. As shown in Fig. 3B, when the cells were infected with JFH1 virus in the presence of aptamers, intracellular viral RNA levels significantly decreased. No effect could be observed with the control library. The 50% effective concentrations (EC50) of aptamer E1E2-2, E1E2-4, E1E2-5, and E1E2-6 in virus-infected cells were 86.12 nM, 146.69 nM, 84.63 nM, and 62.37 nM, respectively (Fig. 3C). The concentration of aptamers used in our experiments showed no apparent toxic effect to the cells (Fig. 3D). The overall cell numbers were similar for cells treated with E1E2 aptamers and the corresponding controls, suggesting that the inhibition of viral replication is not due to the cytotoxic effect of the aptamers.
To confirm that the inhibition of viral RNA synthesis induced by aptamers correlated with the decrease in the production of virus particles, the supernatants of human hepatocytes infected with JFH1 in the presence of the aptamers or the control library were harvested at 48 h postinfection and used to reinfect naive Huh7.5 cells. As shown in Fig. 3E, the levels of extracellular infectious virus decreased in aptamer-treated cells in comparison with the level in the control library group. In accordance with the real-time PCR result, aptamer E1E2-1 had no effect on the production of extracellular infectious virus.
In addition, we examined the effect of aptamers on HCV genotype 1a infection in human hepatocytes. H77-S RNA was in vitro transcribed from the plasmid pH 77-S, and H77-S virus was collected as described previously (20). H77-S virus was used to infect Huh7.5 cells in the presence of aptamers. As shown in Fig. 3F, the viral RNA levels significantly decreased in aptamer-treated cells in comparison with cells treated with the control library. Aptamers do not affect hepatitis B viral DNA replication (see Fig. S2 in the supplemental material), implying that the aptamers against E1E2 specifically inhibit HCV replication. All the data strongly suggest that E1E2 aptamer can prevent and suppress HCV infection.
E1E2-specific aptamer does not trigger an innate response in HCV-infected hepatocytes.
Our data showed that an aptamer against E1E2 inhibits HCV infection. The presence of DNA molecules inside or outside the cells may cause a nonspecific induction of IFN, which is likely to lead to an antiviral effect. To exclude the possibility that the inhibition of HCV infection by E1E2 aptamer is due to aptamer-induced innate immunity, we examined the expression of IFN-β and IFN-stimulated genes (ISGs) in aptamer-treated cells. IFN-β was not induced in aptamer-treated hepatocytes (Fig. 4A). The antiviral activity of IFN is carried out by several ISGs. Our previous study demonstrated that the ISGs G1P3 and 1-8U play a role in intracellular antiviral activity (23, 24). We performed real-time PCR analysis to examine the expression of G1P3 and 1-8U. As shown in Fig. 4B and C, E1E2 aptamers did not stimulate the expression of G1P3 and 1-8U in HCV-infected hepatocytes. The data clearly show that aptamer against E1E2 does not induce IFN-β and ISGs in virus-infected hepatocytes. No induction of IFN-β and ISGs in naive hepatocytes was observed with aptamer or control library treatment (see Fig. S3 in the supplemental material). All the data strongly suggest that inhibition of HCV infection by E1E2 aptamer is not due to the innate immune response.
Fig 4.
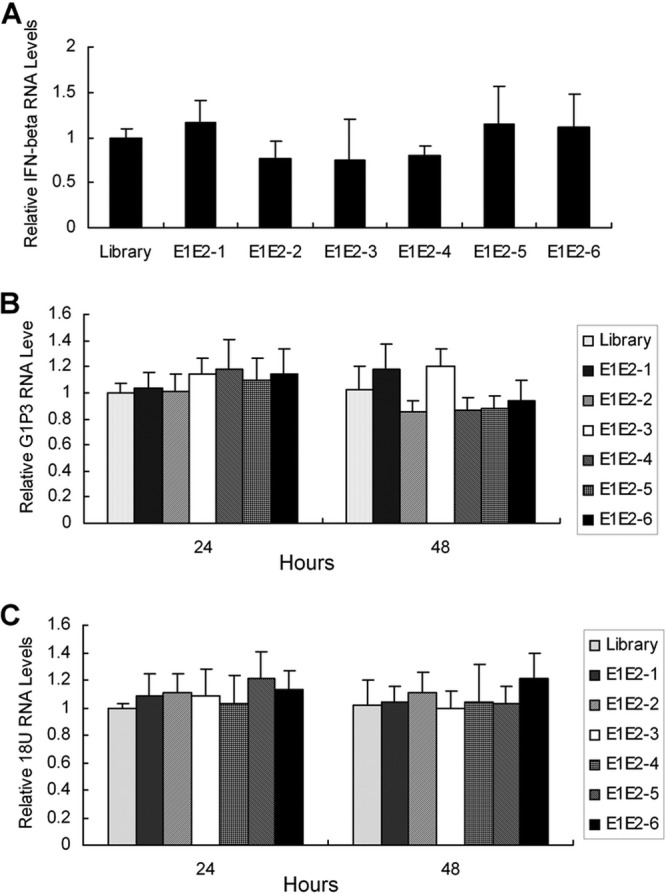
E1E2 aptamers do not trigger an innate immune response in HCV-infected human hepatocytes. (A) E1E2 aptamers do not induce IFN-β in HCV-infected hepatocytes. A virus suspension at a multiplicity of infection of 0.1 containing a 100 nM concentration of each E1E2 aptamer or the control library was used to infect naive Huh7.5 cells. The supernatant was removed at 2 h postinfection. Twenty-four hours later, total cellular RNA was isolated, and IFN-β mRNA was detected by real-time PCR. The expression of IFN-β was normalized to GAPDH. (B and C) E1E2 aptamers do not induce G1P3 and 1-8U in HCV-infected hepatocytes. The cells were treated as described for panel A. The cells were harvested for total cellular RNA extraction at 24 or 48 h postinfection. The expression of G1P3 or 1-8U RNA was examined by real-time PCR and normalized to GAPDH. The data represent the means of three different experiments.
E2 protein is essential for the inhibition of HCV infection by aptamer E1E2-6.
To identify the residues of E1E2 involved in aptamer E1E2-6 binding and inhibition of viral infection, we generated truncated versions of E1E2. As shown in Fig. 5A (left), the expression of E1 or E2 protein could be confirmed using Western blotting. An ELONA was used to determine which truncated version binds to E1E2-6. Aptamer E1E2-6 showed a higher affinity for E2 protein than the control library (Fig. 5A). The data suggest that the binding region of aptamer E1E2-6 is localized inside the E2 protein. To further confirm that aptamer E1E2-6 binds to E2 and inhibits viral infection, we performed competition experiments. E2, E1, or a control protein was mixed with E1E2-6. Then, JFH1 virus containing the mixture was used to infect naive Huh7.5 cells. As shown in Fig. 5B and C, the HCV RNA and NS5A protein levels were markedly higher in Huh7.5 cells infected by virus containing the mixture of E1E2-6 with E2 protein than in cells infected by virus containing the mixture of E1E2-6 with E1 or a control protein. To determine whether E1E2-6 could inhibit binding of HCV virions to cells, we performed virus-cell binding studies at 4°C under conditions in which virus binds to but does not enter the cell. As shown in Fig. 5D and E, when E1E2-6 was present only during virus binding, the viral RNA level was significantly impaired. However, if E1E2-6 was added to cells after virus binding and for the duration of the infection, there was little effect on HCV infection. All the data suggested that aptamer E1E2-6 inhibits HCV infection mainly through blockage of virus binding to cells.
Fig 5.

E2 protein is essential for the inhibition of HCV infection by aptamer E1E2-6. (A) Binding affinity of aptamer E1E2-6 to E2 protein. E1 or E2 was cloned into pET32a. E1 or E2 protein was expressed and purified. The purified protein was detected by Western blotting (left). Biotin-labeled E1E2-6 or the control library was added to the microtiter plate previously coated with streptavidin. Purified E1, E2, or a control protein was added to the plates. An ELONA was performed. The absorbance of each sample was measured at 450 nm and normalized to that of the library. The data represent the means of three different experiments. (B and C) E2 protein is essential for inhibition of HCV infection by aptamer E1E2-6. E1E2-6 aptamer or the control library was mixed with nickel beads conjugated with His-tagged control, E1, or E2 protein. The mixtures were centrifuged, and the supernatant was inoculated with a JFH1 virus suspension at room temperature for 2 h. Huh7.5 cells were infected by the mixtures of JFH1 virus with aptamer or the library, and the supernatant was removed at 2 h postinfection. The cells were harvested at 72 h postinfection. Total cellular RNA was isolated, and HCV RNA was determined by real-time PCR (B). The data were normalized to GAPDH and represent the means of three different experiments. Protein was isolated from the cells (C). NS5A protein was detected with Western blotting. (D and E) E1E2-6 blocks virus binding to the cells. Huh7.5 cells were incubated at 4°C for 2 h in the presence of JFH1 virus at a multiplicity of infection of 0.1 with or without 100 nM E1E2-6. Cells were washed extensively at 4°C and then incubated with or without 100 nM aptamer E1E2-E6 for 48 h at 37°C. Total cellular RNA was isolated from the cells. Viral RNA was quantified by real-time PCR. In panel D, sampling times indicate the following: 0 h, different doses of aptamer only present were during a 2-h adsorption period, and viral RNA was detected immediately after the cells washing; 48 h, aptamer was present only during the 2-h adsorption period, and the cells were cultured at 37°C for 48 h. In panel E, numbers on the x axis indicate the following: −2, aptamer was present only during the 2-h adsorption period, and the cells were cultured at 37°C for 48 h; 0, aptamer was absent during the 2-h adsorption period, and the cells were cultured at 37°C for 48 h with aptamer immediately after cell washing; 1 to 4, aptamer was absent during the 2-h adsorption period and was added at 1, 2, 3, or 4 h, respectively, after cell washing. GE, genome equivalent.
Isolation and characterization of JFH1-resistant variants.
Viral target-based inhibitors allow for the selection of resistant viruses. To identify amino acid substitutions that confer resistance to aptamer E1E2-6, JFH1-infected Huh7.5 cells were treated with 100 nM E1E2-6 for 3 weeks. Control cells were maintained with a 100 nM control library. Substitutions within E1E2 associated with reduced susceptibility to E1E2-6 were selected and identified by sequence analysis of E1E2 cDNA isolated by RT-PCR from control and aptamer-treated cells. A substitution at E1E2 residue 412 (Q412R substitution) was identified.
To evaluate the contribution of the selected specific amino acid substitutions to resistance, the Q412R substitution was introduced into JFH1. The sensitivity of the variant to E1E2-6 was assessed in the infectious cell culture system. The Q412R substitution resulted in a decrease in E1E2-6 potency (Fig. 6A and B). The data suggested that the selected Q412R substitution within E2 is the major resistance substitution identified.
Fig 6.

The Q412R substitution in E2 is the major selective resistance substitution identified. Selection of a resistance-conferring substitution was performed. Huh7.5 cells were infected with medium from Huh7.5 cells transfected with RNA from the wild type (WT) or the selected Q412R viral clone. (A) The effect of aptamer E1E2-6 on RNA replication of the wild-type and the selected Q412R virus was examined as described in the legend of Fig. 3B. (B) The effect of aptamer E1E2-6 on the production of infectious particles of the wild-type or the selected Q412R virus was tested as described in the legend of Fig. 3E. The data are from three independent experiments. Error bars represent means ± standard deviations. *, P < 0.05; **, P < 0.01, versus control cells.
DISCUSSION
Many chronic hepatitis C patients do not respond to the current pegylated-IFN-α-based therapy. Future regimens will incorporate multiple direct-acting antiviral drugs to increase treatment efficacy (25). The protease inhibitors against NS34A protease have been used for the treatment of HCV-infected individuals recently (26), but there are many patients resistant to the therapy (27, 28). It is desirable to seek a combination therapy for HCV infection. It is logical to design antiviral therapies targeting the viral envelope protein essential for virus entry. Antiviral agents inhibiting virus entry may protect cured or healthy hepatocytes. Moreover, inhibition of virus entry can prevent reinfection of liver allografts in HCV-infected patients receiving liver transplants.
Here, we reported the selection of aptamers against E1E2 protein and inhibition of HCV infection by these aptamers. Our aptamers have different sequences from those of other recently described DNA aptamers selected for E2 (29). The difference might be due to the selection technique used. We used SELEX to select aptamers against E1E2 protein in our study although Cell-SELEX against hepatocytes expressing E2 was used in the study of Chen et al. (29). Moreover, the differences may also be attributed to the source of envelope protein since our aptamers were selected by using E1E2 from genotype 2a JFH1, whereas those obtained in the previous study were isolated by using E2 of genotype 1a.
Our study showed that the aptamers selected by using E1E2 of genotype 2a JFH1 not only bound E1E2 protein and inhibited the infection of genotype 2a JFH1 but also blocked genotype 1a H77S infection. Our data indicate that the E1E2 proteins from these two genotypes of HCV may share aptamer binding sites.
Although the aptamer E1E2-6 was found to bind E2 protein, the specific residues involved in the interaction between E1E2-6 and E2 remain to be investigated. Determination of these residues may provide information about the essential functional regions of E2. In addition, aptamers against E1E2 can be used with E1E2 to understand the mechanisms of HCV entry and fusion. The interaction between HIV reverse transcriptase and its aptamers provides a model for the study of the mechanism of how the aptamers act as broad-spectrum inhibitors of reverse transcriptase (30). Anti-HIV Gag protein aptamers can be used to examine Gag and HIV RNA interactions (31). These examples display the potential use of aptamers in the structure-function studies of virology.
One recent study reported that inhibition of viral infection by aptamers might be due to the aptamer-induced innate immune response (32). This was one exceptional example because RIG-I aptamer was designed to have specific motifs to bind and activate RIG-I in this study. However, many studies suggest that inhibition of viral infection by aptamers targeting viral proteins is attributed to the suppression of the function of viral proteins by aptamers (33–36). In accordance with these studies, our study showed that an aptamer for HCV E1E2 does not induce the expression of IFN-β and ISGs, indicating that inhibition of HCV infection by an E1E2-specific aptamer is not due to the innate immune response.
The study showed that aptamers against E1E2 protein block viral infection, thereby inhibiting HCV RNA replication. Although efficient delivery of DNA aptamers across cell membranes remains a challenge, aptamers for HCV E1E2 need not enter into the cells. E1E2 aptamers may act as inhibitors of HCV infection, and they represent powerful drugs for the treatment of HCV infection in the near future.
In summary, our study provides the first evidence of direct antiviral activity of aptamers for HCV E1E2 in an infectious cell culture system. These results demonstrate the power of the SELEX approach for the selection of inhibitors for viral infection and the exploration of the mechanisms of viral infection. The data indicate that an aptamer against HCV E1E2 protein exerts its antiviral effects through inhibition of virus binding to host cells and thus may hold promise for the development of aptamers as therapeutic drugs for hepatitis C patients.
Supplementary Material
ACKNOWLEDGMENTS
We thank Charles M. Rice (Rockefeller University, New York, NY) for the gift of Huh7.5 and FL-Neo cells, Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for pJFH1 and pJFH1/GND plasmids, and Stanley M. Lemon (University of North Carolina, Chapel Hill, NC) for pH 77-S and pH 77-S/ΔE1P7 plasmids.
This work was supported by grants from the National Science and Technology Major Project of the Ministry of Science and Technology of China (2009ZX10004-312), National Natural Science Foundation of China (81271885), and Program for New Century Excellent Talents in University (NCET-09-0339) to H.Z.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00897-13.
REFERENCES
- 1.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int. 29(Suppl 1):74–81 [DOI] [PubMed] [Google Scholar]
- 2.Zeuzem S, Andreone P, Lawwitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Müllhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M, REALIZE Study Team 2011. Telaprevir for retreatment of HCV infection. N. Engl. J. Med. 364:2417–2428 [DOI] [PubMed] [Google Scholar]
- 3.Sakamoto N, Watanabe M. 2009. New therapeutic approaches to hepatitis C virus. J. Gastroenterol. 44:643–649 [DOI] [PubMed] [Google Scholar]
- 4.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 5.von Hahn T, Rice CM. 2008. Hepatitis C virus entry. J. Biol. Chem. 283:3689–3693 [DOI] [PubMed] [Google Scholar]
- 6.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941 [DOI] [PubMed] [Google Scholar]
- 7.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805 [DOI] [PubMed] [Google Scholar]
- 9.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626 [DOI] [PubMed] [Google Scholar]
- 11.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellington AD, Szostak JW. 1990. In vitro selection of RNA molecules that binds specific ligands. Nature 346:818–822 [DOI] [PubMed] [Google Scholar]
- 14.Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510 [DOI] [PubMed] [Google Scholar]
- 15.Tan W, Wang H, Chen Y, Zhang X, Zhu H, Yang C, Yang R, Liu C. 2011. Molecular aptamers for drug delivery. Trends Biotechnol. 29:634–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keefe AD, Pai S, Ellington A. 2010. Aptamers as therapeutics. Nat. Rev. Drug Discov. 9:537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng EW, Shima DT, Calias P, Cunninggham ET, Guyer DR, Adamis AP. 2006. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 5:123–132 [DOI] [PubMed] [Google Scholar]
- 18.Reyes-Reyes EM, Teng Y, Bates PJ. 2010. A new paradigm for aptamer therapeutic AS1411 action: uptake by macropinocytosis and its stimulation by a nucleolin-dependent mechanism. Cancer Res. 70:8617–8629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. 2002. High permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. 2006. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. U. S. A. 103:2310–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang D, Liu N, Zuo C, Lei S, Wu X, Zhou F, Liu C, Zhu H. 2011. Innate host response in primary human hepatocytes with hepatitis C virus infection. PLoS One 6:e27552. 10.1371/journal.pone.0027552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu H, Dong H, Eksioglu E, Hemming A, Cao M, Crawford JM, Nelson DR, Liu C. 2007. Hepatitis C virus triggers cell death through innate intracellular antiviral defense system. Gastroenterology 133:1649–1659 [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Zhao H, Collins CD, Eckerrode SE, Ruan Q, McIndoe RA, Crawford JM, Nelson DR, She JX, Liu C. 2003. Gene expression associated with interferon alfa antiviral activity in an HCV replicon cell line. Hepatology 37:1180–1188 [DOI] [PubMed] [Google Scholar]
- 24.Yao L, Dong H, Zhu H, Nelson DR, Liu C, Lambiase L, Li X. 2011. Identification of IFITM3 gene as an inhibitors of hepatitis C viral translation in a stable STAT1 cell line. J. Viral. Hepat. 18:e523–e529. 10.1111/j.1365-2893.2011.01452.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Targett-Adams P, Graham EJ, Middleton J, Palmer A, Shaw SM, Lavender H, Brain P, Tran TD, Jones LH, Wakenhut F, Stammen B, Pryde D, Pickford C, Westby M. 2011. Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action. J. Virol. 85:6353–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. 2011. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant virus. Gastroenterology 141:1067–1079 [DOI] [PubMed] [Google Scholar]
- 27.Shindo H, Maekawa S, Komase K, Sueki R, Miura M, Kadokura M, Shindo K, Amemiya F, Kitamura T, Nakayama Y, Inoue T, Sakamoto M, Okada SI, Asahina Y, Izumi N, Honda M, Kaneko S, Enomoto N. 2012. Characterization of naturally occurring protease inhibitor-resistance mutations in genotype 1b hepatitis C virus patients. Hepatol. Int. 6:482–490 [DOI] [PubMed] [Google Scholar]
- 28.Halfon P, Locarnini S. 2011. Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 55:192–206 [DOI] [PubMed] [Google Scholar]
- 29.Chen F, Hu Y, Li D, Chen H, Zhang XL. 2009. CS-SELEX generates high-affinity ssDNA aptamers as molecular probes for hepatitis C virus envelope glycoprotein E2. PLoS One 4:e8142. 10.1371/journal.pone.0008142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ditzler MA, Bose D, Shkriabai N, Marchand B, Sarafiano SG, Kvaratskhelia M, Burke DH. 2011. Broad-spectrum aptamer inhibitors of HIV reverse transcriptase closely mimic natural substrates. Nucleic Acids Res. 39:8237–8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramalingam D, Duclair S, Datta SAK, Ellington A, Rein A, Prasad VR. 2011. RNA aptamers directed to human immunodeficiency virus type 1 Gag protein bind to matrix and nucleocapsid domains and inhibit virus production. J. Virol. 85:305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang SY, Sun HY, Lee KH, Oh BH, Cha YJ, Kim BH, Yoo JY. 2012. 5′-Triphosphate-RNA-independent activation of RIG-I via RNA aptamer with enhanced antiviral activity. Nucleic Acids Res. 40:2724–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Melckebeke H, Devany M, Di Primo C, Beaurain F, Toulme JJ, Bryce DL, Boisbouvier J. 2008. Liquid-crystal NMR structure of HIV TAR RNA bound to its SELEX RNA aptamer reveals the origins of the high stability of the complex. Proc. Natl. Acad. Sci. U. S. A. 105:9210–9215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore MD, Cookson J, Coventry VK, Sproat B, Rabe L, Cranston RD, McGowan I, James W. 2011. Protection of HIV neutralizing aptamers against rectal and vaginal nucleases: implications for RNA-based therapeutics. J. Biol. Chem. 286:2526–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wheeler LA, Trifonova R, Vrbanac V, Basar E, McKernan S, Xu Z, Seung E, Deruaz M, Dudek T, Einarsson JI, Yang L, Allen TM, Luster AD, Tager AM, Dykxhoorn DM, Lieberman J. 2011. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using C4 aptamer-siRNA chimeras. J. Clin. Invest. 121:2401–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bentham M, Holmes K, Forrest S, Rowlands DJ, Stonehouse NJ. 2012. Formation of higher-order foot-and-mouth disease virus 3Dpol complexes is dependent on elongation activity. J. Virol. 86:2371–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.